

Abstract
Gnotobiotic mouse model is generally used to evaluate the efficacy of gut microbiota. Sex differences of gut microbiota are acknowledged, yet the effect of recipient’s gender on the bacterial colonization remains unclear. Here we inoculated male and female germ-free C57BL/6J mice with fecal bacteria from a man with short-term vegetarian and inulin-supplemented diet. We sequenced bacterial 16S rRNA genes V3-V4 region from donor’s feces and recipient’s colonic content. Shannon diversity index showed female recipients have higher bacteria diversity than males. Weighted UniFrac principal coordinates analysis revealed the overall structures of male recipient’s gut microbiota were significantly separated from those of females, and closer to the donor. Redundancy analysis identified 46 operational taxonomic units (OTUs) differed between the sexes. The relative abundance of 13 OTUs were higher in males, such as Parabacteroides distasonis and Blautia faecis, while 33 OTUs were overrepresented in females, including Clostridium groups and Escherichia fergusonii/Shigella sonnei. Moreover, the interactions of these differential OTUs were sexually distinct. These findings demonstrated that the intestine of male and female mice preferred to accommodate microbiota differently. Therefore, it is necessary to designate the gender of gnotobiotic mice for complete evaluation of modulatory effects of gut microbiota from human feces upon diseases.
The human gastrointestinal tract carries up to 1014 bacteria more than ten-fold the number of human cells in the body, which is causally related to human health via yielding metabolites, and/or interacting with the immune systems1,2. In the last decade, germ-free mouse model has been widely applied to demonstrate gut microbiota as a contributing factor in some human diseases3,4,5. As known, germ-free mice were resistant to high-fat and high-sugar diet-induced obesity3, and when they were transplanted the gut microbiota from the obese mice or human, they became adiposity4,6. On the contrary, transfer of gut microbiota from lean donors improved insulin sensitivity in individuals with metabolic syndrome7. That is to say that transplantation of the intact gut microbiota to germ-free mice is a good tool to study the function of the gut microbiota in the development and treatment of gut microbiota-related diseases.
However, gut microbiota showed sex bias in either human beings or animals5,8,9,10,11,12. It was identified that Bacteroidetes showed higher abundance in men than women according to an American study8. A cross-sectional study in European countries also showed that Bacteroides-Prevotella group was more prominent in male than female11. Bacteroides thetaiotaomicron was also identified as being higher in men than women in Chinese population10. Moreover, male mice held lower bacterial diversity12, and higher Parabacteroides belonging to Bacteroidetes than females, consistent with the above results of clinical trial5.
Intriguingly, the compositional and metabolic changes of gut microbiota response to diet, exogenous bacteria or genetic deletion also demonstrated sexual dimorphism13,14,15,16. Whether in wild and laboratory fish, laboratory mice or in human beings, the diet made different effects on gut microbiota composition between sexes13. An oligofructose-supplemented diet increased the abundance of Bacteroidetes in female rats, yet it did not change fecal community structure even if the butyrate levels increased in males14. Butyrate has been shown to attenuate gut inflammation by multiple mechanisms17,18,19,20,21. Probiotics also exerted dissimilar responses on female and male mice. After probiotic consumption, two genera Staphylococcus and Roseburia in Firmicutes were overrepresented in females, while butyrate and acetate were increased in males15. Deletion of Bmal1, a gene encoding a core molecular clock component, induced changes in the microbial composition based on sexes. In male mice, it decreased Proteobacteria (including Helicobacter and Sutterella) and increased TM7 (including F16 spp.); while in female mice, Cyanobacteria and Tenericutes were increased, but Proteobacteria and TM7 were unchanged16.
More importantly, sex differences in the gut microbiota play a key role in gender-related diseases5,12. Recently, it was found that the gut microbiome from specific pathogen-free (SPF) nonobese diabetic (NOD) male mice could protect the immature recipients of female from type 1 diabetes (T1D) by altering microbiota, elevating testosterone, and reducing islet inflammation and autoantibody production5.
Hence, sex is an important determinant to gut microbiota, and involves in the development of disease. However, how sex influence human gut microbiota colonization in germ-free mice has not been characterized so far. We wonder whether an intact human gut microbiota colonizes differently between male and female recipient mice, and what the differences are. Herein, we inoculated fecal suspension of a healthy man with short-term vegetarian and inulin-supplemented diet to male and female germ-free mice. By using high-throughput sequencing and bioinformatics analysis, we showed the colonization of gut microbiota was different in diversity, overall structure, and individual phylotype, and explored the interactions among differential bacteria between male and female recipient mice. These results provided a direct evidence for sexual dimorphism of gut microbiota colonization, which would suggest that both sexes of gnotobiotic mice should be applied to investigate the effects of gut microbiota.
Results
Alpha-diversity analysis of gut microbiota of male and female recipient
To investigate whether gut microbiota derived from the same donor colonize in the recipients’ gut in a sex-dependent manner, we performed bacterial 16S rRNA gene V3-V4 region sequencing for the donor, 10 male and 9 female recipient’s colonic content samples on the Illumina MiSeq platform. A total of 698,564 usable high-quality raw reads were obtained. The sequences were binned into 2934 OTUs at the 97% similarity level. After chimeras and singleton removing, there remained 554 OTUs. Among those, 200, 148 ± 31 ((74 ± 5)% of the total OTUs in the donor), and 154 ± 38 ((77 ± 5)% of the total OTUs in the donor) OTUs existed in the gut of the donor, male and female recipients, respectively (Supplementary Figure S1A,B). There were 134 OTUs shared between the donor and the recipients, and 66 and 354 OTUs were unique in the donor and recipients, respectively. Theoretically, all the OTUs in recipient should come from the donor. The unique OTUs found in recipients might be dormant species in the donor’s gut that below the detection limit, and then become dominant after transplanted into mice. Shannon diversity indices were significantly higher in female recipient than male one (Fig. 1A,B), which suggested that more bacteria colonized in the female mice after inoculated with fecal microbiota of the donor. Observed Species curves showed that there was an increase trend in female mice than male mice, but the difference did not reach statistical significance (Supplementary Figure S2A,B).
Figure 1. The colonization of gut microbiota in female recipient have higher alpha-diversity than that in male recipient.
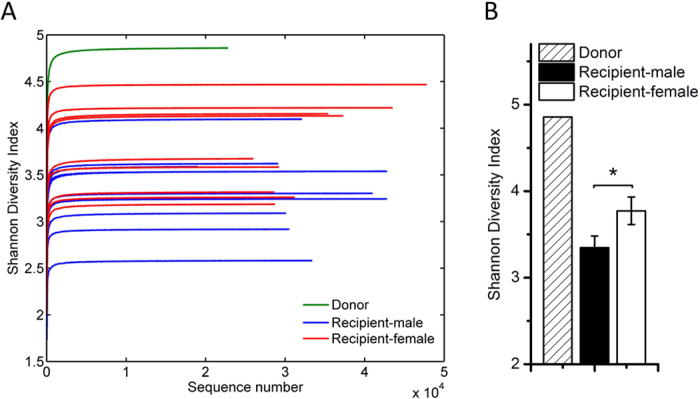
(A) Shannon Diversity Index curves; (B) Shannon diversity index of each group. Data were shown as means ± SEM. Differences were assessed by Mann-Whitney test. *P < 0.05. n = 1 for Donor; n = 10 for Recipient-male group; n = 9 for Recipient-female group.
Taxonomy-based comparisons of gut microbiota at the phylum, family and genus levels
Taxonomic assignment showed that there were 4 phyla, 28 families and 58 genera in the feces of the donor and recipient (Supplementary Figure S3). The 4 phyla included Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria (Supplementary Figure S3A). Actinobacteria decimated in the recipient mice (Supplementary Figure S3A). The dominant families included Lachnospiraceae, Porphyromonadaceae, Ruminococcaceae, Erysipelotrichaceae, Enterobacteriaceae, Prevollaceae, and Bifidobacteriaceae (Supplementary Figure S3B). Among them, Lachnospiraceae and Porphyromonadaceae were increased, and Prevollaceae and Bifidobacteriaceae were decreased in the recipient (Supplementary Figure S3B). The dominant genera included Clostridium XIVa, Parabacteroides, Blautia, Gemmiger, Faecalibacterium, Bifidobacterium, Bacteroides, Ruminococcus, Escherichia/Shigella, Clostridium XVIII, Erysipelotrichaceae_incertae_sedis, Anaerostipes, Butyricicoccus, Clostridium IV and so on (Supplementary Figure S3C).
In the donor’s gut, there were high abundance of Faecalibacterium (14.07%), Prevotella (11.31%), Bifidobacterium (10.69%), Bacteroides (8.32%), Parabacteroides (8.31%), Clostridium IV (2.87%), Blautia (1.22%), and Ruminococcus (1.04%) (Supplementary Figure S4). Nine genera were more than 1% of total bacteria in the donor man, while 12 genera in the recipient mice (Supplementary Figure S4). In these dominant genera, Faecalibacterium, Prevotella, Bifidobacterium, Bacteroides, Dialister, and Clostridium IV were decreased in the recipient’s gut, and Clostridium XIVa, Parabacteroides, Blautia, Gemmiger, Ruminococcus, Escherichia/Shigella, Clostridium XVIII, Erysipelotrichaceae_incertae_sedis, Anaerostipes, and Butyricicoccus were enriched in recipient (Supplementary Figure S4). Therefore, bacteria originating from a human intestinal microbiota selectively colonized in the gut of germ-free mice, and the dominant bacteria in the human donor were replaced by other higher abundant bacteria in recipient mice.
Statistically, the relative abundance of 1 phylum: Proteobacteria; 4 families: Erysipelotrichaceae, Enterobacteriaceae, Clostridiaceae, and Peptostreptococcaceae; and 11 genera: Blautia, Escherichia/Shigella, Clostridium XVIII, Clostridium IV, Flavonifractor, Clostridium sensu stricto, Holdemania, Clostridium XI, Anaerotruncus, Clostridium XlVb, Dorea were found to be significantly higher in the female recipient than the male recipient (p < 0.05, Table 1). These results suggested gut microbial colonization was sex-dependent in different taxonomic level such as phylum, family and genus.
Table 1. List of taxa that were significantly different between male and female recipient.
| Taxon | P value | FDR | Relative abundance (%) |
Fold | |
|---|---|---|---|---|---|
| Recipient-male | Recipient-female | ||||
| Phylum | |||||
| Proteobacteria | 0.0004 | 0.0022 | 0.18 ± 0.12 | 2.31 ± 0.43 | 12.8 |
| Family | |||||
| Erysipelotrichaceae | 0.0090 | 0.0868 | 2.05 ± 0.24 | 3.17 ± 0.23 | 1.60 |
| Enterobacteriaceae | 0.0004 | 0.0129 | 0.18 ± 0.12 | 2.31 ± 0.43 | 12.8 |
| Clostridiaceae | 0.0015 | 0.0210 | 0.06 ± 0.02 | 0.50 ± 0.17 | 8.30 |
| Peptostreptococcaceae | 0.0135 | 0.0976 | 0.01 ± 0.00 | 0.17 ± 0.07 | 17.0 |
| Genus | |||||
| Blautia | 0.0071 | 0.0693 | 2.27 ± 0.49 | 6.79 ± 1.57 | 3.00 |
| Escherichia/Shigella | 0.0004 | 0.0262 | 0.18 ± 0.12 | 2.30 ± 0.43 | 12.8 |
| Clostridium XVIII | 0.0071 | 0.0693 | 0.97 ± 0.11 | 1.51 ± 0.13 | 1.60 |
| Clostridium IV | 0.0019 | 0.0377 | 0.49 ± 0.05 | 1.15 ± 0.21 | 2.30 |
| Flavonifractor | 0.0338 | 0.1532 | 0.41 ± 0.06 | 0.83 ± 0.18 | 2.00 |
| Clostridium sensu stricto | 0.0015 | 0.0377 | 0.06 ± 0.02 | 0.50 ± 0.17 | 8.30 |
| Holdemania | 0.0055 | 0.0693 | 0.15 ± 0.02 | 0.25 ± 0.02 | 1.70 |
| Clostridium XI | 0.0135 | 0.0993 | 0.01 ± 0.00 | 0.17 ± 0.07 | 17.0 |
| Anaerotruncus | 0.0338 | 0.1532 | 0.03 ± 0.01 | 0.06 ± 0.01 | 2.00 |
| Clostridium XlVb | 0.0322 | 0.1532 | 0.02 ± 0.01 | 0.04 ± 0.01 | 2.00 |
| Dorea | 0.0085 | 0.0713 | 0.00 ± 0.00 | 0.03 ± 0.02 | — |
Note: Data were shown as means ± SEM. Differences were assessed in QIIME.
Comparison of overall structural base on OTU level of gut microbiota
To provide an overview of the gut microbiota composition of the donor and the two recipient groups, principal coordinate analysis (PCoA) based on weighted UniFrac distances of OTU relative abundance matrix was performed. Plotted PCoA scores clearly separated the donor, the male recipient group and the female recipient group (Fig. 2A). Permutational multivariate analysis of variance (PERMANOVA) derived from weighted UniFrac distance confirmed a significant separation between the microbiota of male and female mice (p = 0.0149, Fig. 2A). Moreover, the distance to the donor were significantly lower in male recipient than female, suggesting that male recipient had more similar gut microbiota to the donor’s, compared with female recipient (Fig. 2B).
Figure 2. The overall structure of colonic content microbiota of male recipient mice was separated from that of female recipient, and closer to the donor.
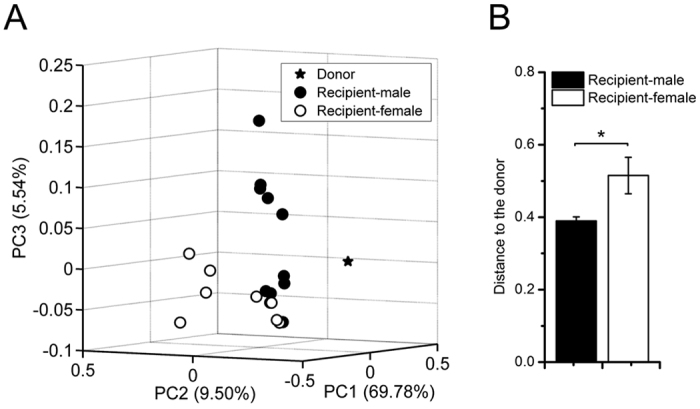
(A) PCoA score plot based on weighted UniFrac metrics. Each point represented the fecal microbiota of a mouse. (B) Weighted UniFrac distances to the donor of the two groups. Differences were assessed by Mann-Whitney test. *P < 0.05. n = 1 for Donor; n = 10 for Recipient-male group; n = 9 for Recipient-female group.
PCoA plot and PERMANOVA based on unweighted UniFrac and Bray-Curtis distances also confirmed that the overall structure of gut microbiota was significantly different between male and female groups (p < 0.05, Figure S5). However, the distances to the donor of the male and female recipient based on these algorithms were not significantly different. It was weighted UniFrac but not Euclidean/Bray-curtis/unweighted UniFrac algorithms that showed differences between the distances to the donor of male and female mice, and this might because abundance and evolutionary distance of the gut bacteria together determined the overall structure deviation from the donor.
Comparison of gut microbiota composition at the phylotype level
Redundancy analysis (RDA) models was employed to identify specific bacterial phylotypes for which the abundance was different between the two groups of mice (Fig. 3). Gut microbiota was significantly different between the male and female recipient (p = 0.008, Fig. 3A). Differential OTUs were revealed along the canonical axis, which explained 8.1% of the variance in OTU abundance (Fig. 3A).
Figure 3. Forty-six bacterial OTUs that were different in abundance between male and female recipient according to redundancy analysis (RDA).

(A) Biplot of the RDA between male and female recipient on relative abundance of OTUs. Constrained explanatory variables were indicated by red triangles. Blue arrows indicated the OTUs with more than 20% variability in values explained by the canonical axis. Upper left showed P-value of Monte Carlo Permutation Test. (B) Heatmap of the abundance of the 46 OTUs. Rows corresponded to 13 OTUs enriched in male recipient (blue), and 33 OTUs more in female recipient (red). Columns represented each mouse in the two groups. The taxonomy of the OTUs (family/genus/species) was shown on the right.
There were 46 OTUs different between male and female recipient mice, with 13 OTUs higher in male mice and 33 OTUs enriched in female mice (Fig. 3B). The male-biased OTUs including the representatives from Parabacteroides distasonis were highly abundant in that group. The OTUs higher in abundance in the female recipient included the bacteria belonging to Clostridium sensu stricto (Clostridium disporicum and Clostridium paraputrificum), Clostridum XIVa (Eisenbergiella tayi, Clostridium lavalense), Clostridium XIVb, and Clostridium XVIII (Erysipelatoclostridium ramousum, Clostridium spiroforme), Clostridium innocuum, Flavonifractor plautii (formerly Clostridium orbiscindens), Holdemania filiformis, and Escherichia fergusonii/Shigella sonnei (Fig. 3B). Blautia and Clostridium IV have different behaviors among OTUs. Blautia faecis and otu1845 from Clostridium IV were higher in male recipient, while Blautia wexlerae and Clostridium leptum (belonging to Clostridium IV) were enriched in female recipient (Fig. 3B). Additionally, the relative abundance of some OTUs (such as otu1770, 923, 749, 71 and 2001) were more similar among the mice in the same cage than those in another cage in each gender, just as shown in the previous studies22,23,24. These results indicated that the gut microbiota of human donor differentially colonized in recipient mice of different sexes at the species level.
Then, a linear discriminant analysis (LDA) effect size (LEfSe) was used to confirm the above results (Supplementary Figure S6). Thirty-four bacteria OTUs were different in abundance between male and female recipient (Supplementary Figure S6B). Ten OTUs were higher in male mice, including Parabacteroides distasonis, Blautia faecis and otu1845 from Clostridium IV; while 24 OTUs were higher in female mice, such as Clostridium paraputrificum, Clostridium lavalense, Clostridium leptum, Flavonifractor plautii, Holdemania filiformis, and Escherichia fergusonii/Shigella sonnei (Supplementary Figure S6B). Among these OTUs, totally 21 OTUs were also identified according to RDA (Fig. 3, Supplementary Figure S6C), and in line with the taxonomy differences (Table 1), which indicated these bacterial phylotypes were indeed closely related to sex. Also, there were 13 and 25 OTUs were uniquely identified by LEfSe and RDA, respectively (Supplementary Figure S6C), because the principles and algorithms were different between the two methods25,26. Although these OTUs were divergent, they belonged to the same taxa, such as Lachnospiraceae, Ruminococcaceae, Clostridium XIVa, and Clostridiales (Supplementary Figure S6C).
Comparison of correlation networks of differential OTUs between two sexes of recipient
To identify the symbiosis and competition relationship among these sexually differential phylotypes, Spearman’s rank correlation test was employed (Fig. 4). In male recipient, a co-abundance cluster was formed among some bacteria with relatively low abundance, such as Holdemania filiformis, Clostridium paraputrificum, Clostridium lavalense, Clostridium XIVb, Clostridium IV, Escherichia fergusonii/Shigella sonnei, etc; and high abundant Parabacteroides distasonis in male was significantly negatively correlated with 9 OTUs, such as Clostridium lavalense, Clostridium XIVb, Clostridium IV, etc, which were in the co-abundance cluster (Fig. 4A). Meanwhile, the co-abundance cluster in the female recipient was composed of highly abundant OTUs including Eisenbergiella tayi, Clostridium disporicum, and Flavonifractor plautii. And Parabacteroides distasonis was significantly negatively correlated with Clostridium disporicum, Rominococcaceae and Oscilibacter (Fig. 4B). All above indicated that the interactions among these differential OTUs were different between the two sexes.
Figure 4. Gut microbial symbiosis and competition were distinct in male and female recipient.

(A) Co-abundance network of differential OTUs in Recipient-male group; (B) Co-abundance network of differential OTUs in Recipient-female group. Blue nodes showed OTUs with higher abundance in male recipient. Red nodes showed OTUs higher in abundance in female recipient. The size of the nodes indicated OTU abundance. Connecting lines represented Spearman correlation coefficient values above 0.5 (red) or below −0.5 (green).
Discussions
Although germ-free mice were applied broadly in studying the etiological effects of gut microbiota, the sexual disparity of human gut microbiota colonization in germ-free mice largely remains unknown. In this study, we found that female recipients had significantly different gut microbiota colonization compared to males. The differences might lie in the following four aspects: (1) Female recipients had higher bacteria diversity than male; (2) Overall structure of gut microbiota of female recipient were significantly separated from those of male recipient, and farther away from the donor’s than male recipient; (3) The differences of gut microbiota colonization between two sexes occurred at the species level; (4) The interactions among the differential OTUs were sexually distinct. Our results indicated that sex was a force shaping gut microbiota.
In this study, we compared the gut microbiota composition of male and female germ-free recipient mice after transplanting fecal bacteria from a healthy adult man. Shannon diversity analysis revealed that female recipient’s gut had bacteria with higher species diversity than that of male. In a previous study, post-pubescent female mice harbored bacteria with higher Shannon diversity index than males, which was in concert with our results12. Additionally, we demonstrated that it was significantly distinct between the gut microbial overall structure of male and female recipient by using unsupervised multivariate analysis. Furthermore, we performed the supervised multivariate statistical analysis to identify the species-level phylotypes causing these differences. In male recipient, the enriched species included some potentially beneficial bacteria, such as Parabacteroides distasonis (belonging to Bacteroidetes) and Blautia faecis, which was consistent with previous researches5,8,10,11. P. distasonis could reduce the severity of acute and chronic colitis in the dextran sulphate sodium-induced mice model by modulating immunity and microbiota composition27. B. faecis, a kind of butyrate-producing bacteria, was significantly reduced in the patients with Crohn’s disease compared to healthy individuals28. While, female recipient mice prefered Clostridium groups (including cluster sensu stricto, XIVa, XIVb, IV, and XVIII), Flavonifractor plautii, and Holdemania filiformis, of which many produced butyrate or acetate29,30,31,32,33,34. Many previous studies showed butyrate and acetate had anti-inflammatory properties17,18,19,20,21,35. However, the female-biased phylotypes also included some opportunistic pathogen, such as Clostridium disporicum (belonging to Clostridium sensu stricto), Clostridium leptum (belonging to Clostridium IV), Clostridium spiroforme (belonging to Clostridium XVIII), Clostridium innocuum, and Escherichia fergusonii/Shigella sonnei, which were implicated in the development of infection or colitis36,37,38,39,40,41,42. Moreover, the interactions among these bacteria were different as well, suggesting that the gut of male and female mice have ecologically distinct environments.
Hereinabove, male and female recipients were colonized by different bacteria, which showed different or even opposite effects on diseases. This fact might account for why male and female individuals reacted differently to the same disease. So fecal microbial transplantation to germ-free mice from more donors should be carried out to verify the above results in the future. In that case, it is recommended to take a comprehensive picture of relationship between gut microbiota and disease through utility of both sexes of germ-free mice model.
Although female recipients had more variety of bacteria than males, especially more butyrate/acetate-producing bacteria and opportunistic pathogen, the overall structure of their gut microbiota was farther away from that of the donor than male recipient. The overgrowth of some bacteria in female mice’s gut might lead to the more significant deviation of the whole gut microbiota from the male donor than male recipient. It might reflect that the gut microbiota colonized in the mice of the same gender was more similar to that from the donor. In order to fully illustrate this point, it would be necessary to compare the colonization pattern of gut microbiota from female donor in two sexes of mice.
Several mechanisms might involve in sex modulation of gut microbiota colonization. Host sex hormones can modulate microbiota composition5,12. Sex differences in gut microbiota did not appear before puberty, and removal of the androgen source by castration would drive male microbiota closer to female microbiota, which suggested that sexual maturation and sexual hormones were major determinants of the gut microbial community structure difference between two genders5,12. In addition, immune function differed between sex, with more macrophage numbers and inflammatory cytokines secretion in male, influenced gut microbiota composition5. Our study did not clarify the mechanisms driving sexual differentiation of gut microbiota colonization at present. Instead, we demonstrated for the first time that sex did control microbiota colonization pattern in adult germ-free mice.
In brief, our results represented an example of microbiota colonization depending on sexes of germ-free mice. Female recipient mice processed higher bacteria diversity, unique overall structure and individual phylotype pattern, and different bacteria interactions in comparison to the male recipient. These findings suggested that the sex of gnotobiotic mice should be taken into account when investigating modulatory effects of gut microbiota on diseases, and it’s better to tailor the host sex in the treatment of diseases through “gut microbiota-targeted” strategies according to the bacterial colonization ability.
Materials and Methods
Animal experiments
The donor was a 25-year-old healthy man, without medication for at least 6 months. Before donating the stool, he had 7 days of vegetarian diet and inulin intake, in order to increase the abundance of potential beneficial bacteria43,44,45. Written informed consent was obtained from this donor. Fresh stool was collected and diluted 5-fold and homogenized in sterile pre-reduced 0.01 M phosphate buffer saline (pH 7.4). Food derived debris of too large size was removed through sterile gauze.
Ten male and ten female germ-free C57BL/6J mice (6–8 weeks old) were purchased from Shanghai Laboratory Animal Center (SLAC), Chinese Academy of Sciences, Shanghai, China. They were fed on normal chow diet provided by SLAC. After two weeks’ acclimatization, the 20 mice were divided into 2 groups: male group and female group. The ten mice in each group were housed in two cages (five per cages). The human fecal suspension was administered orally by gavage to the germ-free mice at a dosage of 100 ul/mice once daily in the first two days. The trial lasted for 7 days. One female mouse died after inoculation. At the end of the trial, colonic contents of each mouse were collected, and frozen in liquid nitrogen immediately for further analysis.
The use of human fecal sample was approved by Medical Ethics Committee of Shanghai General Hospital (No. 2016KY072), and all experiments were performed in accordance with the relevant guidelines and regulations. All the animal experiments were carried out in strict accordance with the Guidelines for Care and Use of Laboratory Animals of SLAC. The protocol was approved by the Institutional Animal Care and Use Committee of SLAC (SLAC-20160320001).
Sequencing of gut bacterial 16S rRNA gene V3-V4 region on the Illumina MiSeq platform
Genomic DNA was extracted from colonic content samples collected from two groups of mice using bead beating and an InviMag® DNA kit (Invitek, Berlin, Germany) method as reported previously46. The extracted DNA from each sample was used as the template to amplify the V3-V4 region of the 16S rRNA gene. The bacterial universal primer pair in the V3-V4 region consisted of the forward primer 5′-TCGTCGGCAGC GTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′and the reverse primer 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGG GTATCTAATCC-3′. A subsequent limited-cycle amplification step was performed to add multiplexing indices and Illumina sequencing adapters. Libraries were normalized and pooled, and sequenced on the MiSeq platform according to the manufacturer’s instructions (Illumina, CA, USA).
Bioinformatics and statistical analysis of sequencing data
All raw reads were sorted into different samples according to the multiplexing indices. High-quality sequences were selected under the following criteria by using Usearch 7.0.1090: 1) sequences at both ends should be cut at the nucleic acid bases whose quality was lower than Q2; 2) the overlap of splicing sequences should be longer than 50 bp, and the length of complete sequences should be longer than 400 bp, and 3) the expected error of the complete sequences should be less than 1 base. PCR primers were truncated out afterwards.
Bioinformatics analysis of sequencing data was performed using QIIME (v1.8.0)47. All high-quality sequences were aligned against Greengenes core dataset with by PyNAST multialigner, and percent identity ≥75% with the reference sequences were regarded as bacterial sequences and used for further analysis. Operational taxonomic units (OTU) delineation was performed with UCLUST at 97% similarity level. Chimera sequences identified by Uchime and singletons were discarded. Bacterial diversity was assessed with the Shannon Diversity Index and rarefaction analysis based on abundance of OTU sequences. Principal coordinate analysis (PCoA) was performed on unweighted and weighted UniFrac, Bray-Curtis and Euclidean distances of the relative abundances (normalized for each sample) of OTUs to visualize whether there was segregation of gut microbiota structure between two animal groups or not. The statistical significance of PCoA plots of two groups was assessed by permutational multivariate analysis of variance (PERMANOVA) test in MATLAB R2010a (The MathWorks, Inc., Natick, MA, USA).
To identify OTUs separating the two groups, redundancy analysis (RDA) models and linear discriminant analysis effect size (LEfSe) were contrasted based on the relative abundance of OTUs. RDA was performed with Canoco for Windows 4.5 (Microcomputer Power, NY, USA) according to the manufacturer’s instructions. Statistical significance was assessed by Monte Carlo Permutation Procedure (MCPP) with 499 random permutations under the full model. In LEfSe, OTUs were picked out when alpha value of the factorial Kruskal–Wallis test was <0.05, and the logarithmic LDA score is >2.0: (http://huttenhower.sph.harvard.edu/galaxy).
Taxonomical assignments of representative sequences were determined using RDP classifier with a bootstrap cutoff of 80%. Species-level classifications were ascertained by using MegaBLAST with a greater than 99% similarity to a reference sequence derived from a cultured isolate, and no similarity at greater than 97% to any other cultured species44. The significance of differences in taxon-level between the two groups was tested by an embedded script in QIIME (group_significance.py). Differences were considered significant when FDR < 0.20.
The symbiosis and competition relationships between the differential OTUs were evaluated by Spearman’s rank correlation based on the relative abundances of OTUs. The network in each group was visualized by using Cytoscape v3.1.148.
Additional Information
Accession codes: The sequence information in this paper has been submitted to the GenBank Sequence Read Archive database under accession number SRP076247. http://www.nature.com/srep
How to cite this article: Wang, J.-j. et al. Sex differences in colonization of gut microbiota from a man with short-term vegetarian and inulin-supplemented diet in germ-free mice. Sci. Rep. 6, 36137; doi: 10.1038/srep36137 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by grants from Shanghai Jiao Tong University School of Medicine (YG2014ZD10), and National Natural Science Foundation of China (81600409).
Footnotes
Author Contributions L.-p.Z., X.-y.P. and J.-j.W. designed the study; J.-j.W. and J.W. performed the experiments and analyzed the data; L.T., X.-p.W. and L.-p.Z. contributed to the discussion for interpreting the data; J.-j.W. and L.T. wrote and revised the manuscript.
References
- Tremaroli V. & Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249 (2012). [DOI] [PubMed] [Google Scholar]
- Kau A. L., Ahern P. P., Griffin N. W., Goodman A. L. & Gordon J. I. Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F., Manchester J. K., Semenkovich C. F. & Gordon J. I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104, 979–984 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P. J., Backhed F., Fulton L. & Gordon J. I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle J. G. et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339, 1084–1088 (2013). [DOI] [PubMed] [Google Scholar]
- Ridaura V. K. et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916 e917 (2012). [DOI] [PubMed] [Google Scholar]
- Dominianni C. et al. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS One 10, e0124599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 105, 2117–2122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S. et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol 72, 1027–1033 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy L. et al. Gender bias in autoimmunity is influenced by microbiota. Immunity 39, 400–412 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolnick D. I. et al. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun 5, 4500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri P., McCarville J., Kalmokoff M., Brooks S. P. & Green-Johnson J. M. Sex differences in gut fermentation and immune parameters in rats fed an oligofructose-supplemented diet. Biol Sex Differ 6, 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunasena E., McMahon K. W., Chang D. & Brashears M. M. Host responses to the pathogen Mycobacterium avium subsp. paratuberculosis and beneficial microbes exhibit host sex specificity. Appl Environ Microbiol 80, 4481–4490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Bushman F. D. & FitzGerald G. A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc Natl Acad Sci USA 112, 10479–10484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). [DOI] [PubMed] [Google Scholar]
- Singh N. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira E. L. et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J Nutr Biochem 23, 430–436 (2012). [DOI] [PubMed] [Google Scholar]
- Zimmerman M. A. et al. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am J Physiol Gastrointest Liver Physiol 302, G1405–G1415 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segain J. P. et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 47, 397–403 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F. et al. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol 14, R4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B. et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody R. N. et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeuwijk F. A. Interpreting genotype-by-environment interaction using redundancy analysis. Theor Appl Genet 85, 89–100 (1992). [DOI] [PubMed] [Google Scholar]
- Kverka M. et al. Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol 163, 250–259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K. et al. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 93, 59–65 (2016). [DOI] [PubMed] [Google Scholar]
- Meslin J. C., Bensaada M., Popot F. & Andrieux C. Differential influence of butyrate concentration on proximal and distal colonic mucosa in rats born germ-free and associated with a strain of Clostridium paraputrificum. Comp Biochem Physiol A Mol Integr Physiol 128, 379–384 (2001). [DOI] [PubMed] [Google Scholar]
- Yang J., Martinez I., Walter J., Keshavarzian A. & Rose D. J. In vitro characterization of the impact of selected dietary fibers on fecal microbiota composition and short chain fatty acid production. Anaerobe 23, 74–81 (2013). [DOI] [PubMed] [Google Scholar]
- Amir I., Bouvet P., Legeay C., Gophna U. & Weinberger A. Eisenbergiella tayi gen. nov., sp. nov., isolated from human blood. Int J Syst Evol Microbiol 64, 907–914 (2014). [DOI] [PubMed] [Google Scholar]
- Domingo M. C. et al. Clostridium lavalense sp. nov., a glycopeptide-resistant species isolated from human faeces. Int J Syst Evol Microbiol 59, 498–503 (2009). [DOI] [PubMed] [Google Scholar]
- Klaring K. et al. Intestinimonas butyriciproducens gen. nov., sp. nov., a butyrate-producing bacterium from the mouse intestine. Int J Syst Evol Microbiol 63, 4606–4612 (2013). [DOI] [PubMed] [Google Scholar]
- Willems A., Moore W. E., Weiss N. & Collins M. D. Phenotypic and phylogenetic characterization of some Eubacterium-like isolates containing a novel type B wall murein from human feces: description of Holdemania filiformis gen. nov., sp. nov. Int J Syst Bacteriol 47, 1201–1204 (1997). [DOI] [PubMed] [Google Scholar]
- Maslowski K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. et al. Clostridium bacteraemia characterised by 16S ribosomal RNA gene sequencing. J Clin Pathol 58, 301–307 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassart C. et al. First case of intra-abdominal infection with Clostridium disporicum. Anaerobe 19, 77–78 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang M. et al. Structural shifts of mucosa-associated lactobacilli and Clostridium leptum subgroup in patients with ulcerative colitis. J Clin Microbiol 45, 496–500 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff M. R. & Boquet P. Clostridium spiroforme toxin is a binary toxin which ADP-ribosylates cellular actin. Biochem Biophys Res Commun 152, 1361–1368 (1988). [DOI] [PubMed] [Google Scholar]
- Borriello S. P. & Carman R. J. Association of iota-like toxin and Clostridium spiroforme with both spontaneous and antibiotic-associated diarrhea and colitis in rabbits. J Clin Microbiol 17, 414–418 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrona A. F., Watanakunakorn C., Schaub C. R. & Jagetia A. Clostridium innocuum endocarditis. Clin Infect Dis 21, 1306–1307 (1995). [DOI] [PubMed] [Google Scholar]
- Sokol H., Lepage P., Seksik P., Dore J. & Marteau P. Temperature gradient gel electrophoresis of fecal 16S rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J Clin Microbiol 44, 3172–3177 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G. D. et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 334, 105–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W. S. F. et al. Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC Biology 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Farias C. et al. Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br J Nutr 101, 541–550 (2009). [DOI] [PubMed] [Google Scholar]
- Wang J. et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J 9, 1–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.