

Abstract
Background
Neoplasms of histiocytic and dendritic cell origin, including follicular dendritic cell sarcoma (FDCS), histiocytic sarcoma (HS) and interdigitating dendritic cell sarcoma (IDCS), are extremely rare, and data on their natural history and treatment outcomes are sparse. We evaluated the impact of surgery, radiation and systemic therapies on overall survival (OS).
Methods
We conducted a retrospective chart review of patients with FDCS, IDCS and HS treated at Memorial Sloan Kettering Cancer Center between 1995 and 2014.
Results
We identified 31, 15 and 7 patients with FDCS, HS and IDCS, respectively. Median age was 48.7, 42.3 and 58.8 years for FDCS, HS and IDCS, respectively. Only a slight disparity in gender distribution existed for FDCS and HS; however, IDCS predominantly affected males (6:1). The most common sites of presentation were abdomen and pelvis (42%), extremities (33%) and head and neck (57%) for FDCS, HS and IDCS, respectively. At diagnosis, 74%, 40% and 86% of patients presented with localised disease in FDCS, HS and IDCS, respectively. Patients with localised disease had significantly improved OS than those with metastatic disease in FDCS (P = 0.04) and IDCS (P = 0.014) but not in HS (P = 0.95). In FDCS and HS, adjuvant or neo-adjuvant therapy was not associated with improved OS compared with observation. In IDCS, surgery alone provided a 5-year overall survival rate of 71%.
Conclusions
Adjuvant or neo-adjuvant treatment in FDCS and HS did not affect OS. Patients with IDCS had an excellent outcome with surgery. In the metastatic setting, chemotherapy and small molecule inhibitors may provide benefit.
Keywords: Dendritic sarcoma, Histiocytic neoplasm, Adjuvant therapy
1. Introduction
Histiocytic and dendritic cells play critical roles in the immune system, contributing to phagocytosis, antigen processing and presentation to B and T cells [1–3]. The broad term histiocytic neoplasms as used by pathologists refers to neoplasms derived from monocytes/macrophages and dendritic cells [1]. Monocytes/macrophages and dendritic cells are derived from a common precursor known as the macrophage-dendritic cell progenitor (MDP). MDP is of myeloid origin and gives rise to the monocytes and a common dendritic cell precursor (CDP), which is a dendritic cell-restricted precursor. The CDP then differentiates into two broad categories of human dendritic cells: plasmacytoid dendritic cells (pDC) and classical dendritic cells (cDC) (Supplementary Fig. 1 and Table 1).
Monocytes/macrophages express the CD14 lineage marker. pDC and cDC constitutively express major histocompatibility complex II proteins (MHC-II) and lack lineage-specific markers (CD3, CD14, CD19, CD20, CD56, and glycophorin A) [4]. Follicular dendritic cells are the exception; these cells are mesenchymal in origin and express CD21, CD23 and CD35 [1]. Neoplasms that arise from these cells are exceedingly rare: they are estimated to make up <1% of neoplasms that arise in lymph nodes, extranodal lymphatic tissues or soft tissues. Since they arise in sustentacular cells in lymph nodes but not from lymphocytes, they are historically classified as “sarcoma”. Haematopoietic tumours arising from CD34+ myeloid progenitor cells include histiocytic sarcoma (HS), Langerhans cell histiocytosis/sarcoma (LCH) and interdigitating dendritic cell sarcoma (IDCS). In contrast, stromal or mesenchymal derived tumours include follicular dendritic cell sarcoma (FDCS) and the rarer fibroblastic reticulum cell tumours. While these are conveniently classified as histiocytic and dendritic neoplasms, their biology, natural history and treatment appear to be distinct [1].
Follicular dendritic cells (FDC) are mesenchymal in origin and found in the germinal center of lymph nodes where they are involved in antigen presentation, proliferation and differentiation of B-cells [1,5]. Although mesenchymal in origin, there have been reports of FDCS sharing clonality with co-existent follicular lymphoma, possibly through trans-differentiation or a common progenitor cell [6]. Tumours usually occur sporadically, but a subset of cases are associated with Castleman disease [7]. FDCS typically presents in middle-aged adults as a slow growing mass in the head and neck or abdominal lymph nodes [1]. For localised disease, surgical resection is the mainstay of treatment and adjuvant chemotherapy and radiation remains controversial. For disseminated disease, lymphoma-type regimens (cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), ifosfamide and etoposide+/−carboplatin (ICE), adriamycin, bleomycin, vinblastine and dacarbazine (ABVD)) are often employed although no prospective data on outcomes are available [5].
Histiocytic sarcoma is an extremely rare tumour of mature tissue histiocytes (non-Langerhans), with fewer than 40 cases reported in the literature [3,5,8–10]. HS typically presents in the gastrointestinal tract, skin, and soft tissues, but lymph node involvement has also been observed. Age varies widely at diagnosis (range, 6 months–89 years) and has a bi-modal distribution with a slight male predominance [11]. A treatment standard for HS does not exist. Localised disease is surgically resected and may additionally receive adjuvant radiation or chemotherapy with non-Hodgkin lymphoma regimens such as CHOP, ICE or ABVD.
Interdigitating dendritic cells are found in the paracortex of lymph nodes and involved in antigen presentation to T cells [1,2,5]. Neoplasms of interdigitating dendritic cells have been reported in less than 100 cases, and may be associated with low grade B-cell neoplasms, mycosis fungoides, and other solid tumours [12]. IDCS is typically diagnosed in middle-aged adults and presents as a solitary lymph node mass, and surgical resection is the mainstay of treatment [5,12]. A pooled-analysis of published case reports reported 2-year survival rates for localised and disseminated disease of 68.1% and 15.8%, respectively [12]. These reports showed conflicting outcomes on the impact of multi-modal therapy following surgery [5,12]. Disseminated disease is often treated with lymphoma-like regimens with variable success.
In an effort to improve the current understanding of the natural history and treatment outcomes of FDCS, HS, and IDCS, we conducted a retrospective analysis of patients evaluated at our institution. In this study, we evaluated patient demographics, pathological and clinical characteristics, and treatment modalities, and correlated these factors with survival outcomes.
2. Materials and methods
Following approval from the Institutional Review Board (MSK, WA0209), we conducted a retrospective review of all patients with FDCS, HS, and IDCS who were evaluated at the Memorial Sloan Kettering Cancer Center (MSK; New York, NY) between 1995 and 2014. Pathology was confirmed at MSKCC in all patients at time of initial diagnosis by those with expertise in sarcoma (16/53, 30%), haematology (17/53, 32%) and other (20/53, 37%). 9 pathologists reviewed 87% of the cases. However, pathology was not re-reviewed for this analysis. Pathology reports were reviewed, and when available, results of the immunohistochemical stains performed were collected. The following patient characteristics were reviewed: age at diagnosis, gender, race, Karnofsky Performance Status (KPS), stage (localised, locally advanced or metastatic), sites of presentation, size, lymph node status, date of surgery for primary tumour, margin status, type of adjuvant or neoadjuvant therapy, use and type of radiotherapy, type and response to systemic therapies and overall survival (OS). To update survival status, the online Social Security Death Index was queried in cases where the outcome was not available in medical records.
2.1. Statistical analysis
Patient characteristics are presented by frequencies and percentages for categorical variables; median and range for continuous variables. Missing data are reported as unknown. The primary endpoint of the analysis was overall survival, calculated as time from diagnosis to death or last follow-up. Patients alive at last follow-up were censored. Recurrence-free survival was calculated amongst patients with primary disease from date of surgery to date of recurrence, death or last follow-up. The Kaplan–Meier method was used to calculate survival probabilities; the log-rank test was used to compare survival between groups; and the Cox regression score test was used to assess the association with survival for continuous variables such as age. P-values <0.05 were considered significant. All statistical analysis was done using R version 3.1.1 (cran.r-project.org).
3. Results
3.1. Follicular dendritic cell sarcoma
We identified 31 patients with FDCS. Patient demographics, disease characteristics, stage and treatment modalities are listed in Table 1. Immunohistochemistry was positive for CD35 (92.8%), CD21 (84.6%), S-100 (50%), CD68 (66%), and clusterin (80%) (Table 2). All patient with localised disease underwent surgical resection and surgical margins were negative in 19/23 (82%). Within that subset of patients, 11/23 (48%) patients underwent either adjuvant radiation (9/11, 82%) or neo-adjuvant chemotherapy with doxorubicin and ifosfamide-based regimens (2/11, 18%). Adjuvant radiation consisted of doses ranging from 3000 to 6300 cGy delivered over 30–35 fractions typically using intensity modulated radiation therapy (IMRT) to areas involving neck, trachea, mediastinum, spine and chest wall. In univariate analysis, age, gender, performance status or location of primary disease had no impact on survival (Table 3). The 5-year overall survival for patients who received adjuvant or neo-adjuvant therapies (n = 11) and those who were observed (n = 12) were 39% and 69%, respectively; however, this difference was not statistically significant (P = 0.58). The 5-year overall survival (OS) rate in patients was superior for localised compared with metastatic disease at 55% and 38%, respectively (P = 0.04), Fig. 1. The median and 5-year recurrence-free survival (RFS) after surgery was 2.9 years and 34%, respectively (Table 3). Other neoplasms including lymphoma, solid tumours, and Castleman disease were noted in 3%, 35% and 6% of patients, respectively (Table 2).
Table 1.
Patient demographics and treatment characteristics.
| FDCS | HS | IDCS | |
|---|---|---|---|
| Number of patients | 31 | 15 | 7 |
| Age in years (range) | 48.7 (28.7–83) | 42.3 (1.7–76.7) | 58.8 (30.3–75.6) |
| Gender (% total) | |||
| Female | 18 (58) | 6 (40) | 1 (14) |
| Male | 13 (42) | 9 (60) | 6 (86) |
| Race | |||
| White, non-hispanic | 22 (71) | 12 (80) | 6 (86) |
| Hispanic | 4 (13) | 1 (7) | 1 (14) |
| Black | 1 (3) | 1 (7) | – |
| Asian | 3 (10) | – | – |
| Unknown/other | 1 (3) | 1 (7) | – |
| KPS (% total) | |||
| ≤80% | 10 (74) | 5 (33) | – |
| 90% | 12 (26) | 4 (27) | 2 (29) |
| Unknown | 9 (29) | 6 (40%) | 5 (71) |
| Primary site (% total) | |||
| Unknown | 1 (3) | 2 (13) | 0 (0) |
| Head and neck | 8 (26) | 3 (20) | 4 (57) |
| Thoracic/chest wall | 8 (26) | 0 (0) | 2 (29) |
| Abdomen/pelvis | 13 (42) | 4 (27) | 1 (14) |
| Extremities | 0 (0) | 5 (33) | 0 (0) |
| Paraspinal/CNS/other | 1 (3) | 0 (0) | 0 (0) |
| Stage | |||
| Localised or locally advanced | 23 (74) | 6 (40) | 6 (86) |
| Metastatic | 8 (26) | 9 (60) | 1 (14) |
| Treatment characteristics | |||
| Localised disease | n = 23 | n = 6 | n = 6 |
| Surgery alone | 12 (52) | 4 (66) | 5 (83) |
| Surgery + adjuvant or neo-adjuvant therapy | 11 (48) | 2 (34) | – |
| Radiation alone | – | – | 1 (17) |
| Margin Status after surgery | N = 23 | N = 6 | N = 6 |
| Positive | 2 (9) | – | 1 (17) |
| Negative | 19 (82) | 6 (100) | 2 (33) |
| Unknown | 2 (9) | – | 3 (50) |
| Type of adjuvant/neo-adjuvant (% of N) | N = 11 | N = 2 | N = 0 |
| Neo-adjuvant Chemotherapy | 2/11 (18) | 1 (50) | – |
| Adjuvant chemotherapy | – | 1(50) | – |
| Adjuvant radiation | 9/11 (82) | – | – |
FDCS, follicular dendritic cell sarcoma; HS, histiocytic sarcoma; IDCS, interdigitating dendritic cell sarcoma; CNS, central nervous system; KPS, Karnofsky Performance Status.
Table 2.
Immunohistochemical (IHC) profile for select markers and secondary cancers and infections.
| Positive staining | FDCS n = 31 |
HS n = 15 |
IDCS n = 7 |
|---|---|---|---|
| CD35 | 26/28 | 1/4 | 1/4 |
| CD21 | 22/26 | 0/7 | 0/4 |
| S-100 | 9/18 | 8/12 | 7/7 |
| CD68 | 4/6 | 15/15 | 4/5 |
| Clusterin | 4/5 | – | 1/2 |
| CD163 | – | 7/7 | 0/1 |
| Lysozyme | 0/1 | 4/6 | 1/2 |
| CD1a | – | 0/8 | 0/3 |
| Vimentin | 11/12 | 4/4 | 3/3 |
| EMA | 4/10 | 0/2 | 0/1 |
| HLA-DR | – | – | – |
| CD45 | 0/4 | 5/5 | 1/2 |
| Other Neoplasms (Age at diagnosis of other neoplasms, Age at diagnosis of FDCS or HS) | |||
| Lymphoma | Hodgkin lymphoma | Diffuse large B cell lymphoma (37, 37) Chronic lymphocytic leukaemia (76, 76) Marginal zone lymphoma (51, 54) Follicular lymphoma (76, 76) |
|
| Solid tumours | Melanoma (43, 53; 39, 34) Uterine (56, 72) Endometrial (53, 57) Transitional cell (64, 72; 69, 82) Breast (47, 53) Gastric (NA, 71) Laryngeal (NA, 82) Papillary thyroid (49, 49) Hurthle cell thyroid (47, 44) |
Bladder (40, 50) Melanoma (60, 57) |
|
| Infections | Castleman disease (n = 2) | HIV/AIDS (n = 1) | – |
| Other | Multiple scrotal and cutaneous lipomas | Sarcoidosis | – |
FDCS, follicular dendritic cell sarcoma; HS, histiocytic sarcoma; IDCS, interdigitating dendritic cell sarcoma; CNS, central nervous system; NA, not available.
Table 3.
Univariate survival analysis for median and 5-year overall survival (OS) in FDCS and HS.
| Follicular dendritic cell sarcoma
|
Histiocytic sarcoma
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Number of patients (number of events) | Median overall survival | 5-year overall survival | P | Number of patients (number of events) | Median overall survival, years | 5-year overall survival | P | |
| Age | 31 (17) | – | – | 0.19* | 15 (4) | – | – | 0.82* |
| Gender | ||||||||
| Female | 18 (10) | 9.8 | 0.53 | 0.37 | 6 (0) | NR | 1 | 0.04 |
| Male | 13 (7) | 4.85 | 0.44 | 9 (4) | 2.52 | 0.45 | ||
| Race | ||||||||
| Asian | 3 (2) | 3.27 | 0.33 | 0.49 | 0 | – | – | 0.24 |
| Black | 1 (0) | NR | NE | 1 (0) | NR | NE | ||
| Caucasian | 22 (13) | 5.88 | 0.52 | 12 (3) | NR | 0.74 | ||
| Hispanic | 4 (2) | 3.27 | NE | 1 (1) | 2.49 | NE | ||
| Unknown | 1 (0) | HR | NE | 1 (0) | NR | NE | ||
| KPS | ||||||||
| ≤80% | 10 (6) | 5.24 | 0.5 | 0.36 | 5 (3) | 2.52 | 0.4 | 0.49 |
| 90 | 12 (5) | 9.8 | 0.7 | 4 (1) | NR | 0.75 | ||
| Location | ||||||||
| Unknown | 1 (0) | NR† | NE‡ | 2 (1) | 0.61 | 0.5 | 0.65 | |
| Head/Neck | 8 (5) | 9.8 | 0.67 | 3 (0) | NR | 1 | ||
| Thoracic | 8 (4) | 4.41 | 0.47 | – | – | – | ||
| Abdomen and pelvis | 13 (7) | 4.85 | 0.44 | 0.51 | 4 (1) | NR | NE | |
| Extremities | – | – | – | 5 (2) | NR | 0.6 | ||
| Other | 1 (1) | 3.27 | NE | – | – | – | ||
| Stage | 0.95 | |||||||
| Localised | 23 (11) | 9.8 | 0.55 | 0.04 | 6 (2) | NR | 0.67 | |
| Metastatic | 8 (6) | 2.69 | 0.38 | 9 (2) | NR | 0.75 | ||
| Adjuvant or 0.45 | Neoadjuvant | 11 (6) | 4.77 | 0.39 | 0.58 | 2 (1) | 2.49 | NE |
| None | 12 (5) | 9.8 | 0.69 | 4 (1) | NR | 0.75 | ||
FDCS, follicular dendritic cell sarcoma; HS, histiocytic sarcoma; IDCS, interdigitating dendritic cell sarcoma; CNS, central nervous system; KPS, Karnofsky Performance Status.
Bold signifies that it has statistical significance with a p value <0.05.
P-value from score test.
NR indicates that the median OS time is not reached.
NE indicates that the 5-year OS is not estimable.
Fig. 1.
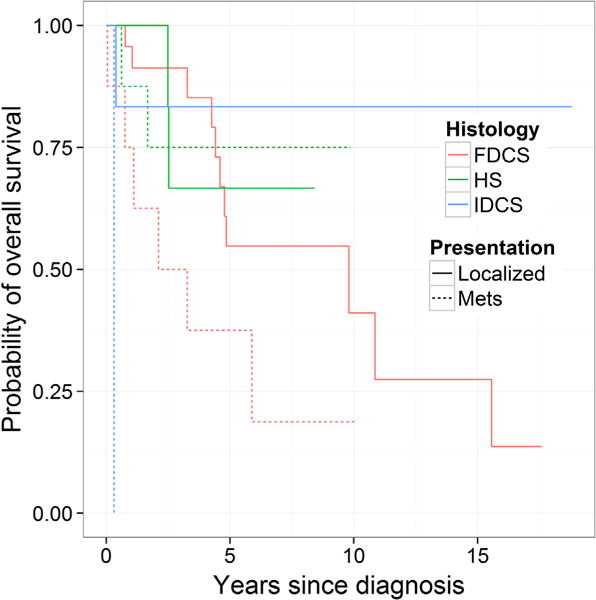
Kaplan–Meier curve of overall survival by histology and stage. The log rank p-value comparing all six curves is <0.001. The log-rank p-value comparing localised and metastatic disease in follicular dendritic cell sarcoma (FDCS), histiocytic sarcoma (HS) and interdigitating dendritic cell sarcoma (IDCS) patients is 0.04, 0.94, and 0.01, respectively.
3.2. Histiocytic sarcoma
We identified 15 patients as noted in Table 1. Tumours were positive for CD68 (100%), S-100 (66%), CD163 (100%), and lysozyme (66%) (Table 2). Amongst the 15 patients, haematological malignancies, solid tumours and HIV/AIDS were identified in 4/15 (26%), 1/15 (6%), and 1/15 (6%) patients, respectively (Table 2). All patient with localised disease (n = 6) that underwent surgery had negative surgical margins. Of those patients with surgical resection of the tumour, 2/6 (33%) underwent adjuvant or neo-adjuvant chemotherapy and no adjuvant radiation was delivered. In univariate survival analysis, median overall survival was not reached. Amongst patients with localised disease, the median OS was not reached in the observation alone group (n = 4). Median OS in the adjuvant or neo-adjuvant (n = 2) group was 2.5 years. OS did not differ between patients who had localised disease (67%) or metastatic disease (75%, P = 0.94). Age, performance status or primary site of disease did not impact OS. Interestingly, female patients (n = 6) had a better OS than males (n = 9, P = 0.04) (Table 4).
Table 4.
Univariate analysis of recurrence-free survival (RFS) for patients with localised disease.
| Number of patients | Number of events | Median RFS | 5-year RFS | P-value | |
|---|---|---|---|---|---|
| Histology | |||||
| FDCS | 23 | 16 | 2.86 | 0.34 | |
| HS | 6 | 2 | NR* | 0.67 | 0.49 |
| IDCS | 5 | 2 | NR | 0.6 |
NR: not reached FDCS, follicular dendritic cell sarcoma; HS, histiocytic sarcoma; IDCS, interdigitating dendritic cell sarcoma; CNS, central nervous system.
3.3. Interdigitating dendritic cell sarcoma
IDCS patients as listed in Table 1 had diagnoses confirmed by morphology and immunohistochemical staining for vimentin (100%), CD68 (80%), and S-100 (100%) (Table 2). Five patients with localised disease underwent surgery with only 2/5 (40%) patients having negative surgical margins. One patient received definitive radiation of 7020 cGy consisting of external beam radiation to the left neck. These patients received no additional therapy and had a 5-year recurrence-free survival of 60%. Those with localised disease had a significantly improved 5-year OS compared to those with metastatic disease (P = 0.01). Given the small sample size, multivariate analysis was not performed.
3.4. Systemic therapy in FDCS, HS, and IDCS
Twenty-four patients presented with locally recurrent or metastatic disease with available start and end dates of each therapy. In Fig. 2a and b, the systemic therapies utilised and time to next therapy are graphically represented for the 13 patients with FDCS (patients 1–13), the single patient with features of both IDCS and HS (patient 14), and the 10 patients with HS (patients 15–24). Re-resection, radiation, and a wide range of systemic therapies were used in patients with locally recurrent or metastatic disease (n = 10). Given variations in treatment regimens and the limited sample size for each regimen, we evaluated time to change in therapy for each unique patient instead of response rates for a specific regimen. Systemic regimens included doxorubicin and ifosfamide (AIM); vincristine, doxorubicin and cyclophosphamide (VAC); ifosfamide and etoposide+/−carboplatin (ICE/IE); cyclophosphamide, doxorubicin, vincristine, and prednisone (CH); gemcitabine with or without docetaxel, carboplatin and irinotecan; interferon-alfa; alemtuzumab (anti-CD52 antibody) and kinase inhibitors such as sunitinib, pazopanib, sorafenib, brivanib and sirolimus.
Fig. 2.

Type and duration of benefit of systemic therapies in patients with metastatic or recurrent disease with (A) follicular dendritic cell sarcoma (FDCS) and (B) histiocytic sarcoma (HS) and interdigitating dendritic cell sarcoma (IDCS). RT, radiation; A, doxorubicin; AIM, doxorubicin, ifosfamide and mesna; C, cyclophosphamide; CP, standard CHOP regimen, cyclophosphamide, doxorubicin, vincristine and prednisone; CLD, cladribine; IF, ifosfamide; ICE, ifosfamide, carboplatin and etoposide; GD, gemcitabine and docetaxel; GC, gemcitabine and carboplatin; GI, gemcitabine and irinotecan; VB, vinblastine; VC, vincristine; VAC, vincristine, doxorubicin and cyclophosphamide; † = dead at last follow-up.
4. Discussion
FDCS, HS and IDCS are extremely rare tumours for which current data are sparse. To our knowledge, this is the largest single institution case series of these diseases. As such, this report sheds light on the natural history and impact of multimodal treatments on 5-year overall survival. Our data show that these diseases have distinct natural histories, and therefore, prognosis and treatment should be individualised.
We confirmed diagnosis of FDCS by demonstrating positive staining for the FDC markers CD21 and CD35 in a majority of cases, variable staining of S-100 and CD68, and lack of staining for CD1a. The median age, gender distribution, and localised presentation in the head and neck and abdominal area observed in our series are similar to those reported in the literature [1,5,12]. We did not find these factors to impact overall survival in FDCS. We found two patients with Castleman disease (angiofollicular lymph node hyperplasia), which has also been reported in other studies. The pathophysiology between Castleman and FDCS diseases remains unclear; however, epidermal growth factor receptor (EGFR) expression has been suggested as a common link [13]. Interestingly, we found that ~35% of patients had other solid tumours, and only one patient (3%) had lymphoma. While our sample size is small, the frequency of other cancers appears unusually high in a cohort with a median age of 47 years. In most cases the solid tumour preceded the development of FDCS by several years, suggesting immunosuppression as a possible risk factor.
We found that patients with localised disease had a significantly improved median OS (9.8 years) compared to those with metastatic or recurrent disease (2.69 years, P = 0.04). Our finding differs from a pooled analysis of published case reports that showed no difference in 5-year OS between localised and metastatic disease; however, the follow-up period in that study was only 20 months [12]. Surgery was the mainstay of treatment in FDCS patients with localised disease. Adjuvant radiation or neo-adjuvant chemotherapy was not associated with improvement in median or 5-year OS when compared to surgery alone. The definitive role of adjuvant radiation or chemotherapy can only be ascertained in a prospective study. Furthermore, given that our study was performed as a single institution, certain principles that assess risk and prognosis may not have been accounted for when given adjuvant therapy. In our study, when we evaluated some well recognised risk factors, we were not able to discern a significant difference between the two cohorts (Supplementary Table 2). In the published pooled analysis of case reports, patients who received adjuvant RT (n = 51) did not have improved OS compared to surgery alone (n = 78) [12]. Systemic therapies used in the metastatic setting include doxorubicin, ifosfamide, or gemcitabine based regimens, which provided prolonged stable disease as outlined in Fig. 2a and b. Several patients were treated with tyrosine kinase inhibitors such as sorafenib, sirolimus, sunitinib, brivanib, imatinib and pazopanib and had prolonged benefit in the first-line or later therapy. In pre-clinical studies, maturation and activation of dendritic cell was inhibited by vascular endothelial growth factor (VEGF) and this effect was reversed with inhibitors of VEGF such as bevacizumab, sorafenib and sunitinib [14]. In a Phase II trial, investigators reported a durable partial response in a patient with metastatic FDCS treated with ridaforolimus, an mTOR inhibitor [15]. The precise mechanism of action of tyrosine kinase inhibitors in FDCS is unknown. Since these small molecules target multiple signalling pathways such as MAPK, PDGF, VEGF, mTOR and others, the precise mechanism of action in FDCS remains unclear. We hypothesise that genomic interrogation of these pathways may provide important clues on the pathogenesis and therapeutic targets.
HS was confirmed in our study with universal positivity for CD68 and variable staining for CD163, S-100, and lysozyme. Like others, we observed a wide age distribution from infancy to elderly; however, the typical presentation was in middle-aged adults. There was also a slight male preponderance (1.5:1). The most common site of presentation was in the extremities; this finding is different from that of other series, where intestines and skin were most commonly involved. Other lymphomas and solid tumours were seen in 4/15 (26%) and 2/15 (13%) of patients, respectively. This is in contrast to FDCS where haematologic malignancies were only noted in 3% of patients. Recently, several studies have shown identical immunoglobulin gene rearrangements and chromosomal translocations t(14;18) in patients metachronously diagnosed with both HS and either follicular lymphoma, chronic lymphocytic leukaemia (CLL) or acute lymphoblastic leukaemia [16–19]. We did not investigate whether our patients with other malignancies underwent a process of trans-differentiation, de-differentiation, or had a common clonal origin. Surprisingly, we did not see any difference in median or 5-year overall survival between those with localised or metastatic disease, with approximately 70% of all patients being alive at 5 years. These observations led us to ask whether chemotherapy can improve survival in patients with localised disease. In our series, albeit small, HS patients with localised disease had no survival difference between those who received chemotherapy (n = 2) or observation (n = 4). There were no significant differences between the two cohorts in terms of some commonly recognised risk factors (Supplementary Table 2). Patients with metastatic disease had prolonged benefit with gemcitabine, cladribine, anthracycline, and ifosfamide-based regimens. One patient had prolonged benefit with sirolimus after progressing on sunitinib, and another patient had durable stable disease on imatinib after progressing on CHOP regimen. Like FDCS, the pathogenesis in HS is unknown. Experimental models in mice have shown that loss of PTEN and INK4A/ARF results in histiocytic sarcoma [20]. Interestingly, genomic sequencing studies of Langerhans cell histiocytosis, which is a histiocytic neoplasm that arises from a specialised dendritic cell known as the Langerhans cell, has revealed mutually exclusive driving mutations of BRAF V600E and MAP2K1 mutations that implicate MAP kinase signalling cascade in the pathogenesis of this disorder [21–24]. Furthermore, genomic sequencing studies of Erdheim–Chester disease, a non-Langerhans cell histiocytic (LCH) neoplasm of monocyte/macrophage origin, has demonstrated mutations in BRAF V600E and NRAS, which can activate the MAP kinase pathway [25,26]. Therefore, the benefit of sirolimus, an mTOR inhibitor, may be further explained by investigating the PTEN/Akt/mTOR signalling pathway in these patients. CD52 is a cell surface glycoprotein expressed on T-lymphocytes and circulating monocyte-derived dendritic cells but absent in dermal (LCH) and mucosal dendritic cells [27]. Alemtuzumab, a monoclonal antibody against CD52, was shown to induce complete response for >5 years in two patients who were critically ill and failed multiple lines of chemotherapy [9]. Currently, we are undertaking broad efforts to evaluate CD52 expression in HS along with genomic profiling for therapeutic targets.
In our series, patients with IDCS presented at a mean age of 58.8 years and were predominantly male (6:1). In other reports patients presented at similar age; however, the male preponderance was much lower at 1.4:1. A majority of patients presented with disease in the head and neck region as a solitary mass. Those with localised disease underwent surgery without any additional therapy and had a 5-year relapse-free survival of 60%. Those with localised disease had a significantly improved survival compared to those with metastatic disease (P = 0.014). This difference has also been observed by other investigators. The patient with features of both IDCS and HS and locally relapsed disease had disease control with interferon for more than 10 years (Fig. 2b).
This study highlights the natural history and treatment outcomes of some rare monocyte/macrophage and dendritic cell neoplasms. Interestingly, although labelled as “sarcomas”, the data suggest that HS and IDCS are haematologic disorders while FDCS is disorder of mesenchymal cells and thus more of a true sarcoma. We believe that ongoing efforts to map the genomic aberrations in these tumours will shed light on their lineage and help properly reclassify these tumours. In such rare diseases, randomised or prospective studies are hardly possible, and case series will likely continue to be the backbone to inform prognosis and guide treatment decisions. Diagnosis is challenging and requires the expertise of an experienced pathologist. One limitation of this study is that diagnosis was not re-confirmed for this study and potentially introducing inconsistencies since pathologist from multiple disciplines made the diagnosis in a twenty year time frame. When possible, patients should be referred to a tertiary care center for multidisciplinary care. In FDCS and IDCS, localised disease had better prognosis than metastatic disease; however, this difference was not seen in HS. In patients with FDCS and HS, adjuvant or neo-adjuvant therapies did not provide a survival advantage however our data has to be taken in context of our small sample size. Patients with IDCS generally had localised disease and had long-term benefit with surgery alone. Chemotherapy regimens similar to those used in treating lymphoma and sarcoma provided durable stable disease in those with metastatic disease. Many patients derived prolonged benefit from multi-targeted tyrosine kinase and mTOR inhibitors. These observations serve as an interesting area for the genomic sequencing of other related monocyte/macrophage and dendritic cell neoplasms in efforts to determine molecular targets for therapy.
Supplementary Material
Acknowledgments
We would like to thank Mr. and Mrs. Gary and Cynthia Porter for their support of sarcoma research.
Funding source
None.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ejca.2015.06.109.
Footnotes
Conflict of interest statement
None declared.
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th. Lyon, France: IARC Press; 2008. [Google Scholar]
- 2.Perkins SM, Shinohara ET. Interdigitating and follicular dendritic cell sarcomas: a SEER analysis. Am J Clin Oncol. 2013;36:395–8. doi: 10.1097/COC.0b013e31824be22b. [DOI] [PubMed] [Google Scholar]
- 3.Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol. 2004;28:1133–44. doi: 10.1097/01.pas.0000131541.95394.23. [DOI] [PubMed] [Google Scholar]
- 4.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002;41:1–29. doi: 10.1046/j.1365-2559.2002.01418.x. [DOI] [PubMed] [Google Scholar]
- 6.Feldman AL, Arber DA, Pittaluga S, et al. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood. 2008;111:5433–9. doi: 10.1182/blood-2007-11-124792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cokelaere K, Debiec-Rychter M, De Wolf-Peeters C, Hagemeijer A, Sciot R. Hyaline vascular Castleman’s disease with HMGIC rearrangement in follicular dendritic cells: molecular evidence of mesenchymal tumorigenesis. Am J Surg Pathol. 2002;26:662–9. doi: 10.1097/00000478-200205000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Schlick K, Aigelsreiter A, Pichler M, et al. Histiocytic sarcoma – targeted therapy: novel therapeutic options? A series of 4 cases Onkologie. 2012;35:447–50. doi: 10.1159/000340066. [DOI] [PubMed] [Google Scholar]
- 9.Shukla N, Kobos R, Renaud T, et al. Successful treatment of refractory metastatic histiocytic sarcoma with alemtuzumab. Cancer. 2012;118:3719–24. doi: 10.1002/cncr.26712. [DOI] [PubMed] [Google Scholar]
- 10.Dalia S, Jaglal M, Chervenick P, Cualing H, Sokol L. Clinicopathologic characteristics and outcomes of histiocytic and dendritic cell neoplasms: the moffitt cancer center experience over the last twenty five years. Cancers (Basel) 2014;6:2275–95. doi: 10.3390/cancers6042275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi E, Nakamura S. Histiocytic sarcoma: an updated literature review based on the 2008 WHO classification. J Clin Exp Hematopathol. 2013;53:1–8. doi: 10.3960/jslrt.53.1. [DOI] [PubMed] [Google Scholar]
- 12.Saygin C, Uzunaslan D, Ozguroglu M, Senocak M, Tuzuner N. Dendritic cell sarcoma: a pooled analysis including 462 cases with presentation of our case series. Crit Rev Oncol Hematol. 2013;88:253–71. doi: 10.1016/j.critrevonc.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Chang KC, Abruzzo LV, Lai R, Younes A, Jones D. Epidermal growth factor receptor expression in follicular dendritic cells: a shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum Pathol. 2003;34:835–40. doi: 10.1016/s0046-8177(03)00356-3. [DOI] [PubMed] [Google Scholar]
- 14.Alfaro C, Suarez N, Gonzalez A, et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br J Cancer. 2009;100:1111–9. doi: 10.1038/sj.bjc.6604965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mita MM, Poplin E, Britten CD, et al. Phase I/IIa trial of the mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) administered orally in patients with refractory or advanced malignancies and sarcoma. Ann Oncol. 2013;24:1104–11. doi: 10.1093/annonc/mds602. [DOI] [PubMed] [Google Scholar]
- 16.Brunner P, Rufle A, Dirnhofer S, et al. Follicular lymphoma transformation into histiocytic sarcoma: indications for a common neoplastic progenitor. Leukemia. 2014;28:1937–40. doi: 10.1038/leu.2014.167. [DOI] [PubMed] [Google Scholar]
- 17.Stoecker MM, Wang E. Histiocytic/dendritic cell transformation of B-cell neoplasms: pathologic evidence of lineage conversion in differentiated hematolymphoid malignancies. Arch Pathol Lab Med. 2013;137:865–70. doi: 10.5858/arpa.2012-0104-RS. [DOI] [PubMed] [Google Scholar]
- 18.Wang E, Papalas J, Hutchinson CB, et al. Sequential development of histiocytic sarcoma and diffuse large b-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: a divergent (bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol. 2011;35:457–63. doi: 10.1097/PAS.0b013e3182098799. [DOI] [PubMed] [Google Scholar]
- 19.Kumar R, Khan SP, Joshi DD, Shaw GR, Ketterling RP, Feldman AL. Pediatric histiocytic sarcoma clonally related to precursor B-cell acute lymphoblastic leukemia with homozygous deletion of CDKN2A encoding p16INK4A. Pediatr Blood Cancer. 2011;56:307–10. doi: 10.1002/pbc.22810. [DOI] [PubMed] [Google Scholar]
- 20.Carrasco DR, Fenton T, Sukhdeo K, et al. The PTEN and INK4A/ARF tumor suppressors maintain myelolymphoid homeostasis and cooperate to constrain histiocytic sarcoma development in humans. Cancer Cell. 2006;9:379–90. doi: 10.1016/j.ccr.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 21.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–23. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211:669–83. doi: 10.1084/jem.20130977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown NA, Furtado LV, Betz BL, Kiel MJ, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. 2014;124:1655–8. doi: 10.1182/blood-2014-05-577361. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124:3007–15. doi: 10.1182/blood-2014-05-577825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emile JF, Diamond EL, Helias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim–Chester disease. Blood. 2014;124:3016–9. doi: 10.1182/blood-2014-04-570937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–3. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 27.Buggins AG, Mufti GJ, Salisbury J, et al. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood. 2002;100:1715–20. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.