

Significance
Nature has evolved several strategies for using dioxygen (O2) for chemical oxidations. These enzymatic processes typically use metal cofactors to impart selectivity and minimize production of deleterious reactive oxygen species. In particular, hormone biosynthesis and neurotransmitter regulation are accomplished by copper- and O2-dependent enzymes possessing a unique copper active site; however, the mechanism has been debated and the role of the unique active site structure in enabling the chemistry is unclear. To understand this, structural and spectroscopic data are used to computationally model the full reaction mechanism to define the molecular basis for O2 reactivity and define the role of the active site structure in controlling proper enzymatic function.
Keywords: O2 activation, copper, reaction coordinates, electron transfer, reorganization energy
Abstract
Peptidylglycine α-hydroxylating monooxygenase (PHM) and dopamine β-monooxygenase (DβM) are copper-dependent enzymes that are vital for neurotransmitter regulation and hormone biosynthesis. These enzymes feature a unique active site consisting of two spatially separated (by 11 Å in PHM) and magnetically noncoupled copper centers that enables 1e– activation of O2 for hydrogen atom abstraction (HAA) of substrate C–H bonds and subsequent hydroxylation. Although the structures of the resting enzymes are known, details of the hydroxylation mechanism and timing of long-range electron transfer (ET) are not clear. This study presents density-functional calculations of the full reaction coordinate, which demonstrate: (i) the importance of the end-on coordination of superoxide to Cu for HAA along the triplet spin surface; (ii) substrate radical rebound to a CuII hydroperoxide favors the proximal, nonprotonated oxygen; and (iii) long-range ET can only occur at a late step with a large driving force, which serves to inhibit deleterious Fenton chemistry. The large inner-sphere reorganization energy at the ET site is used as a control mechanism to arrest premature ET and dictate the correct timing of ET.
Copper is an essential cofactor for many cellular processes requisite for life (1). In particular, one or multiple coppers are found in active sites of enzymes that bind, activate, and reduce O2 using the biologically accessible CuII/CuI redox couple (2, 3). One important class of Cu-dependent O2-activating enzymes is responsible for the stereospecific C–H α-hydroxylation of hormones and glycine-extended neuropeptides for proper neurotransmitter regulation and hormone biosynthesis. These enzymes [peptidylglycine α-hydroxylating monooxygenase (PHM), dopamine β-monooxygenase (DβM), and tyramine β-monooxygenase (TβM)] feature two distinct Cu sites (designated CuM and CuH) separated in the protein by an ∼11-Å distance (Fig. 1) (4–6). The lack of magnetic coupling between these sites distinguishes this class as “noncoupled” binuclear copper monooxygenases, in contrast to coupled binuclear copper enzymes (tyrosinase, hemocyanin, etc.). Intriguingly, a recent structure of dimeric DβM (7) shows an “open” conformation reminiscent of PHM in one apo monomer (predicted Cu–Cu distance ∼14 Å) and a “closed” conformation in the second half-apo monomer with a significantly contracted core (predicted Cu–Cu distance ∼5 Å). This suggests that the domains containing CuH and CuM have some conformational flexibility; however, it is currently unknown whether this is relevant to turnover. The CuM site, featuring a 2His/1Met ligand set, is the center involved in O2 activation, and the CuH site, supported by 3His coordination, is an unusual electron transfer (ET) site that provides the second electron required for turnover.
Fig. 1.

Structure of the noncoupled binuclear copper site in the ternary complex of PHM (O2 bound to CuM and substrate in binding pocket) using coordinates from PDB ID code 1SDW (5). CuH and CuM are located in different domains of the protein and are separated by 11 Å.
Kinetic isotope studies on PHM, DβM, and TβM have established several important parameters of the mechanism of C–H hydroxylation. A large intrinsic substrate H/D isotope effect (10.6) on the C–H cleavage step in PHM is consistent with homolytic C–H cleavage [i.e., via H-atom abstraction (HAA)] (8); however, the small isotope effect on kcat (∼1.5) (9) indicates this step is not overall rate-limiting. Originally it was thought that O2 was reduced by 2e– to a intermediate that was responsible for HAA from substrate (10); however, the half-occupied frontier molecular orbital (FMO) in CuII–OOH does not have significant OOH character and thus is not activated for H-atom abstraction (11). Also, apparent HAA from a model CuII–OOH complex has been shown to instead proceed via Fenton-type chemistry, eliminating results that had supported a CuII–OOH intermediate in HAA (12). Thus, a 1e–-reduced superoxo-CuII intermediate was implicated as the reactive intermediate responsible for HAA by Klinman (13) and by Chen and Solomon (14). This was subsequently supported by synthetic systems that perform HAA reactions (15, 16), and analysis that shows a half-occupied FMO lies entirely on the and is activated toward HAA (17). This is supported by heavy atom kinetic isotope analysis on PHM, where the magnitude of the 18O isotope effect is coupled to the substrate H/D isotopologue, implying that O–O cleavage must occur after HAA (8, 18). More recent work has identified a magnetostructural correlation in complexes, in which side-on (η2) coordination of leads to a singlet ground state because strong Cu–O σ-bonding causes a large splitting, whereas end-on (η1) coordination of leads to a triplet ground state because the small splitting is insufficient to overcome the spin pairing energy (Fig. 2) (3, 19).
Fig. 2.

Magnetostructural correlation of end-on triplet (Left, blue) to side-on singlet (Right, red).
In this study, we evaluate the ability of an end-on triplet to perform HAA in PHM. This leads to a full turnover mechanism consistent with the experimentally determined turnover rate. Additionally, we evaluate rates of ET from the reduced CuHI site to oxidized intermediates, and show that the inherently large inner-sphere reorganization energy of the CuH site is an important regulatory feature of the enzyme that controls the timing of ET to avoid uncoupled turnover via deleterious Fenton chemistry.
Results and Analysis
O2 Binding, Activation, and H-Atom Abstraction: Role of Superoxo .
O2 binding to reduced CuM.
The structure of optimized features a tetrahedral CuI center, which is consistent with spectroscopic data indicating a four-coordinate CuMI reduced site. Removal of the water ligand to give a three-coordinate was calculated to be uphill (ΔG = +1.4 kcal/mol). Dioxygen binding to CuM was investigated by replacing the water ligand with either side-on or end-on coordinated O2. Both O2 binding geometries gave a CuII/superoxo electronic structure description, where one electron transfers from Cu to O2 upon binding. Consistent with the magnetostructural correlation observed in cupric superoxos (Fig. 2) (19), side-on was calculated to have a singlet ground spin state, whereas end-on gave a triplet ground state. The lowest energy superoxo isomer is the end-on triplet, which is favored by ΔG = 6.3 kcal/mol over the end-on singlet. O2 binding through water displacement from to give is only modestly uphill (ΔG = +1.3 kcal/mol). Associative binding of O2 without OH2 loss to give five-coordinate is unfavorable (ΔG = +17.5 kcal/mol); therefore, O2 likely displaces coordinated OH2.
H-atom abstraction by CuII-superoxo.
Once O2 is bound and reduced to superoxide, is activated to abstract H• from the substrate and form the hydroperoxo (Fig. 3D). The transition state (TS) for HAA from formylglycine (FmG) on the triplet spin surface is shown in Fig. 3C, where the large imaginary frequency (–1,773 cm−1) shows almost exclusively H motion along the linear (175.8°) Odistal···H···Csubstrate vector. The calculated HAA barrier height of ΔG‡ = +22.2 kcal/mol was corrected by calibrating the B3LYP method (Becke three-parameter functional with Lee–Yang–Parr exchange correction) to the known HAA barrier in the model superoxo complex LPheCuO2 (20). The calculated barrier in the model system using the same computational method is ΔG‡ = +28.2 kcal/mol, which is 7.8 kcal/mol higher than that experimentally determined (20.4 kcal/mol). Thus, a correction of –7.8 kcal/mol was applied to the HAA ΔG‡ in PHM to give a corrected barrier height of ΔG‡ = +14.4 kcal/mol, which is consistent with the ∼14 kcal/mol obtained experimentally from the temperature dependence of the intrinsic isotope effect. Similarly, the calculated thermodynamics of HAA from FmG were calibrated to experimental bond dissociation enthalpies (21, 22), giving a corrected value of ΔGHAA = +6.7 kcal/mol in PHM (see SI Appendix for details).
Fig. 3.

Structures of CuM (A–I) and CuH (J and K) stationary points in this study. Only the hydrogen atoms derived from the substrate or H2O are shown for clarity.
The orientation of the C–H bond during HAA is close to perpendicular to the CuO2 plane (with the Cu–O–O–H dihedral angle = 137°), suggesting that HAA uses the superoxide π*-orbital perpendicular to the CuO2 plane ( in Fig. 2). To quantify the preference of HAA into , a series of TS structures was optimized at various fixed ∠Cu–O–O–H, and the orientation dependence on the HAA barrier (SI Appendix, Fig. S1) shows that HAA into is favored over (i.e., when ∠Cu–O–O–H is close to 180°) by at least 10 kcal/mol.
Rebound to the Protonated Versus Nonprotonated Oxygen of .
Previous studies have invoked rebound of the substrate radical (FmG•) with (i.e., the two products of HAA) to form the C–O bond in the alcohol product (14), but this reaction barrier has not been evaluated. The rebound TS where FmG• rebounds to the distal, protonated OH in was located at relative to the HAA products (+27.9 kcal/mol relative to ) on the triplet surface, and the TS structure is shown in Fig. 3E. This rebound leads directly to uncoordinated FmG–OH product and the cupric oxyl (Fig. 3I) by homolytic O–O cleavage with significant driving force (ΔGrebound = –29.7 kcal/mol); however, the large barrier suggests this may not be the most accessible pathway. An alternative TS (Fig. 3G) was located where FmG• rebounds to the nonprotonated O in with a much lower barrier , leading to the alternative product (Fig. 3H) in which the one electron oxidized alkoxyl product is bound to . Following the intrinsic reaction coordinate of this alternative rebound to the nonprotonated O back to the reactant complex reveals that it is accessed from the side-on–coordinated hydroperoxo (Fig. 3F), a structure that provides better substrate access to the nonprotonated O than in . Even accounting for the energy required to isomerize the end-on hydroperoxo to side-on (ΔG = +5.8 kcal/mol), rebound to the nonprotonated, proximal O occurs with (TS in Fig. 3G) relative to the HAA products in Fig. 3D (and +23.8 kcal/mol relative to , a ∼4-kcal/mol lower overall barrier than rebound to the protonated, distal OH (TS in Fig. 3E).
Proton-Coupled Electron Transfer from CuH to CuM.
A major unanswered question in the mechanism of the noncoupled binuclear Cu monooxygenases is the timing of ET from CuH to CuM that provides the second electron for the overall reaction, and specifically if this ET occurs prior or subsequent to substrate hydroxylation. ET and proton-coupled ET (PCET) rates were calculated for the transfer of an electron from to the various oxidized CuM/O2-derived intermediates to evaluate the mechanistic possibilities and identify the favored timing of ET. In the Marcus framework of ET (Eq. 1) (23), the rate of an ET reaction is dependent on three parameters in addition to temperature: reorganization energy (λ), driving force (ΔG°), and the magnitude of the electronic coupling between donor and acceptor (HDA).
| [1] |
The reorganization energy λ is the sum of inner-sphere (λi) and outer-sphere (λo) contributions, which represent the energy required to reorient the ligands and the solvent, respectively, from initial to final coordinates. Outer-sphere reorganization is assumed to be 0.4 eV (24, 25), and inner-sphere reorganization energies are obtained from density-functional theory (DFT)-calculated electronic energies (SI Appendix).
Oxidation of the CuH site.
The CuH site was optimized in reduced (Fig. 3J) and oxidized (Fig. 3K) states, using crystallographic coordinates of the three ligating histidines (His107, His108, and His172). The optimized structures are consistent with the solution spectroscopic data (26, 27), where the lowest energy forms were trigonal (H2O binding to is unfavorable, ΔG = +11.3 kcal/mol) and tetragonal (H2O loss from is unfavorable, ΔG = +6.6 kcal/mol). The change in coordination number of CuH upon redox results in an inherently large inner-sphere reorganization energy (calculated λi(CuH) = 0.93 eV; λtotal(CuH) ∼ 1.33 eV). This is significantly larger than other typical ET sites in biology, which generally feature low inner-sphere reorganization due to the structural similarity between reduced and oxidized states (for example: blue copper, CuA, and rubredoxins, each with λi ∼0.2–0.4 eV). However, this unusually large reorganization energy at CuH may be functionally required to the control correct timing of ET (vide infra).
Reduction of the CuM site.
It has been proposed that PCET may occur to to generate and H2O (the mechanism in Fig. 4A, which starts from the structure in Fig. 3D), which could couple directly with FmG• to form . Alternatively, PCET could occur at the step following rebound, to (the mechanism in Fig. 4B, which starts from the structure in Fig. 3H). Calculated values of ΔG and λi(CuM) for each permutation of mechanism (i.e., protonation following ET, ET following protonation, or concerted) and for each possible site of protonation for PCET to and (•OFmG) are given in Fig. 4 A and B, respectively. For , the most favorable mechanism for CuM reduction (Fig. 4A, Left) is initial protonation of the proximal O to the hydrogen peroxide complex (reaction b; ΔG = +8.7 kcal/mol) followed by ET from CuH (reaction c; ΔG = –9.5 kcal/mol). This would generate H2O2 in the presence of CuI, which is known experimentally to lead to Fenton chemistry via homolytic O–O cleavage and release of •OH (12). Indeed, O–O cleavage in to produce •OH would be thermodynamically accessible in PHM with calculated ΔG = –2.2 kcal/mol (Fig. 4A, Bottom; reaction f). The tight coupling between O2 reduction and substrate hydroxylation observed in these enzymes suggests the reactive O2-derived intermediates are tightly regulated, which would be inconsistent with production of free •OH and subsequent Fenton chemistry that would likely lead to uncoupled turnover and oxidative damage to the enzyme.
Fig. 4.

PCET from CuH to (A) and (B) and the calculated values of inner-sphere reorganization energy at the CuM site (λi in blue; values in electron volts) and free energies (ΔG in red; values in kilocalorie per mole at 298 K) for each step. The total free energy for each ET and PCET reaction includes the oxidative half-reaction , and for PT includes the proton desolvation free energy of +262 kcal/mol. The most kinetically favorable pathway in each mechanism is shaded (stepwise PT/ET followed by homolytic O–O cleavage in A, and concerted PCET in B). The corresponding diagram for PCET to is shown in SI Appendix, Fig. S2.
Alternatively, PCET to (•OFmG) following the rebound step is significantly more favorable than PCET to before rebound. The most favorable mechanism for reducing (•OFmG) is concerted PCET with the protonation occurring at the hydroxo ligand (Fig. 4B, reaction p). This reaction would occur with significant driving force (ΔG = –14.9 kcal/mol) along with a favorable inner-sphere reorganization energy at CuM (λi(CuM) = 0.22 eV). A similar PCET mechanism but with protonation at the alkoxyl is also favorable (Fig. 4B, reaction k), but has slightly less driving force reflecting the slightly higher basicity of hydroxo versus alkoxyl ligands. Stepwise PT/ET is also thermodynamically accessible (ΔGPT = +3.2 kcal/mol; ΔGET = –11.1 kcal/mol; reactions q→ r in Fig. 4B), but with less driving force and a higher reorganization energy, thus making PCET the overall most favorable mechanism.
Quantification of ET and PCET rates.
The Marcus equation (Eq. 1) was used to quantify the expected (PC)ET rates in these processes from the calculated values of ΔG and λ (Table 1). Due to the noncoupled nature of the active site positioning CuH and CuM to be ∼11 Å distant with no magnetic coupling between the CuII centers, HDA must be very small (but nonzero), and a value of 0.01 cm−1 is assumed for the purpose of calculating ET rates here based on previous molecular dynamics simulations that calculated a conformationally averaged HDA across the water-filled cleft and through the substrate of ∼0.015 cm−1 (28). The most favorable pathway for ET among all of the calculated permutations is PCET to (•OFmG) (reaction p in Fig. 4B), which predicts a rate on the order of 101 s–1. This is consistent with turnover (kcat = 39 s−1) (8) and is thus kinetically competent. Alternatively, the most favorable accessible ET to CuMII–OOH has a much slower calculated rate (on the order of 10−5⋅s–1 for reaction c in Fig. 4A), a factor of 106 slower than PCET to (•OFmG) and also ∼106-fold slower than kcat. ET to (reaction i) would be fast enough to be competitive with turnover (calculated rate ∼101⋅s−1); however, it cannot be accessed due to the ΔG = +30.6 kcal/mol required to protonate and cleave the O–O bond. This suggests that ET cannot occur to (i.e., before substrate hydroxylation) because the driving force is not sufficient to overcome the large reorganization energy and low HDA to result in an ET rate that is compatible with turnover. This is important as it precludes premature ET, which would lead to uncoupled turnover via Fenton chemistry.
Table 1.
ET and PCET rates calculated from Eq. 1 at 298 K
| ET reaction* | ΔG, kcal/mol | λi(CuM), eV | λtotal†, eV | HDA, cm–1 | kET, s–1 |
| ET and PCET to CuMII–OOH | |||||
| (a) CuMII–OOH + e– + H+ → CuMI–O(H)OH | −0.5 | 0.78 | 2.51 | 0.01 | 10−6 |
| (c) CuMII–O(H)OH + e– → CuMI–O(H)OH | –9.5 | 0.82 | 2.55 | 0.01 | 10−5 |
| (d) CuMII–OOH + e– → CuMI–OOH | +40.1 | 1.10 | 2.83 | 0.01 | 10−29 |
| (g) CuMII–OOH + e– + H+ → CuMII–O•– + H2O | +8.7 | 1.80 | 3.53 | 0.01 | 10−14 |
| (i) CuMIII–O•– + e– → CuMII–O•– | −22.5 | 0.57 | 2.30 | 0.01 | 101 |
| ET and PCET to CuMII(OH)(•OFmG) | |||||
| (k) CuMII(OH)(•OFmG) + e– + H+ → CuMII(OH)(O(H)FmG) | −7.9 | 0.67 | 2.40 | 0.01 | 10−3 |
| (m) CuMII(OH)(•O(H)FmG) + e– → CuMII(OH)(O(H)FmG) | −11.1 | 1.10 | 2.83 | 0.01 | 10−4 |
| (n) CuMII(OH)(•OFmG) + e– → CuMII(OH)(OFmG) | +14.8 | 0.61 | 2.34 | 0.01 | 10−12 |
| (p) CuMII(OH)(•OFmG) + e– + H+ → CuMII(OH2)(OFmG) | –14.9 | 0.22 | 1.95 | 0.01 | 101 |
| (r) CuMII(OH2)(•OFmG) + e– → CuMII(OH2)(OFmG) | −6.7 | 1.00 | 2.73 | 0.01 | 10−5 |
| ET and PCET to CuMII–O•– | |||||
| (t) CuMII–O•– + e– + H+ → CuMII–OH | −6.5 | 0.04 | 1.77 | 0.01 | 10−1 |
| (v) CuMIII–OH + e– → CuMII–OH | –17.8 | 0.48 | 2.21 | 0.01 | 100 |
| (w) CuMII–O•– + e– → CuMI–O•– | +35.3 | 0.46 | 2.19 | 0.01 | 10−23 |
The ET or PCET reaction that is along the favored pathway is highlighted in bold. Note that reaction i is faster than favored reaction c, but i is not accessible due to the highly endergonic PT that would precede it (reaction h).
Only the reductive half-reaction is shown. In each case, this is coupled to CuHI + H2O → CuHII(OH2) + e–. The reaction letters refer to Fig. 3A (a–i), Fig. 3B (k–r), or SI Appendix, Fig. S2 (t–w).
λtotal = [λi(CuH) + λo(CuH)] + [λi(CuM) + λo(CuM)]. Here we assume λo(CuH) = λo(CuM) = 0.4 eV.
Product Release from Reduced CuM.
After hydroxylation and PCET, product release from CuM completes the reaction. Some studies have suggested product release may be rate-limiting (29–31). From the product of PCET, (OFmG), intramolecular H+ transfer from OH2 to FmG–O– is moderately uphill (ΔG = +6.9 kcal/mol); however, this proton transfer proceeds with a low barrier of ΔG‡ = +10 kcal/mol, which is significantly lower than the radical rebound step (+23.8 kcal/mol), and thus not rate-limiting. The subsequent loss of protonated FmG–OH from the active site pocket (to give ) is thermoneutral (ΔG = +0.3 kcal/mol). Other mechanisms of product release were also considered, but each is less favorable than intramolecular protonation (SI Appendix, Fig. S3).
Discussion
Overall Mechanism of PHM.
Taken together, the calculations presented here have identified a single overall lowest energy mechanism of PHM. This pathway for O2 binding and activation, HAA and rebound from substrate, PCET from CuH to CuM, and product release is shown in Fig. 5. The overall rate-determining step in turnover is calculated to be rebound of the substrate radical to the nonprotonated oxygen of . This is consistent with the small substrate H/D kinetic isotope effect on kcat (1.5) (9), where the calculated H/D isotope effect on this rebound step is 1.1. Furthermore, the isotope effect on rebound is calculated to be 1.023, similar to the observed isotope effect on kcat/Km (1.017) (8). The overall calculated barrier of ΔG‡ = +23.8 kcal/mol [i.e., from to the rebound TS] is somewhat higher than that measured in turnover [∼17 kcal/mol, calculated from kcat = 39 s–1 at 37 °C (8)]; however, this difference can be attributed to the constraints imposed on the active site in the computational model. In particular, removing the positional constraint on the β-carbon of the substrate anchor Arg240 lowers the rebound barrier by ΔΔG‡ = –4.1 kcal/mol (overall ΔG‡ = +19.1 kcal/mol) by allowing a more favorable orientation of substrate at the TS.
Fig. 5.

Lowest energy mechanism of O2 binding and activation, substrate hydroxylation, PCET, and product release in PHM. Free energies (ΔG and ΔG‡) are given in kilocalorie per mole at 298 K. The HAA barrier is calibrated to experimental values in a model complex, and HAA thermodynamics are calibrated to experimental bond dissociation enthalpies.
H-Atom Abstraction by End-On Triplet Through Its Low-Lying .
The 1e– reduction of O2 by CuM is the key first step in PHM that leads to its monooxygenase activity. Consistent with the apparent end-on–bound O2 in the crystal structure of the PHM ternary complex, calculations predict an end-on as the lowest energy isomer, and this end-on binding leads to a triplet ground state due to the weak superoxo σ-donor bonding to that cannot overcome the spin pairing energy (Fig. 2). This leads to a low-lying unoccupied superoxide orbital (Fig. 2, Left) with very high oxygen character (49% on the distal oxygen, Fig. 6) that is primed for H-atom abstraction. The availability of in the end-on triplet in also results in a lower HAA barrier than the side-on singlet, in which stronger superoxo donation results in spin pairing and double occupation of (Fig. 2, Right).
Fig. 6.
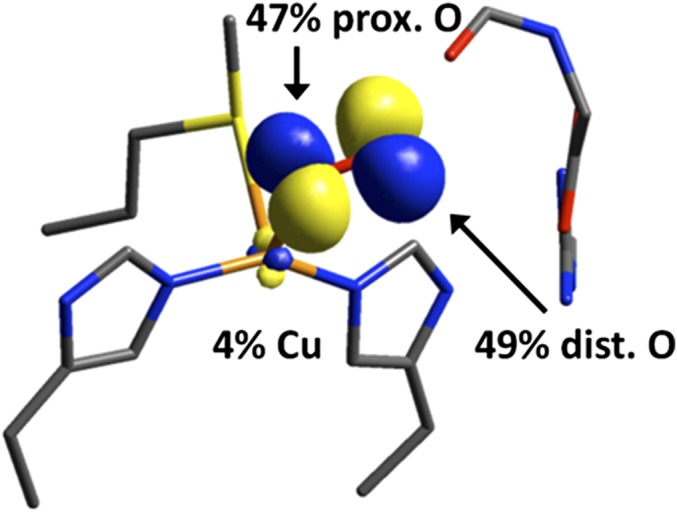
Unoccupied, low-lying β FMO in end-on triplet activated toward H-atom abstraction.
Rebound of Substrate Radical: Compensation for O–O Cleavage and Preference for the Nonprotonated Oxygen.
Homolytic cleavage of the O–O bond in to give and •OH is calculated to be 56 kcal/mol uphill; however, the calculated barrier for O–O cleavage via rebound attack by the substrate radical occurs with a much lower barrier (+10.1 kcal/mol). Thus, the substrate radical intercepts the incipient •OH early along the O–O cleavage coordinate and provides a large driving force by forming a strong C–O bond in the product. This compensates for the energy penalty of breaking the O–O bond and brings the barrier down to a kinetically accessible height. The O–O distance at the rebound TS (1.68 Å) shows only a 0.22-Å distortion from the reactant (O–O = 1.46 Å), which contributes ∼7 kcal/mol of the +10.1-kcal/mol rebound barrier.
The lower barrier for rebound to the nonprotonated O than rebound to the protonated OH reflects the amount of O–O distortion to reach the TS: The nonprotonated O rebound TS is earlier in O–O cleavage (1.68 Å) than the corresponding protonated OH rebound TS (1.72 Å). The origin of the earlier TS can be seen from the FMOs for substrate radical rebound. SI Appendix, Fig. S4 compares the α-spin FMOs of end-on (A) and side-on (B) in both the reactant complexes and TSs, where the filled α-highest occupied molecular orbital (HOMO, the radical on FmG•) and empty α-lowest unoccupied molecular orbital (LUMO, O–O σ* on the hydroperoxo) form a C···O σ/σ* pair along the reaction coordinate. Population analysis shows that the O–O σ* LUMO is polarized toward the nonprotonated O of the hydroperoxo in both end-on and side-on cases, a result of polarizing the filled O–O σ toward the H+. Thus, rebound to the nonprotonated O results in better overlap between O–O σ* and FmG• and a lower barrier by forming a stronger FmG···O bonding interaction at an earlier TS.
CuH as an ET Site.
The lack of exchange coupling between CuH and CuM has been explained as the key factor directing the reactivity of the noncoupled binuclear copper enzymes toward 1e– HAA/hydroxylation, as opposed to 2e– electrophilic aromatic attack in the coupled binuclear Cu enzymes (32). This control allows the overall two-electron oxidation of substrate to be accomplished by delivering electrons to the active CuM site one at a time, which permits one-electron reduction of O2 to the superoxide intermediate to “turn on” substrate HAA rather than two-electron reduction of O2 to a (hydro)peroxo intermediate that is not activated toward HAA. However, an inherent mechanistic challenge of the enzyme using two sequential ETs is controlling the timing of the second ET to only reduce the correct CuM intermediate that leads to a productive reaction.
A significant constraint on ET rate is due to the large inner-sphere reorganization at CuH (calculated λi = 0.93 eV). Therefore, rapid ET can only occur at a step in the mechanism with significant driving force. Of the possible ETs (Fig. 4 and Table 1), the most favorable is PCET to the intermediate that derives from FmG• rebound to , where the large PCET driving force (ΔG = –14.9 kcal/mol) is the minimum required for kET ≥ 10 s–1. PCET directly to before rebound does not have this driving force (ΔG = +0.8 kcal/mol), thus preventing PCET at this early stage of the reaction. ET to the protonated hydroperoxo is slightly more favorable (ΔG = –4.4 kcal/mol) than PCET to ; however, this is still not sufficient to result in ET rapid enough to be consistent with the turnover rate. Preventing this ET to is functionally required because it would generate CuI–O(H)OH, which would lead to deleterious Fenton chemistry. The requirement for large λi offers an explanation for why Nature chose the counterintuitive CuH site for electron transfer, especially because type 2 His3Cu sites are generally used directly for substrate binding and oxidation/reduction in other enzymes (for example, in copper amine oxidase, nitrite reductase, and superoxide dismutase) (33). In particular, if CuH were replaced by a type 1, blue, copper ET site (with λi ∼0.35), premature ET leading to Fenton chemistry would be accessible with a rate approaching 0.1 s–1 (SI Appendix, Fig. S5). Therefore, the noncoupled binuclear copper monooxygenases exploit an unusual ET site with large λi to control the correct timing of ET.
Conclusions
The noncoupled nature of the binuclear copper active site in PHM is the key distinguishing feature that leads to its hydroxylase function. By separating the two copper sites ∼11 Å in the protein, Nature provides a mechanism to accomplish the 2e– oxidation of peptide and catecholamine prohormones by separating it into two 1e– steps. The first step uses the one-electron reduction of O2 to at CuM to activate the triplet, end-on superoxide intermediate toward substrate H-atom abstraction through the channel. The incipient substrate radical is reactive toward rebound to the , and in particular, to the nonprotonated O to achieve an earlier transition state along O–O cleavage and thus a lower barrier. The large reorganization energy at the unusual CuH ET site, along with the low HDA between CuH and CuM, serve an important functional role in arresting ET until there is a sufficiently large driving force. This not only prevents premature ET that would lead to uncoupled turnover via Fenton chemistry, but also allows the enzyme to generate a powerful oxidant at CuM by preventing the reduction of all but the most reactive CuM intermediate. This represents a distinct strategy of gating ET in the noncoupled binuclear copper monooxygenase family, and appears critical for proper enzymatic function.
Materials and Methods
DFT calculations used the Gaussian 09 software package. Initial structures of the CuM and CuH sites were generated from crystallographic coordinates [Protein Data Bank (PDB) ID code 1SDW] (5). In addition to the primary CuM coordination sphere comprising His242, His244, and Met314, the model also included a truncated peptide substrate (FmG), and Arg240 as a substrate anchor. Each residue was truncated at the α-carbon, and all α-carbons were fixed during optimization to preserve the constraints imposed by the protein's secondary and tertiary structure. Free energies (ΔG, ΔG‡) are reported at 298 K. For computational details and coordinates of optimized species, see SI Appendix.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (R01DK31450 to E.I.S. and Ruth L. Kirschstein National Research Service Award F32GM105288 to R.E.C.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1614807113/-/DCSupplemental.
References
- 1.Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63(5):797S–811S. doi: 10.1093/ajcn/63.5.797. [DOI] [PubMed] [Google Scholar]
- 2.Solomon EI, et al. Copper active sites in biology. Chem Rev. 2014;114(7):3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon EI, et al. Copper dioxygen (bio)inorganic chemistry. Faraday Discuss. 2011;148:11–39, discussion 97–108. doi: 10.1039/c005500j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: The structure of peptidylglycine α-hydroxylating monooxygenase. Science. 1997;278(5341):1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 5.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen binds end-on to mononuclear copper in a precatalytic enzyme complex. Science. 2004;304(5672):864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 6.Prigge ST, Mains RE, Eipper BA, Amzel LM. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell Mol Life Sci. 2000;57(8-9):1236–1259. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vendelboe TV, et al. The crystal structure of human dopamine β-hydroxylase at 2.9 Å resolution. Sci Adv. 2016;2(4):e1500980. doi: 10.1126/sciadv.1500980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francisco WA, Merkler DJ, Blackburn NJ, Klinman JP. Kinetic mechanism and intrinsic isotope effects for the peptidylglycine α-amidating enzyme reaction. Biochemistry. 1998;37(22):8244–8252. doi: 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]
- 9.Francisco WA, Blackburn NJ, Klinman JP. Oxygen and hydrogen isotope effects in an active site tyrosine to phenylalanine mutant of peptidylglycine α-hydroxylating monooxygenase: Mechanistic implications. Biochemistry. 2003;42(7):1813–1819. doi: 10.1021/bi020592t. [DOI] [PubMed] [Google Scholar]
- 10.Miller SM, Klinman JP. Secondary isotope effects and structure-reactivity correlations in the dopamine β-monooxygenase reaction: Evidence for a chemical mechanism. Biochemistry. 1985;24(9):2114–2127. doi: 10.1021/bi00330a004. [DOI] [PubMed] [Google Scholar]
- 11.Chen P, Fujisawa K, Solomon EI. Spectroscopic and theoretical studies of mononuclear copper(II) alkyl- and hydroperoxo complexes: Electronic structure contributions to reactivity. J Am Chem Soc. 2000;122(41):10177–10193. [Google Scholar]
- 12.Kim S, et al. Amine oxidative N-dealkylation via cupric hydroperoxide Cu-OOH homolytic cleavage followed by site-specific Fenton chemistry. J Am Chem Soc. 2015;137(8):2867–2874. doi: 10.1021/ja508371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klinman JP. The copper-enzyme family of dopamine β-monooxygenase and peptidylglycine α-hydroxylating monooxygenase: Resolving the chemical pathway for substrate hydroxylation. J Biol Chem. 2006;281(6):3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 14.Chen P, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J Am Chem Soc. 2004;126(15):4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 15.Peterson RL, et al. Cupric superoxo-mediated intermolecular C-H activation chemistry. J Am Chem Soc. 2011;133(6):1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JY, et al. Mechanistic insights into the oxidation of substituted phenols via hydrogen atom abstraction by a cupric-superoxo complex. J Am Chem Soc. 2014;136(28):9925–9937. doi: 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woertink JS, et al. Spectroscopic and computational studies of an end-on bound superoxo-Cu(II) complex: Geometric and electronic factors that determine the ground state. Inorg Chem. 2010;49(20):9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian G, Berry JA, Klinman JP. Oxygen-18 kinetic isotope effects in the dopamine β-monooxygenase reaction: Evidence for a new chemical mechanism in non-heme metallomonooxygenases. Biochemistry. 1994;33(1):226–234. doi: 10.1021/bi00167a030. [DOI] [PubMed] [Google Scholar]
- 19.Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Correlation of the electronic and geometric structures in mononuclear copper(II) superoxide complexes. Inorg Chem. 2013;52(22):12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunishita A, et al. Active site models for the Cu(A) site of peptidylglycine α-hydroxylating monooxygenase and dopamine β-monooxygenase. Inorg Chem. 2012;51(17):9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
- 21.Zhao R, Lind J, Merenyi G, Eriksen TE. Significance of the intramolecular transformation of glutathione thiyl radicals to α-aminoalkyl radicals. Thermochemical and biological implications. J Chem Soc, Perkin Trans. 1997;2(3):569–574. [Google Scholar]
- 22.Ruscic B, et al. Active Thermochemical Tables: Accurate enthalpy of formation of hydroperoxyl radical, HO2. J Phys Chem A. 2006;110(21):6592–6601. doi: 10.1021/jp056311j. [DOI] [PubMed] [Google Scholar]
- 23.Marcus RA. On the theory of oxidation‐reduction reactions involving electron transfer. I. J Chem Phys. 1956;24(5):966–978. [Google Scholar]
- 24.Solomon EI, Szilagyi RK, DeBeer George S, Basumallick L. Electronic structures of metal sites in proteins and models: Contributions to function in blue copper proteins. Chem Rev. 2004;104(2):419–458. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- 25.Hadt RG, Gorelsky SI, Solomon EI. Anisotropic covalency contributions to superexchange pathways in type one copper active sites. J Am Chem Soc. 2014;136(42):15034–15045. doi: 10.1021/ja508361h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott RA, Sullivan RJ, DeWolf WE, Jr, Dolle RE, Kruse LI. The copper sites of dopamine β-hydroxylase: An X-ray absorption spectroscopic study. Biochemistry. 1988;27(15):5411–5417. doi: 10.1021/bi00415a005. [DOI] [PubMed] [Google Scholar]
- 27.Blackburn NJ, Hasnain SS, Pettingill TM, Strange RW. Copper K-extended x-ray absorption fine structure studies of oxidized and reduced dopamine β-hydroxylase. Confirmation of a sulfur ligand to copper(I) in the reduced enzyme. J Biol Chem. 1991;266(34):23120–23127. [PubMed] [Google Scholar]
- 28.de la Lande A, Martí S, Parisel O, Moliner V. Long distance electron-transfer mechanism in peptidylglycine α-hydroxylating monooxygenase: A perfect fitting for a water bridge. J Am Chem Soc. 2007;129(38):11700–11707. doi: 10.1021/ja070329l. [DOI] [PubMed] [Google Scholar]
- 29.Miller SM, Klinman JP. Magnitude of intrinsic isotope effects in the dopamine β-monooxygenase reaction. Biochemistry. 1983;22(13):3091–3096. doi: 10.1021/bi00282a011. [DOI] [PubMed] [Google Scholar]
- 30.Brenner MC, Klinman JP. Correlation of copper valency with product formation in single turnovers of dopamine β-monooxygenase. Biochemistry. 1989;28(11):4664–4670. doi: 10.1021/bi00437a023. [DOI] [PubMed] [Google Scholar]
- 31.Brenner MC, Murray CJ, Klinman JP. Rapid freeze- and chemical-quench studies of dopamine β-monooxygenase: Comparison of pre-steady-state and steady-state parameters. Biochemistry. 1989;28(11):4656–4664. doi: 10.1021/bi00437a022. [DOI] [PubMed] [Google Scholar]
- 32.Chen P, Solomon EI. O2 activation by binuclear Cu sites: Noncoupled versus exchange coupled reaction mechanisms. Proc Natl Acad Sci USA. 2004;101(36):13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacPherson IS, Murphy MEP. Type-2 copper-containing enzymes. Cell Mol Life Sci. 2007;64(22):2887–2899. doi: 10.1007/s00018-007-7310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.