

ABSTRACT
Tumor necrosis factor (TNF) is known as an important regulator of tumor microenvironment and inflammation. TNF levels are markedly elevated in the bronchoalveolar lavage fluid (BALF) of patients with chronic obstructive pulmonary disease (COPD), which is an independent risk factor for lung cancer. We have previously shown that COPD-like airway inflammation promotes lung cancer in a K-ras mutant mouse model (CC-LR mouse). This was associated with a significant increase of neutrophils in BALF, accompanied by a marked increase in TNF level, suggesting a link between COPD, TNF, and lung cancer promotion. Therefore, we first overexpressed TNF in the airway epithelium of CC-LR mice, which promoted lung cancer by ∼2-fold. This was associated with increased numbers of Ki67 and CD31 positive cells in lung tumors of CC-LR/TNF-Tg mice. We also found a robust increase in NF-κB activation, and numbers of neutrophils and myeloid-derived suppressor cells (MDSCs) in lung. Accordingly, we depleted MDSCs in CC-LR/TNF-Tg mice, which lead to significant tumor suppression emphasizing on the role of TNF-induced MDSCs in K-ras induced lung tumorigenesis. Finally, we targeted TNF expression by crossing CC-LR mice with TNF knock-out mice (CC-LR/TNF-KO), which resulted in a significant decrease in lung tumor burden in the absence or presence of COPD-like airway inflammation. Interestingly, there were less MDSCs and lower Ki67 and CD31 expression in the lung of the CC-LR/TNF-KO mice. We conclude that TNF links COPD to lung cancer promotion by induction of an immunosuppressive MDSC response, and subsequent amplification of proliferation and angiogenesis in tumors.
KEYWORDS: COPD, K-ras, lung cancer, MDSC, NF-κB, TNF
Introduction
Cancer-related inflammation is an essential process in malignant progression, which became one of the cancer hallmarks with enabling effects.1,2 It is known now that both intrinsic (induced by a genetic event) and extrinsic (e.g., infection-induced) inflammation could promote cancer by inducing tumor cells to produce various cytokines and chemokines and subsequently attracting leukocytes to further amplify inflammation in the tumor microenvironment.1,3 Among the wide range of cytokines produced by tumor and stromal cells, TNF has been reported as an important regulator in the tumor microenvironment.4 The role of TNF in tumor immunity is complex and remains controversial. Research studies using high doses of exogenous TNF or gene modified tumor cell lines secreting TNF demonstrate an antitumor activity for TNF.5-7 However, TNF has also shown tumor-promoting effects in various tumor models. In murine models of colitis, it has been linked to colon and liver tumorigenesis through NF-κB activation and induction of inflammation-associated cytokines and anti-apoptotic proteins.8-10 TNF is also involved in the induction, maturation, differentiation and recruitment of myeloid derived suppressor cells (MDSCs) that may result in a pro-tumor immunosuppressive inflammatory condition.11-14
Lung cancer is the leading cause of cancer death worldwide both because of a high incidence and a low cure rate.15 Accumulating evidence has shown a role for inflammation in the pathogenesis of lung cancer, especially when induced by cigarette smoke, that develops in patients with chronic obstructive pulmonary disease (COPD).16 COPD is characterized by chronic lung inflammation17,18 and is considered an independent risk factor for lung cancer.19,20 We have previously established a COPD-like mouse model of airway inflammation21 and shown that this type of airway inflammation,22 but not asthma-like airway inflammation,23 promotes lung cancer in a K-ras mutant mouse model of lung cancer (CC-LR). CC-LR mice have airway epithelial-restricted expression of an activated form of K-ras. The significance of K-ras mutations in lung tumorigenesis can be highlighted by the fact that K-ras mutations are found in up to about 30–50% of lung adenocarcinomas, and that lung cancers harboring K-ras mutations are more aggressive with worse prognosis.24,25 We found that tumor promotion in CC-LR mice was associated with NF-κB pathway activation, increased levels of inflammatory cytokines including TNF, and recruitment of myeloid (macrophage, neutrophils, and MDSCs) and lymphoid (T helper 17 and T regulatory) cells.22,23,26,27 Here, using a genetic targeting strategy, we showed autocrine and paracrine roles for TNF, which is transcriptionally regulated by NF-κB, in lung cancer promotion through regulating tumor cell proliferation and angiogenesis, and induction of a MDSCs associated immunosuppressive inflammatory response.
Results
Overexpression of TNF in the airway epithelium causes lung remodeling and promotes lung cancer
To verify the role of TNF on lung tumor promotion, we have crossed our previously developed airway epithelial specific overexpressing TNF transgenic mouse (CCSP-TNF-Tg)21 with a CC-LR mouse22 to develop a lung specific K-ras mutant TNF overexpressing mouse (CC-LR/TNF-Tg mouse). Interestingly, the TNF overexpressing mouse has developed an emphysematous lung phenotype with alveolar wall destruction, air trapping, along with abnormally increased total lung capacity (TLC) and functional residual capacity (FRC) (Fig. S1) further confirming a role for TNF in COPD pathogenesis as we and others previously described.21 Macroscopic and microscopic examination of the lungs from epithelial-restricted TNF overexpressing mice (CC-LR/TNF-Tg mice) showed development of larger and advanced tumors (Fig. 1A) with local invasion to the vessel walls (Fig. 1A, arrow head). Overexpression of TNF in the airway epithelium also resulted in ∼50% (∼1.9-fold) increase in lung surface tumor number in CC-LR mice (Fig. 1B) and total tumor volume (∼3.8-fold) (Fig. 1C). No distant metastasis was found. Lung tumors from CC-LR/TNF-Tg mice have also shown increased expression of proliferation marker; Ki67 (Fig. 1D and E), angiogenic/invasion markers; CD31 (Fig. 1F and G), and MMP9 (Fig. 1H and I), with no changes in apoptosis marker; cleaved caspase 3 (data not shown).
Figure 1.
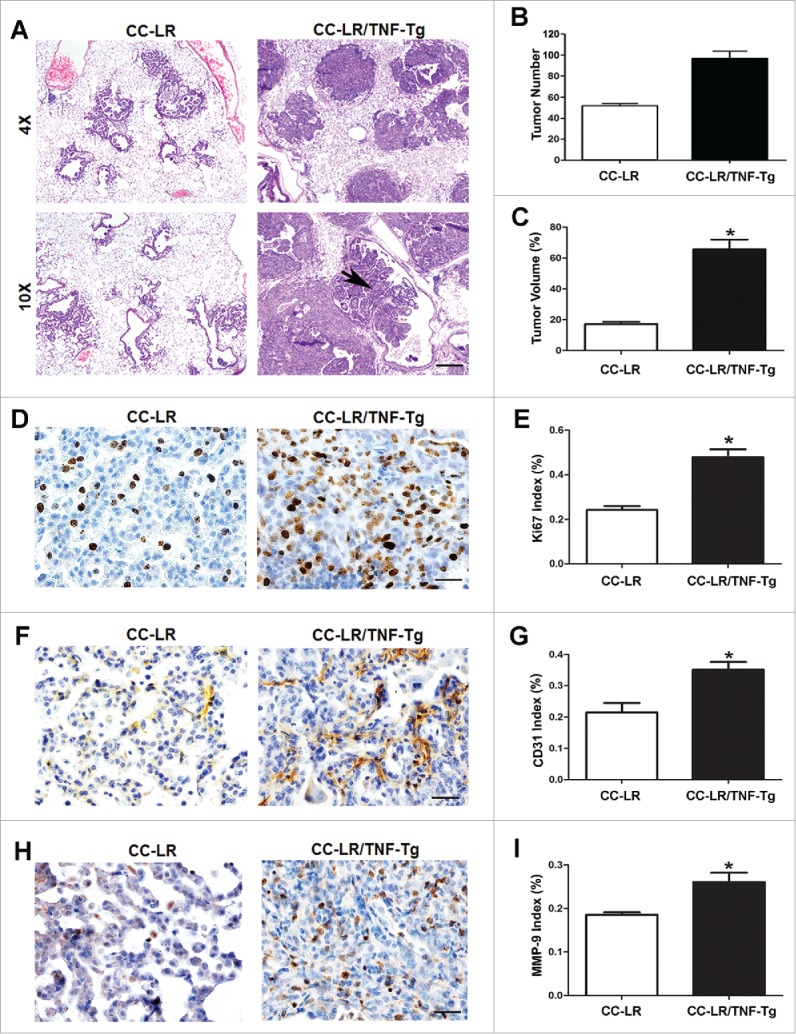
TNF overexpression in lung epithelium promotes lung cancer. (A) Histopathological appearance of lung tissue from CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (4× magnification, scale bar = 250 μm, 10× magnification, scale bar = 100 μm, applicable to all panels). (B) Lung surface tumor number, and (C) lung tumor volume in CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05). (D) Representative photomicrographs (40× magnification, scale bar = 25 μm, applicable to all panels), and (E) quantitative analysis of Ki-67 positive cells in lung tumor tissue of CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05). (F) Representative photomicrographs (40× magnification, scale bar = 25μm, applicable to all panels), and (G) quantitative analysis of CD-31 positive cells in lung tumor tissue of CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05). (H) Representative photomicrographs (40× magnification, scale bar = 25 μm, applicable to all panels), and (I) quantitative analysis of MMP-9 positive cells in lung tumor tissue of CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05).
TNF overexpression induces MDSCs recruitment in the lung tumor microenvironment
Macrophages are the predominant cells in the BALF of CC-LR mice at the age of 14 weeks. By contrast, there was around a 10-fold increase in the number of infiltrated neutrophils in the BALFs from CC-LR/TNF-Tg mice (Fig. 2A) similar to the inflammatory profile that is present in our previously developed NTHi-induced COPD-like mouse model.21 It has been reported that TNF signaling drives MDSCs accumulation in the tumor microenvironment.14 We therefore analyzed the proportion of MDSCs and MDSC subsets in the BALFs and whole lung by flow cytometry. The Gr1+CD11b+ MDSCs population increased significantly after TNF overexpression in the BALFs (∼7.4-fold) (Fig. 2B and C) and whole lungs (∼12.1-fold) (Fig. 2D and E) of CC-LR mice. However, this increase in total number of MDSCs was mostly due to an increase in M-MDSCs subtype, because there was no change in number of G-MDSCs in CC-LR/TNF-Tg mice compared to CC-LR mice (Fig. 2F). To investigate the functional role of MDSCs in the TNF-mediated promotion of lung tumors, we depleted these cells by intraperitoneal (i.p.) injection of RB6.8C5 monoclonal antibody in CC-LR/TNF-Tg mice, which resulted in a significant reduction in lung surface tumor number (∼1.8-fold) (Fig. 2G).
Figure 2.
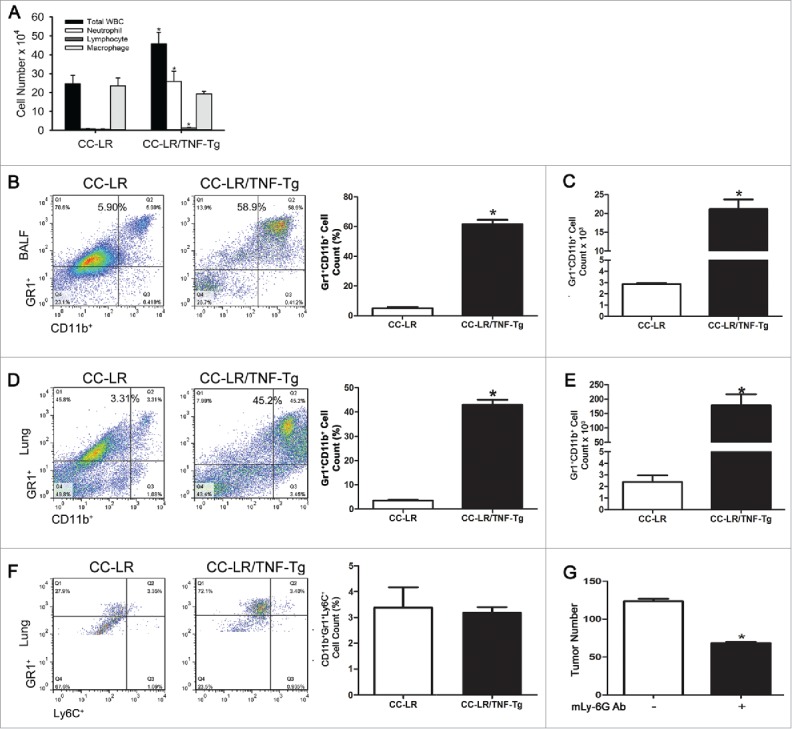
TNF overexpression in lung epithelium results in recruitment of neutrophils and myeloid derived suppressor cells (MDSCs). (A) Total and lineage-specific leukocyte numbers in bronchoalveolar lavage fluids (BALFs) of CC-LR and CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05). (B, C) Percentage and absolute number of Gr1+CD11b+ cells in BALFs of CC-LR and CC-LR/TNF-Tg mice (mean ± SE; *p < 0.05). (D, E) Percentage and absolute number of Gr1+CD11b+ cells in whole lung of CC-LR and CC-LR/TNF-Tg mice (mean ± SE; *p < 0.05). (F) Percentage and absolute number of Gr-1+Ly6C+ cells in lung of CC-LR and CC-LR/TNF-Tg mice (mean ± SE; *p < 0.05). (G) Lung surface tumor number after MDSC depletion in CC-LR/TNF-Tg mice at the age of 14 weeks (mean ± SE; *p < 0.05).
TNF deficiency inhibits K-ras mutant lung cancer
To further confirm the promoting role of TNF in lung cancer, we crossed CC-LR mice with TNF-KO mice28 to generate a K-ras mutant mouse with lack of TNF (CC-LR/TNF-KO mice), then exposed them to a lysate of non-typeable Haemophilus influenzae (NTHi) once a week for 8 weeks to induce a COPD phenotype, as previously described.21 Lack of TNF significantly inhibited lung cancer promotion in the absence (∼2.4-fold, or ∼60%) and presence (∼1.3-fold, or ∼25%) of COPD-type airway inflammation (Fig. 3A and B). Tumor size and total tumor volume also decreased robustly (∼2.8-fold) (Fig. 3C). Histopathologic examination of the lung sections from CC-LR/TNF-KO mice showed that most lung tumors remain at the early stage and have less inflammatory cell infiltrate compared to CC-LR mice. This was associated with significant reduction in tumor cell proliferation detected by reduced Ki67 expression (Fig. 3D and E). Immunohistochemical staining of angiogenesis/invasion markers CD31 and MMP9 did not show any significant decrease in the CC-LR/TNF-KO group compared to CC-LR mice (Fig. 3F). However, there was a significant decrease in expression of these markers in CC-LR/TNF-KO mice exposed to NTHi compared to NTHi-exposed CC-LR mice (Fig. 3G). Lack of TNF did not affect apoptosis of tumor cells in CC-LR mice in the absence or presence of COPD-type airway inflammation (data not shown).
Figure 3.
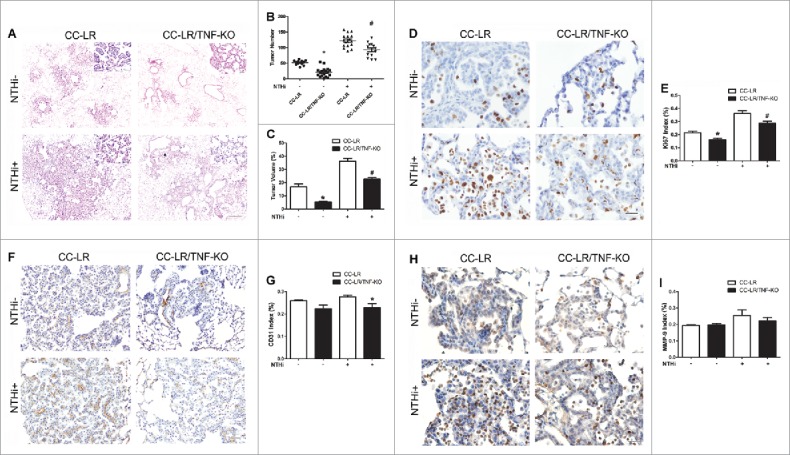
TNF deficiency inhibits lung cancer promotion. (A) Histopathological appearance of lung tissue from CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (10× magnification, scale bar = 100 μm, applicable to all panels). (B) Lung surface tumor number in CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05 for CC-LR vs CC-LR/TNF-KO mice). (C) Lung tumor volume in CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05 for CC-LR vs CC-LR/TNF-KO mice). (D) Representative photomicrographs (40× magnification, scale bar = 25 μm, applicable to all panels), and (E) quantitative analysis of Ki-67 positive cells in lung tissue of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05). (F) Representative photomicrographs (20× magnification, scale bar = 50 μm, applicable to all panels), and (G) quantitative analysis of CD-31 positive cells in lung tissue of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05). (H) Representative photomicrographs (40× magnification, scale bar = 25 μm, applicable to all panels), and (I) quantitative analysis of MMP-9 positive cells in lung tissue of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05).
TNF deficiency inhibits MDSCs recruitment
We have previously shown that lung cancer promotion by COPD-type airway inflammation is associated with a robust MDSC response.27 Therefore, we compared the inflammatory cell populations in the lung of CC-LR/TNF-KO mice with age and sex match control CC-LR mice in the absence and presence of NTHi-induced COPD-type airway inflammation. Surprisingly, lack of TNF did not change the BALF inflammatory cell profile of CC-LR mice at baseline or after inducing COPD-type airway inflammation (Fig. 4A and B). To further verify the potential role of TNF on MDSCs during lung tumor promotion, flow cytometry were done on BALF and total lung of CC-LR and CC-LR/TNF-KO in the absence and presence of COPD-type airway inflammation. There were no differences in the percentage and number of Gr1+CD11b+ MDSC population in the BALF and the whole lung of CC-LR/TNF-KO at the baseline compared to CC-LR mice (Fig. 4C). However, we found significant reduction in the number and percentage of lung infiltrated (BALF and whole lung) Gr1+CD11b+ MDSCs in the CC-LR/TNF-KO mice compared to CC-LR mice after induction of COPD phenotype by repetitive NTHi exposure (Fig. 4D). It was interesting that unlike IL-6 blockade which inhibited lung G-MDSCs infiltration,27 lack of TNF mostly affected the M-MDSC population (Fig. 4E).
Figure 4.
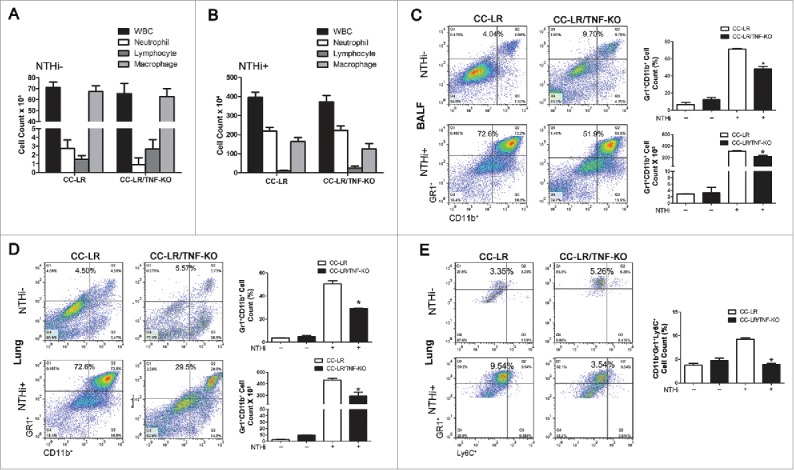
TNF deficiency inhibits MDSCs recruitment to the lung. (A) Total and lineage-specific leukocyte numbers in BALFs of CC-LR and CC-LR/TNF-KO mice at the age of 14 weeks (mean ± SE; *p < 0.05 for CC-LR vs CC-LR/TNF-KO). (B) Total and lineage-specific leukocyte numbers in BALFs of CC-LR and CC-LR/TNF-KO mice after 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05 for CC-LR vs CC-LR/TNF-KO). (C) Percentage and absolute number of Gr1+CD11b+ cells in BALFs of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05). (D) Percentage and absolute number of Gr1+CD11b+ cells in lung of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05). (E) Percentage of Gr1+Ly6C+ cells in lung of CC-LR and CC-LR/TNF-KO mice with and without 8 weeks of NTHi exposure at the age of 14 weeks (mean ± SE; *p < 0.05).
TNF possibly induces M-MDSC response via the NF-κB pathway activation
The NF-κB pathway has been reported to be involved in the activation of MDSCs.29 IHC staining of lung section from CC-LR/TNF-Tg mice showed nuclear translocalization of p65 subunit of NF-κB in most of normal and tumoral airway epithelial cells, as well as inflammatory cells (Fig. 5A). Western blot analysis of nuclear protein extracted from the whole lung showed an increased level of p65 protein in the CC-LR/TNF-Tg mice (Fig. 5B). This was associated with a decreased level of endogenous NF-κB inhibitor, IκBα, in cytoplasmic protein extract of whole lung from this transgenic mouse (Fig. 5C). Surprisingly, immunofluorescence staining of lung sections for NF-κB did not show a difference in overall NF-κB pathway activation between the CC-LR mice and CC-LR/TNF-Tg mice at tumor site (Fig. 5D). However, co-immunofluorescence staining of infiltrated macrophages for NF-κB, showed around a 1.8-fold increase in NF-κB activation level in CC-LR/TNF-Tg compared to CC-LR control mice (Fig. 5E). This was associated with an increased level of NF-κB target cytokines, IL-6 and TGF-β in BALF of CC-LR/TNF-Tg mice (Fig. S2).
Figure 5.
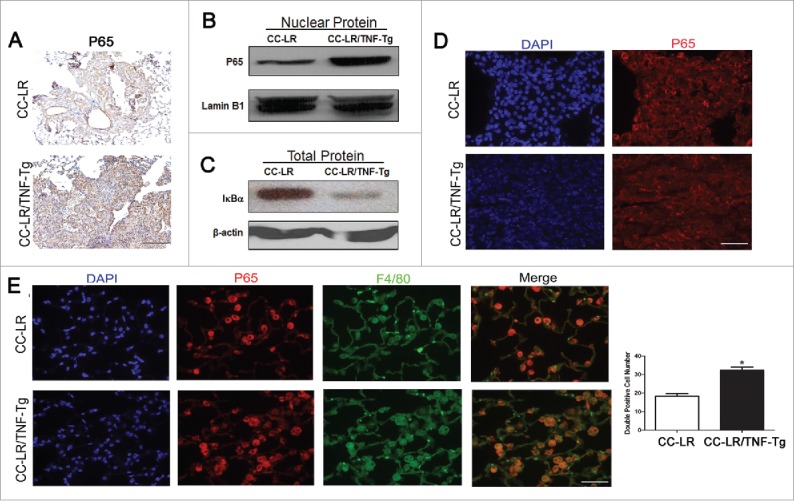
TNF induces activation of NF-κB pathway in lung infiltrating macrophages and M-MDSCs. (A) Representative photomicrographs of P65 positive cells in lung tissue of CC-LR and CC-LR/TNF-Tg mice (20× magnification, scale bar = 50 μm, applicable to all panels). (B) Western blot analysis of P65 protein on the nuclear protein extracted from whole lung tissue of CC-LR and CC-LR/TNF-Tg mice. (C) Western blot analysis of IκBα on the total protein extracted from whole lung tissue of CC-LR and CC-LR/TNF-Tg mice. (D) Representative immunofluorescence staining showing P65 positive cells in lung tumors from CC-LR and CC-LR/TNF-Tg mice (40× magnification, scale bar = 25 μm, applicable to all panels). (E) Representative immunofluorescence photomicrograph (40× magnification, scale bar = 25 μm, applicable to all panels) and absolute number of P65 and F4/80 double positive cells in lungs of CC-LR and CC-LR/TNF-Tg mice (mean ± SE; *p < 0.05).
Discussion
Cancer-related inflammation is an essential process in malignant progression.2 The tumor microenvironment in solid tumors is characterized by a reactive stroma with an abundance of inflammatory mediators and leukocytes, and proteolytic enzymes produced by these cells.3 In this study, we investigated the potential contribution of TNF in the pro-tumor inflammatory lung microenvironment. TNF is highly elevated in tissues and lung lining fluid of patients with COPD,30,31 and it is thought to play a role in COPD pathogenesis.32,33 We have previously shown that COPD-like airway inflammation promotes K-ras mutant lung cancer, and TNF is capable of recruiting leukocytes, predominantly neutrophils and macrophages, into BALF of mice when instilled intra-tracheally or overexpressed in mouse airways.21 A marked increase in MDSCs infiltrating the lungs and in circulation has been reported in smokers and COPD patients.34 We have found here that there is a robust accumulation of MDSCs in the lung of TNF overexpressing K-ras mutant mice or CC-LR mice with NTHi-induced COPD-like airway inflammation. This finding is in agreement with a recent study, which showed TNF involvement in enhancement of MDSCs response during chronic inflammation.13 MDSCs are a heterogeneous population of myeloid progenitor cells, including immature macrophages, granulocytes, and dendritic cells (DCs), that are endowed with a robust immunosuppressive activity.35 These cells engage in cross-talk with each other, resulting in the release of proinflammatory cytokines (e.g., IL-1, IL-6, IL-17, and TNF), chemokines (e.g., CCL2, CXCL5, and CXCL12), growth factors (e.g., TGF-β, GM-CSF, and VEGF), and other effector molecules (e.g., S100A8/A9, high-mobility group box 1).36 These factors, in turn, induce the accumulation and enhance the function of immune-suppressive cells, such as regulatory T cells, plasmacytoid DCs, tumor-associated macrophages, and MDSCs. It has been recently shown that TNF expression during chronic inflammatory conditions lead to enhanced intrinsic MDSC-suppressive function by increasing the activity of both NOS2 and arginase 1 (Arg-1) via the S100A8 and S100A9 proinflammatory proteins and their receptor RAGE.13,36 M-MDSCs were also reported to suppress CTL-mediated cytotoxicity via the induction of NO synthase (iNOS) and Arg-1, and the production of reactive nitrogen species, IL-10, and TGF-β.12,37 They also suppress NK cell cytotoxicity by inhibiting perforin production in a cell–cell contact dependent manner.38 MDSCs could also subvert macrophages toward a pro-tumor M2 phenotype through their production of IL-10.36 Note that MDSCs share properties and gene expression profiles with M2-polarized TAMs, and it is likely that, by nature, they skew toward an M2 polarization.39 Recent data clearly indicate that the pro-tumoral role of MDSCs is not limited to generating a suppressive niche around the tumor but, rather, these cells also play an important role in tumor progression even through immune-independent mechanisms.40 In our studies, lack of TNF expression partially inhibited the recruitment of MDSCs, particularly M-MDSCs, in lung of CC-LR mice with COPD-like lung inflammation, which is considered a chronic inflammatory condition. This was associated with reduced tumor cell proliferation. Meanwhile, over expression of TNF and induction of COPD-type inflammation in the lung of CC-LR mice resulted in tumor promotion and increased tumor cell proliferation. This prompted us to deplete MDSCs in CC-LR/TNF-Tg mice, which lead to significant tumor suppression, further verifying the effect of TNF-induced MDSCs on lung cancer promotion.
Angiogenesis is an important step in lung cancer progression. It is reported that MDSCs and TAMs infiltration correlates with increased vascular density and worse clinical outcomes in human cancer.41,42 It is reported that VEGF blocks DC development and function while simultaneously driving MDSC accumulation.43-45 In addition to tumor cells, MDSCs themselves produce VEGF, thereby, creating an autocrine feedback loop that sustains MDSC accumulation.46 Soluble MMP9 produced by tumor cells also promotes MDSC accumulation and tumor angiogenesis.47 We have found strong evidence of tumor angiogenesis and invasion in CC-LR/TNF-Tg mice, characterized by increased CD31 and MMP-9 expression which was reduced in CC-LR/TNF-KO mice.
As a major signaling pathway regulating inflammation, cell survival, proliferation, and angiogenesis, abnormal activation of NF-κB has been linked to the development of many cancer types, including lung cancer.48 Recent studies in various cell lines and mouse models showed that NF-κB is required for K-ras induced lung tumor progression. In our previous studies, we have also found significant activation of NF-kB and STAT3 signaling in our K-ras mutant lung cancer model (CC-LR) which was amplified after inducing COPD-like inflammation.23 As lung cancer progresses, NF-κB activation is maintained primarily by TNF that induces activation of IKKβ via the canonical pathway,49 which we and others have shown its targeting in the airway epithelium inhibits lung tumorigenesis. However, in this study, we observed activation of the NF-κB pathway in the inflammatory cells more than the tumor site in the CC-LR/TNF-Tg mice, which could promote MDSCs accumulation and function as previously described.29,50
An interesting crosstalk between MDSCs and tumor cells also exists. Tumor cells increase MDSC production of IL-6 and, in turn, MDSCs enhance IL-6 production by tumor cells. IL-6 also increases MDSC suppressive activity, but inhibits MDSC production of IL-10.36 In our previous studies, we have shown that IL-6 blockade in CC-LR mice not only inhibits intrinsic lung cancer development but also inhibits the promoting effect of extrinsic COPD-like airway inflammation by a tumor cell intrinsic mechanism and through paracrine effects by reprogramming the tumor microenvironment.27 It is also reported that MDSC-derived production of TGFβ results in increased tumor burden in lung.51 Interestingly, we have also found increased levels of TGFβ and IL-6 that could induce an immuno-suppressive T regulatory response in our TNF overexpressing K-ras mutant model.
In summary, we demonstrated that TNF promotes lung cancer by recruiting MDSCs infiltration and induction of an immunosuppressive, proliferative and proangiogenic microenvironment probably via NF-κB signaling. Therefore, our results introduce TNF as a potential target with preventive and therapeutic benefits for K-ras mutant lung cancer patients with COPD using already available TNF neutralizing antibody,52 and antagonist,13 or its receptor inhibitor.14
Materials and methods
Animals
CCSPCre/LSL-K-rasG12D mice (CC-LR) were generated as previously described.22 Briefly, this is a mouse generated by crossing a mouse harboring the LSL-K-rasG12D allele with a mouse containing Cre recombinase inserted into the Clara cell secretory protein (CCSP) locus. CC-LR mice were crossed with TNF transgenic (TNF-Tg) mice previously generated by us21 and TNF knock out (TNF-KO) mice purchased from the Jackson Laboratory.28 All mice were housed under specific pathogen-free conditions and handled in accordance with the Institutional Animal Care and Use Committee of M. D. Anderson Cancer Center. Mice were monitored daily for evidence of disease or death.
NTHi lysate aerosol exposure
A lysate of NTHi strain 12 was prepared as previously described,21 the protein concentration was adjusted to 2.5 mg/mL in phosphate buffered saline (PBS), and the lysate was frozen in 10 mL aliquots at −80°C. To deliver the lysate to mice by aerosol, a thawed aliquot was placed in an AeroMist CA-209 nebulizer (CIS-US, Bedford, MA) driven by 10 L/min of room air supplemented with 5% CO2 for 20 min.
Histochemistry
The tracheas of euthanized mice were cannulated with PE-50 tubing and sutured into place. The lungs were infused with 10% buffered formalin (Sigma, St. Louis, MO) and then removed and placed in 10% buffered formalin for 18 h. Tissues were then transferred to 75% ethanol, embedded in paraffin blocks, and sectioned at 5-mm thickness. The sections on glass slides were dried at 60°C for 15 min and then were deparaffinized and stained with hematoxylin and eosin (H&E) by incubating the tissues in Harris hematoxylin (Sigma) followed by serial eosin (Sigma) and graded ethanol steps. The H&E-stained slides were examined by a pathologist blinded to genotype and treatment, and the proliferative lesions of the lungs were evaluated in accordance with the recommendations of the Mouse Models of Human Cancer Consortium.53
Immunohistochemistry
Previously sectioned lung samples on slides were immunohistochemically (IHC) stained and evaluated for expression of Ki67 (1:200; abcam, MA, USA), CD31 (1:10; BD Biosciences, CA, USA), MMP9 (1:500; Santa Cruz, CA, USA), cleaved caspase-3 (1:500, abcam), p65 (1:100, abcam), F4/80 (1:100, ThermoFisher Scientific, MA, USA), and DAPI (Sigma, MO, USA), Alexa Fluor 488 Dye and Alexa Fluor 594 (Invitrogen, MA, USA). All antibodies reacted with mouse antigens. Heat-induced antigen retrieval was performed using 10 mmol/L of citrate buffer (pH 6) in a pressure cooker for 20 min. Endogenous peroxidase was quenched with 3% hydrogen peroxide for 30 min at room temperature. Blocking was performed with non-immune normal serum. Immunoreactivity for immunohistochemistry was detected using biotinylated IgG secondary antibodies specific for each primary antibody followed by incubation with ABC kit (Vector Laboratory, Burlingame, CA) for 30 min, and stained with diaminobenzidine chromogenic substrate for 4–10 min. Slides were counter-stained with Harris hematoxylin for 30 sec, followed by dehydration and mounted with cytoseal 60 (ThermoFisher Scientific, MA, USA). Images were obtained by an OLYMPUS BX 60 inverted microscope at 4×, 20×, or 40× magnification with Image-Pro Plus, version 4.5.1.22. The numbers of labeled positive cells for any of these markers were quantitated as a fraction of total tumor nuclei per high power field (40×) in 10 fields from three mice of each group. Results were expressed as percentage of positive cells ± SE.
Assessment of lung tumor burden and inflammation
On the first day after the final NTHi exposure, animals were euthanized by i.p. injection of a lethal dose of avertin (Sigma). In all mice (n = 8 per group per time point) lung surface tumor numbers were counted and then in some of them (n = 8 per group per time point) the lungs were prepared for histologic analysis as described above. H&E sections were prepared from 3 to 5 animals per group. Five randomly selected microscopic fields from peripheral and central regions of the lungs were photographed, and the percentage of the lung field occupied by tumors measured by overlaying of these images on a dotted grid as we previously described.54 In other mice (n = 8 per group per time point), bronchoalveolar lavage fluid (BALF) was obtained by sequentially instilling and collecting two aliquots of 1 mL PBS through a tracheostomy cannula. Total leukocyte count was determined using a hemacytometer, and cell populations were determined by cytocentrifugation of 300 mL of BALF followed by Wright–Giemsa staining. The remaining BALF (1,400 mL) was centrifuged at 1,250g for 10 min, and supernatants were collected and stored at −80°C.
Cytokines and chemokines measurement
The levels of inflammatory mediators in the BALF were assessed using MCYTOMAG-70K assay (Millipore, St Charles, MO), according to the manufacturer's instructions. Due to reagent incompatibilities, TGF-β was assayed separately from the other cytokines using the TGFB-64K-01 (Millipore) assay. Data were collected using a Luminex 100 (Luminex Corporation, TX, USA). Standard curves were generated using a five-parameter logistic curve-fitting equation weighted by 1/y (StarStation V 2.0; Applied Cytometry Systems, CA, USA). Each sample reading was interpolated from the appropriate standard curve.
Western blot analysis
Total proteins were prepared from each group of mouse lungs. Lung samples were removed and immediately placed in RIPA buffer and a protease inhibitor mixture. The samples were then homogenized and centrifuged at 14,000g for 20 min at 4°C. The supernatants were collected as the total proteins. Nuclear proteins were extracted using NE-PER Nuclear Protein Extraction Kit (Pierce, IL, USA) according to the instructions. Protein concentrations were measured using the Bradford protein assay (Bio-Rad Laboratories, CA, USA). Equal amounts of the proteins (40 μg for total proteins and 20 μg for nuclear proteins) were boiled for 5 min in the presence of loading buffer, loaded on each lane, and separated by 10% SDS-PAGE. The gels were then transferred to nitrocellulose membranes. Equal amounts of protein loading for each lane were checked by Ponceau (Sigma) staining. The anti-p65, anti-IκBα, anti-Lamin-B1 and anti-β-actin (abcam) antibody were diluted to the concentration according to commercial recommendations (1:1,000). Immunoreactive bands were detected with Pierce ECL Western Blotting Substrate (Pierce, IL, USA).
Isolation of lung resident mononuclear cells and flow cytometry
Lungs were harvested after perfusion with PBS. Lungs were first inflated with 0.1 mg/mL collagenase IV and DNase1 for 15 min at 37°C. Single cell suspensions were prepared by mechanical dissociation of lung tissue through a 70-μm nylon mesh. Lung cells were suspended in PBS and layered on LSM Lymphocyte Separation Medium (MP Biomedical, CA, USA). Cells were centrifuged at room temperature for 20 min at 900g. Mononuclear cells were harvested from the gradient interphase. Cells were stained with fluorescently labeled antibodies using CD45 (30-F11), CD11b (M1/70), F4/80 (BM8), CD11c (N418), Ly-6C (AL-21), Ly-6G (1A8), and I-A/I-E (M5/114.15.2) markers on ice for 30 min.55 Cells were analyzed and gated on an LSRII machine. Data were further analyzed by FlowJo.
Statistical methods
Summary statistics for cell counts in BALF and IHC positive cells in lung tissue were computed within treatment groups, and analysis of variance with adjustment for multiple comparisons was conducted to examine the differences between the control group and each of the treatment groups in the presence or absence of NTHi exposure. For tumor counts, comparisons of groups were made using Student's t-test. Differences were considered significant for p < 0.05.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest.
Acknowledgment
We thank Ms. Linda Foot for her editorial assistance in the completion of this project.
Funding
This work was supported by grant [RSG-11-115-01-CNE] from American Cancer Society awarded to Seyed Javad Moghaddam, grant [81501994] from National Natural Science Foundation of China, and grant [14KG143] from Key Grant for Health and Family Planning Commission of Tianjin awarded to Lei Gong.
References
- 1.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436-44; PMID:18650914; http://dx.doi.org/ 10.1038/nature07205 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/ 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009; 9:361-71; PMID:19343034; http://dx.doi.org/ 10.1038/nrc2628 [DOI] [PubMed] [Google Scholar]
- 5.Brouckaert PG, Leroux-Roels GG, Guisez Y, Tavernier J, Fiers W. In vivo anti-tumour activity of recombinant human and murine TNF, alone and in combination with murine IFN-gamma, on a syngeneic murine melanoma. Int J Cancer 1986; 38:763-9; PMID:3095251; http://dx.doi.org/ 10.1002/ijc.2910380521 [DOI] [PubMed] [Google Scholar]
- 6.Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med 1988; 167:1067-85; PMID:3351434; http://dx.doi.org/ 10.1084/jem.167.3.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang YQ, Zhang Z, Cai HR, Zhou H. Killing effect of TNF-mediated by conditionally replicating adenovirus on esophageal cancer and lung cancer cell lines. Int J Clin Exp Pathol 2015; 8:13785-94; PMID:26823692 [PMC free article] [PubMed] [Google Scholar]
- 8.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 2004; 6:297-305; PMID:15380520; http://dx.doi.org/ 10.1016/j.ccr.2004.08.012 [DOI] [PubMed] [Google Scholar]
- 9.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis 2011; 70 Suppl 1:i104-8; PMID:21339211; http://dx.doi.org/ 10.1136/ard.2010.140145 [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J et al.. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014; 26:331-43; PMID:25132496; http://dx.doi.org/ 10.1016/j.ccr.2014.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, Pietryga-Krieger A, Muller N, Ritter U, Mostbock S, Mannel DN. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun Inflamm Dis 2014; 2:121-30; PMID:25400932; http://dx.doi.org/ 10.1002/iid3.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X, Li B, Li X, Zhao X, Wan L, Lin G, Yu M, Wang J, Jiang X, Feng W et al.. Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol 2014; 192:1320-31; PMID:24379122; http://dx.doi.org/ 10.4049/jimmunol.1203195 [DOI] [PubMed] [Google Scholar]
- 13.Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 2013; 38:541-54; PMID:23477736; http://dx.doi.org/ 10.1016/j.immuni.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P et al.. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 2012; 122:4094-104; PMID:23064360; http://dx.doi.org/ 10.1172/JCI64115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 16.Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer 2013; 13:233-45; PMID:23467302; http://dx.doi.org/ 10.1038/nrc3477 [DOI] [PubMed] [Google Scholar]
- 17.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004; 364:709-21; PMID:15325838; http://dx.doi.org/ 10.1016/S0140-6736(04)16900-6 [DOI] [PubMed] [Google Scholar]
- 18.Barnes PJ. Small airways in COPD. N Engl J Med 2004; 350:2635-7; PMID:15215476; http://dx.doi.org/ 10.1056/NEJMp048102 [DOI] [PubMed] [Google Scholar]
- 19.de-Torres JP, Wilson DO, Sanchez-Salcedo P, Weissfeld JL, Berto J, Campo A, Alcaide AB, Garcia-Granero M, Celli BR, Zulueta JJ. Lung cancer in patients with chronic obstructive pulmonary disease. Development and validation of the COPD lung cancer screening score. Am J Respir Crit Care Med 2015; 191:285-91; PMID:25522175; http://dx.doi.org/ 10.1164/rccm.201407-1210OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takiguchi Y, Sekine I, Iwasawa S, Kurimoto R, Tatsumi K. Chronic obstructive pulmonary disease as a risk factor for lung cancer. World J Clin Oncol 2014; 5:660-6; PMID:25300704; http://dx.doi.org/ 10.5306/wjco.v5.i4.660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moghaddam SJ, Clement CG, De la Garza MM, Zou X, Travis EL, Young HW, Evans CM, Tuvim MJ, Dickey BF. Haemophilus influenzae lysate induces aspects of the chronic obstructive pulmonary disease phenotype. Am J Respir Cell Mol Biol 2008; 38:629-38; PMID:18096867; http://dx.doi.org/ 10.1165/rcmb.2007-0366OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, Kurie JM, Dickey BF, DeMayo FJ. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol 2009; 40:443-53; PMID:18927348; http://dx.doi.org/ 10.1165/rcmb.2008-0198OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ochoa CE, Mirabolfathinejad SG, Ruiz VA, Evans SE, Gagea M, Evans CM, Dickey BF, Moghaddam SJ. Interleukin 6, but not T helper 2 cytokines, promotes lung carcinogenesis. Cancer Prev Res (Phila) 2011; 4:51-64; PMID:21098042; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008; 359:1367-80; PMID:18815398; http://dx.doi.org/ 10.1056/NEJMra0802714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starmans MH, Pintilie M, Chan-Seng-Yue M, Moon NC, Haider S, Nguyen F, Lau SK, Liu N, Kasprzyk A, Wouters BG et al.. Integrating RAS status into prognostic signatures for adenocarcinomas of the lung. Clin Cancer Res 2015; 21:1477-86; PMID:25609067; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1749 [DOI] [PubMed] [Google Scholar]
- 26.Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, Dong C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA 2014; 111:5664-9; PMID:24706787; http://dx.doi.org/ 10.1073/pnas.1319051111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, Daliri S, Chang SH, Ochoa CE, Hanash S et al.. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-mutant lung cancer. Cancer Res 2016; PMID:27197187; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med 1996; 184:1397-411; PMID:8879212; http://dx.doi.org/ 10.1084/jem.184.4.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res 2015; 128:95-139; PMID:26216631; http://dx.doi.org/ 10.1016/bs.acr.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153:530-4; PMID:8564092; http://dx.doi.org/ 10.1164/ajrccm.153.2.8564092 [DOI] [PubMed] [Google Scholar]
- 31.Tangedal S, Aanerud M, Persson LJ, Brokstad KA, Bakke PS, Eagan TM. Comparison of inflammatory markers in induced and spontaneous sputum in a cohort of COPD patients. Respir Res 2014; 15:138; PMID:25398249; http://dx.doi.org/ 10.1186/s12931-014-0138-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundblad LK, Thompson-Figueroa J, Leclair T, Sullivan MJ, Poynter ME, Irvin CG, Bates JH. Tumor necrosis factor-alpha overexpression in lung disease: a single cause behind a complex phenotype. Am J Respir Crit Care Med 2005; 171:1363-70; PMID:15805183; http://dx.doi.org/ 10.1164/rccm.200410-1349OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eurlings IM, Dentener MA, Mercken EM, de CR, Bracke KR, Vernooy JH, Wouters EF, Reynaert NL. A comparative study of matrix remodeling in chronic models for COPD; mechanistic insights into the role of TNF-alpha. Am J Physiol Lung Cell Mol Physiol 2014; 307:L557-65; PMID:25106431; http://dx.doi.org/ 10.1152/ajplung.00116.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scrimini S, Pons J, Agusti A, Soriano JB, Cosio BG, Torrecilla JA, Nunez B, Cordova R, Iglesias A, Jahn A et al.. Differential effects of smoking and COPD upon circulating myeloid derived suppressor cells. Respir Med 2013; 107:1895-903; PMID:23993707; http://dx.doi.org/ 10.1016/j.rmed.2013.08.002 [DOI] [PubMed] [Google Scholar]
- 35.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol 2014; 96:1109-18; PMID:25170116; http://dx.doi.org/ 10.1189/jlb.3A0414-210R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/ 10.4049/jimmunol.0802740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang HG. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007; 109:4336-42; PMID:17244679; http://dx.doi.org/ 10.1182/blood-2006-09-046201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res 2007; 67:425; PMID:17210725; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155-66; PMID:17476345; http://dx.doi.org/ 10.1172/JCI31422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casulo C, Arcila M, Bohn OL, Teruya-Feldstein J, Maragulia J, Moskowitz CH. Tumor associated macrophages in relapsed and refractory Hodgkin lymphoma. Leuk Res 2013; 37:1178-83; PMID:23706570; http://dx.doi.org/ 10.1016/j.leukres.2013.03.021 [DOI] [PubMed] [Google Scholar]
- 42.Bergenfelz C, Larsson AM, von SK, Gruvberger-Saal S, Aaltonen K, Jansson S, Jernstrom H, Janols H, Wullt M, Bredberg A et al.. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS One 2015; 10:e0127028; PMID:25992611; http://dx.doi.org/ 10.1371/journal.pone.0127028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998; 92:4150-66; PMID:9834220 [PubMed] [Google Scholar]
- 44.Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, Gabrilovich DI. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol 1998; 160:1224-32; PMID:9570538 [PubMed] [Google Scholar]
- 45.Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res 1999; 5:2963-70; PMID:10537366 [PubMed] [Google Scholar]
- 46.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 2008; 118:3367-77; PMID:18776941; http://dx.doi.org/ 10.1172/JCI35213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res 2007; 67:11438-46; PMID:18056472; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci (Landmark Ed) 2011; 16:1172-85; PMID:21196225; http://dx.doi.org/ 10.2741/3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai Z, Tchou-Wong KM, Rom WN. NF-kappaB in lung tumorigenesis. Cancers (Basel) 2011; 3:4258-68; PMID:24213137; http://dx.doi.org/ 10.3390/cancers3044258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 2008; 181:4666-75; PMID:18802069; http://dx.doi.org/ 10.4049/jimmunol.181.7.4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shvedova AA, Kisin ER, Yanamala N, Tkach AV, Gutkin DW, Star A, Shurin GV, Kagan VE, Shurin MR. MDSC and TGFbeta are required for facilitation of tumor growth in the lungs of mice exposed to carbon nanotubes. Cancer Res 2015; 75:1615-23; PMID:25744719; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karabela SP, Kairi CA, Magkouta S, Psallidas I, Moschos C, Stathopoulos I, Zakynthinos SG, Roussos C, Kalomenidis I, Stathopoulos GT. Neutralization of tumor necrosis factor bioactivity ameliorates urethane-induced pulmonary oncogenesis in mice. Neoplasia 2011; 13:1143-51; PMID:22241960; http://dx.doi.org/ 10.1593/neo.111224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, Fraire AE, Gabrielson EW, Gunning WT, Haines DC et al.. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res 2004; 64:2307-16; PMID:15059877; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3376 [DOI] [PubMed] [Google Scholar]
- 54.Moghaddam SJ, Barta P, Mirabolfathinejad SG, Ammar-Aouchiche Z, Garza NT, Vo TT, Newman RA, Aggarwal BB, Evans CM, Tuvim MJ et al.. Curcumin inhibits COPD-like airway inflammation and lung cancer progression in mice. Carcinogenesis 2009; 30:1949-56; PMID:19793800; http://dx.doi.org/ 10.1093/carcin/bgp229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, Blackwell TS. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol 2013; 49:180-9; PMID:23492192; http://dx.doi.org/ 10.1165/rcmb.2012-0366MA [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.