

ABSTRACT
In tumor biology, nitric oxide (NO) is generally regarded as an immunosuppressive molecule that impedes T-cell functions and activation of endothelial cells. Contrasting with this view, we here describe a critical role for NO derived from inducible nitric oxide (iNOS)-expressing tumor macrophages in T-cell infiltration and tumor rejection as shown by iNOS gene deletion, inhibition of iNOS, or NO donors. Specifically, macrophage-derived NO was found to induce on tumor vessels adhesion molecules that were required for T-cell extravasation. Experiments with human endothelial cells revealed a bimodal dose-dependent effect of NO. High doses of NO donors were indeed suppressive but lower, more physiological concentrations, induced adhesion molecules in an NFkB-dependent pathway and preferentially activated transcription of genes involved in lymphocyte diapedesis. iNOS+ macrophages in tumors appear to generate precisely the amount of NO that promotes endothelial activation and T-cell infiltration. These results will be valuable for the development of strategies designed to overcome the paucity of T-cell infiltration into tumors that is a major obstacle in clinical cancer immunotherapy.
KEYWORDS: Adoptive T-cell transfer, endothelial adhesion molecules, endothelial barrier, iNOS+ macrophage, NO, tumor therapy, T-cell infiltration
Introduction
Despite the impressive advances in cancer immunotherapy obtained with immune checkpoint inhibitors and other novel treatment modalities, the clinical success is still limited. A major obstacle seems to be an insufficient infiltration of T cells into tumors. Thus, numerous clinical data have demonstrated that a low degree of T-cell infiltration usually correlates with poor prognosis, whereas high T-cell infiltration is indicative of prolonged survival.1
Most human and animal tumors are characterized by the formation of an aberrant tumor vasculature, which was found to be responsible for a dramatic decrease in T-cell adhesion and extravasation.2,3 Further studies demonstrated that the tumor endothelial barrier can be overcome by treatment with pro-inflammatory mediators such as radiation or toll-like receptor (TLR) ligands, which resulted in activation of tumor vessels and strong enhancement of T-cell transmigration.4-6 Investigations on the mechanism revealed that local tumor radiation combined with adoptive transfer of tumor-specific T cells activated tumor-associated macrophages (TAMs) and stimulated expression of inducible or type 2 nitric oxide synthase (iNOS or NOS2), which promoted T-cell influx and tumor destruction.7 This result was puzzling because in tumor biology nitric oxide (NO), the primary product generated by iNOS is usually perceived as an immunosuppressive molecule that inhibits T-cell proliferation and functions by a number of mechanisms including induction of apoptosis, direct cytocidal activity against T cells, nitrosylation of the TCR, and inhibiton of production of cytokines and chemokines.8-10 Moreover, NO is known to inhibit the expression of adhesion molecules on endothelial cells11-14 and to induce endothelial cell anergy.15 Another study suggested that the efficacy of radiotherapy of tumors depended on T cells. Whereas radiation alone induced suppression of T cells, blockade of NOS improved T-cell responsiveness and the success of radiotherapy.16 Furthermore, it was reported that NOS expression in melanoma promoted dysfunctional type I IFN signaling.17 In view of these immunosuppressive properties of NO, the mechanism by which radiation-induced iNOS+ macrophages exerted a positive effect and promoted T-cell infiltration and tumor destruction remains elusive.
Although TLR ligands such as bacterial unmethylated cytosine-phosphorothioate-guanine oligodeoxynucleotides (CpG-ODN) represent a very different type of proinflammatory stimulator as compared to radiation, the application of CpG-ODN in combination with adoptive T-cell transfer also strongly enhanced T-cell infiltration into tumors, similar to the effect of tumor radiation.4 Since CpG-ODN is known to bind to TLR-9 in macrophages and to induce iNOS, we addressed in the present study whether or not iNOS, and NO derived thereof, would be involved in the observed enhancement of T-cell transmigration, and if so, by which mechanism. For this purpose, we used different mouse tumor models, namely transplantable tumors and genetically modified Rip.Tag 5 (RT5) mice. Owing to the expression of the SV40 large T antigen (Tag) under control of the rat insulin promoter (Rip), RT5 mice develop at the age of 20 weeks pancreatic neuroendocrine tumors (PNET) that closely resemble human PNETs.18 The resulting tumors exhibit an aberrant vasculature that acts as a barrier against T-cell infiltration. Therefore, these mice represent a useful model for studies on mechanisms regulating T-cell diapedesis into tumors. Our results show that CpG-ODN polarizes TAMs toward iNOS+ macrophages. Importantly, NO derived from these macrophages was not immunosuppressive but directly induced adhesion molecules on tumor endothelia, thereby initiating T-cell diapedesis and tumor rejection.
Results
Combination of adoptive T-cell transfer with CpG-ODN increases T-cell infiltration and survival of RT5 mice
Using the RT5 mouse tumor model, we have previously shown that the adoptive transfer of activated Tag-specific CD8+ T cells alone has only a minor effect on tumor growth and survival. In contrast, transfer of CD8+ T cells in combination with application of the TLR-9 ligand CpG-ODN resulted in rejection of RT5 tumors and dramatic prolongation of survival4 (Fig. 1a). Flow cytometric analysis of tumors from mice treated with combination therapy revealed strong infiltration by CD8+ T cells, CD4+ T cells, B cells, NK cells, DCs, and macrophages into the tumor (Fig. 1b). TAMs resemble the so-called M2 macrophages, which are known to be pro-tumorigenic through production of angiogenesis factors, such as VEGF, and generation of an immunosuppressive microenvironment. In contrast, the so-called M1 macrophages are immunostimulatory.19,20 Since TAMs express TLR-9 and can bind CpG-ODN, their phenotype in RT5 tumors was investigated by flow cytometry for expression of the M1 marker iNOS and the M2 marker mannose receptor 1 (Mrc1, CD206). CpG-ODN was found to polarize TAMs toward an M1-like phenotype as shown by an increase in iNOS expression and a decrease in CD206 levels. The combination of CpG-ODN and adoptive CD8+ T-cell transfer resulted in the augmented expression of iNOS (Fig. 1c).
Figure 1.

Combination of adoptive T-cell transfer and CpG-ODN increases T-cell infiltration and prolongs survival of RT5 mice and 24 week-old tumor-bearing Rip1-Tag5 (RT5) mice were injected with 25 μg of CpG-ODN 1668 i.v. on days 0, 6, and 10. Where indicated, the mice received adoptive transfers of activated TCR8 CD8+ T cells on day 1 and 11. Mice were sacrificed on day 13 and analyzed by flow cytometry. For survival experiments, mice were injected as described above. CpG-ODN injection and adoptive transfers were repeated every 10 d. (a) Kaplan−Meier survival curves of mice treated with CpG-ODN alone, adoptive transfer of TCR8 CD8+ T cells alone and combination of CpG-ODN with adoptive transfer of TCR8 CD8+ T cells. (b) Quantification by flow cytometry of infiltrating leukocyte subpopulations into RT5 tumors. (C) Representative flow cytometric analysis of iNOS (M1 marker) and CD206 (MRC1) (M2 marker) expression on CD11b+ Gr-1− F4/80+ tumor macrophages. Results are shown as mean ±SEM of 10 mice per group from two independent experiments. *p < 0 .05, **p < 0 .01, ***p < 0 .001, ns = not significant.
Macrophages and iNOS activity promote VCAM-1 expression and T cell homing into the tumor
In order to investigate the potential role of iNOS+ macrophages in tumor rejection, clodronate containing liposomes (CLIP) were employed for depletion of macrophages. In addition, L-N6-(1-iminoethyl)lysine9 (L-NIL) was used for the specific inhibition of iNOS activity in tumor-bearing RT5 mice. Both treatments strongly reduced the prolonged survival obtained after combination therapy (Fig. 2a). The reduced survival in the macrophage-depleted group correlated with reduced infiltration by CD8+ T cells, CD4+ T cells, NK cells, and DCs (Fig. 2b). iNOS blockade with L-NIL also impaired leukocyte infiltration, but less efficiently than macrophage ablation, leaving open the possibility that other macrophage-derived factors are involved or that iNOS blockade was not complete.
Figure 2.

Macrophage depletion and iNOS blockade impairs T cell infiltration and RT5 tumor rejection 24 week-old Rip1-Tag5 (RT5) mice were injected with 25 μg of CpG-ODN 1668 and activated TCR8 CD8+ T cells as described in Fig. 1. (a) Kaplan-Meier survival curves. (b) Quantification by cytofluorometry of infiltrating leukocyte subpopulations in RT5 tumors. (c) Representative immunofluorescence microscopy analysis of tumor vessels of the indicated groups of mice stained with antibodies against CD31 (red) and VCAM-1 (green). Yellow color shows colocalization. Size bar indicates 100 μm. (d) Quantification of VCAM-1 expression on CD31+ tumor vessels. Tumors of comparable size were investigated. Three-6 random sections were analyzed from each tumor. (e) Protein concentration of IFNγ, CCL5, CXCL9 and CXCL10 in tumor lysates as determined by Multiplex analysis. Results are shown as mean ±SEM of 10 mice per group from two independent experiments. *p < 0 .05, ** p < 0 .01, ***p < 0 .001, ns = not significant.
Expression of cellular adhesion molecules such as VCAM-1 on tumor vessels is known to be important for T-cell diapedesis. Treatment with CpG-ODN led to an increase in VCAM-1 expression that was strongly enhanced by transferred tumor-specific CD8+ T cells suggesting that CpG-ODN and CD8+ T cells act in concert (Fig. 2a and d). Macrophage depletion or iNOS blockade prevented VCAM-1 expression indicating that VCAM-1 induction required iNOS activity in macrophages. (Fig. 2c and d). The combined treatment with CpG-ODN and T cells induced additional changes in the tumor microenvironment including upregulation of proinflammatory cytokines and chemokines such as IFNγ, CCL5, CXCL9, and CXCL10, which also depended on the presence macrophages and NO production (Fig. 2e). These findings suggest that iNOS-expressing macrophages are required for endothelial activation and T-cell trafficking into the tumor, but they do not rule out the possibility that iNOS expressed by other cell types contributes to this effect.
Adoptive transfer of iNOS-expressing macrophages promotes antitumor immunity
In order to ascertain that iNOS expression in macrophages was indeed required and sufficient for successful tumor therapy, transfer studies with Nos2−/− (iNOS-deficient) macrophages were performed. Macrophages derived from wild type and Nos2−/− bone marrow cells were activated with lipopolysaccharide (LPS) and IFNγ to yield M1 macrophages, or left untreated (M0 macrophages), and compared for the expression of M1 and M2 markers. RT-PCR analysis demonstrated that activation of both wild type and NOS2−/− macrophages led to M1 polarization as indicated by comparable upregulation of the genes encoding the Th1 cytokines TNF, IL-1β, and IL-12, the chemokines CCL2, CCL5, CXCL9, CXCL10, and other M1 markers, such as MHC-II, and downregulation of genes encoding M2 markers, such as arginase (Arg1), CD163, Mrc1, PPAR, VEGF-α, Angpt2, and Fgf1 (Fig. S1a). As expected, Nos2 expression was only induced in wt macrophages that could be confirmed at the protein level by flow cytometry (Fig. S1b). In conclusion, wild-type and Nos2−/− macrophages displayed similar gene expression profiles indicating that the lack of iNOS activity does not have profound effects on the expression of genes encoding cytokines, chemokines, and macrophage markers.
Since RT5 mice are on the C3HeBFe background and Nos2−/− mice are on the C57Bl/6 background, we used for the macrophage transfer experiments C57Bl/6 mice bearing large s.c.-transplanted B16.OVA tumors. Previous studies using this tumor model have shown that the adoptive transfer of tumor-specific OT-I CD8+ T cells alone resulted only in mild T-cell infiltration into B16.OVA tumors21). Macrophages derived from C57Bl/6 CD45.2 wt or Nos2−/− mice were activated with LPS and IFNγ (M1) or untreated (M0), and then injected into congenic CD45.1 mice bearing B16-OVA tumors. One day later, homing of activated wt and Nos2−/− macrophages was followed by staining for the CD45.2 marker. Both types of macrophages were found to migrate into the tumor to the same degree, with wt macrophages maintaining their iNOS expression suggesting that iNOS deficiency has no impact on the homing capacity (Fig. 3a and b). Non-activated M0 macrophages showed lower infiltration probably due to the lower expression of chemokines or chemokine receptors. Importantly, wt macrophages but not Nos2−/− macrophages induced VCAM-1 expression on tumor endothelia (Fig. 3c and d).
Figure 3.

Homing of adoptively transferred macrophages into RT5 tumors Bone marrow-derived CD45.2+ macrophages were isolated from CD45.2 C57Bl/6 wild-type and Nos2−/– mice and activated with LPS plus IFNγ (M1) or left untreated (M0). CD45.1 C57Bl/6 mice bearing B16-OVA tumors of about 300 mm3 were injected i.v. with CD45.2+ macrophages. One day later, mice were sacrificed for analysis. (a) Representative flow cytometric analysis of CD45.2+ transferred macrophages in tumors of CD45.1 mice. (b) Quantification by flow cytometry of CD45.2+ transferred macrophages and CD45.1+ endogenous macrophages in B16-OVA tumors. (c and d) VCAM-1 expression on CD31+ tumor endothelial cells following macrophage transfer as determined by immunohistology. Results are shown as mean ±SEM of 10 mice per group from two independent experiments. *p < 0 .05, **p < 0 .01, ***p < 0 .001, ns = not significant.
We next investigated whether iNOS-expressing macrophages promoted T-cell infiltration and tumor destruction. For this, one day after macrophage transfer the mice received OT-I CD8+ T cells, and the immune infiltrate was examined 2 d later. Transfer with OT-I T cells alone resulted only in moderate T-cell infiltration, whereas the combination of wt M1 macrophages strongly promoted leukocyte influx, in particular, infiltration of CD8+ T cells. (Fig. 4a–c). In contrast, co-transfer of Nos2−/− M1 macrophages and CD8+ T cells yielded only a low degree of T-cell homing into the tumor, similar to co-transfer with M0 macrophages that express only low levels of iNOS. Importantly, and in agreement with the observed VCAM-1 induction and T-cell infiltration, transfer of OT-I T cells alone, or of iNOS-expressing macrophages alone, failed to have a strong effect on tumor growth. Only co-transfer of OT-I T cells with iNOS-expressing macrophages resulted in tumor rejection (Fig. 4d). Analysis of the tumor microenvironment showed that co-transfer of CD8+ T cells with wt macrophages, but not with Nos2−/− macrophages, strongly modulated the microenvironment by induction of genes encoding the cytokines IFN-α, IFN-β, IFNγ, TNF, IL-12, IL-1, IL-2 IL-6, IL-10, IL-4, and IL-13, and the chemokines CCL2, CCL5, CXCL9, and CXCL10 (Fig. S2). In contrast, genes associated with angiogenesis, including Vegf-α, Angpt2, Fgf1, and regulator of G-protein signaling 5 (Rgs5)3 were downregulated. Thus, the co-transfer with iNOS-expressing macrophages induced a proinflammatory tumor microenvironment that supports tumor rejection.
Figure 4.

Co-transfer of iNOS+ macrophages with tumor-specific CD8+ T cells promotes infiltration of T cells and tumor rejection Bone marrow-derived CD45.2+ macrophages from wild-type and Nos2−/– mice and activated with LPS and IFNγ (M1) and injected i.v. into B16-OVA-bearing CD45.1 mice. One day later the mice were injected with activated OT-I Thy1.1+CD8+ T cells and sacrificed 2 d after CD8+ T cell transfer. (a) Quantification of total CD45.2+ T cell infiltration. (b) Lymphocyte infiltration of innate cells. (c) Infiltration of transferred OT-I Thy1.1+CD8+ T cells. (d) Tumor growth curves after co-transfer of macrophage and T cells. Data are shown as mean ±SEM of 10 mice per group from two independent experiments. *p < 0 .05, **p < 0 .01, ***p < 0 .001, ns = not significant.
Macrophage-derived NO stimulates adhesion molecule expression on human umbilical vein endothelial cells (HUVECs)
So far, the findings establish that iNOS expression by polarized M1-like macrophages is essential for induction of adhesion molecules and support of T-cell infiltration. Several possibilities could account for the observed induction of VCAM-1. First, macrophage-derived NO could act directly on endothelial cells and stimulate expression of adhesion molecules. Second, the effect of NO could be indirect and cause other cell types in the tumor microenvironment to upregulate VCAM-1 expression. In order to study the underlying mechanism, as well as the relevance for human endothelial cells, HUVECs were co-cultured with the human monocytic cell line THP-1 that had been differentiated by PMA into macrophages and polarized by treatment with LPS plus IFNγ. Fig. 5a shows that activated THP-1-derived macrophages induce VCAM-1 expression on HUVECs. Inhibition of iNOS activity by L-NIL prevented expression of adhesion molecules. These findings were confirmed with human primary macrophages derived from blood monocytes. Human M1 like macrophages activated with LPS plus IFNγ stimulated the expression of adhesion molecules VCAM-1, ICAM-1, and E-Selectin on HUVECs, whereas M0 macrophages failed to do so. Again, induction of adhesion molecules was inhibited by L-NIL (Fig. 5b). Together, these data suggest that NO derived from M1 like human macrophages can stimulate expression of adhesion molecules on human endothelial cells in the absence of third-party cells.
Figure 5.

Induction of adhesion molecules on human umbilical vein endothelial cells (HUVECs) by co-culture with M1-polarized human macrophages or synthetic NO donor (a) THP-1 human monocytes were differentiated into THP-1 macrophages by phorbol 12-myristate 13-acetate (PMA) treatment for 24 h. After 6 d in culture, THP-1 macrophages were activated by LPS plus IFNγ for 18 h (M1) or left untreated (M0) and co-cultured with HUVECs in the presence or absence of the iNOS inhibitor L-NIL. VCAM-1 expression was determined by flow cytometry. (b) Human macrophages were differentiated from blood monocytes in the presence of M-CSF for 7 d, activated by LPS plus IFNγ for 18 h (M1) or left untreated (M0), and co-cultured with HUVECs. Expression of VCAM-1, ICAM-1, and E-Selectin on HUVECs was determined by flow cytometry. (c) HUVECs were cultured for 18 h with indicated doses of NO donor GTN without or with TNF. VCAM-1, ICAM-1, and E-Selectin expression was measured by flow cytometry. Results are shown as mean ±SEM of six samples per group from two independent experiments. (d) NFkB dependency of induction of adhesion molecules on HUVECs by low dose GTN (100 μM). Inhibition by 10 μM IkB-a inhibitor BAY11-7082. Results are shown from a least two independent experiments.*p < 0 .05, **p < 0 .01, ***p < 0 .001, ns = not significant.
Only low but not high doses of NO induce expression of adhesion molecules in endothelial cells
NO is usually regarded as a suppressive molecule which, among other activities, has been reported to prevent expression of adhesion molecules on endothelial cells.12-14,22 In order to unravel the cause for these discrepant observations, we treated HUVECs with different doses of the NO donor glyceryl trinitrate (GTN). Low amounts of GTN ( 10 μM and 50 μM) were found to induce VCAM-1, ICAM-1, and E-selectin expression on HUVECs, whereas higher concentrations (250 μM and 500 μM) failed to induce adhesion molecules (Fig. 5c). When HUVECs were stimulated with TNF, which is a potent inducer of adhesion molecule expression on endothelial cells, the lower concentrations of GTN enhanced the expression of VCAM-1, ICAM-1, and E-selectin induced by TNF. In contrast, the higher GTN concentrations suppressed the TNF-dependent induction of adhesion molecules (Fig. 5c).
TNF is known to activate the NFkB sigalling pathway. Therefore, we used the specific IkBa inhibitor Bay 11–7082 in order to assess whether or not the NFkB pathway was involved in induction of adhesion molecules by low amounts of NO. Bay 11–7082 efficiently blocked the induction of adhesion molecules by both, NO and TNF, indicating involvement of the NFkB pathway (Fig. 5d). Together, these findings show that low amounts of NO can stimulate adhesion molecule expression in an NFkB-dependent manner, whereas higher doses are suppressive. This concentration dependency explains the discrepancy between our and other studies, in which comparable titrations of NO donors in the absence of TNF apparently have not been performed.
Transcriptional profiling of HUVECs stimulated with low doses of NO or TNF demonstrates preferential induction of genes involved in leukocyte diapedesis
The transcriptional profile of HUVECs treated with low quantities of NO donor (GTN, 100 μM) was compared to HUVECs treated with TNF (10 ng/mL), by microarray analysis using the Human HT-12 expression beadchip (Fig. 6a and b). Analysis of more than 47.000 genes demonstrated that 572 genes were differentially expressed between NO-treated HUVECs and untreated HUVECs. 589 genes were differentially expressed between TNF-treated HUVECs and untreated HUVECs. Of these, 473 genes were upregulated by both, NO or TNF treatment. Interestingly, in this latter group many of the top genes induced by either NO or TNF were among those that encode adhesion molecules, such as E-Selectin (SELE) and VCAM-1 (VCAM1), and chemokines, including CX3CL1, CCL20, CXCL10, CXCL5, CXCL2, and IL-8 (CXCL8) (Table 1). Pathway analysis showed that the leukocyte adhesion and diapedesis gene clusters belonged to the major pathways activated in HUVECs by either NO or TNF treatment (Table 2).
Figure 6.
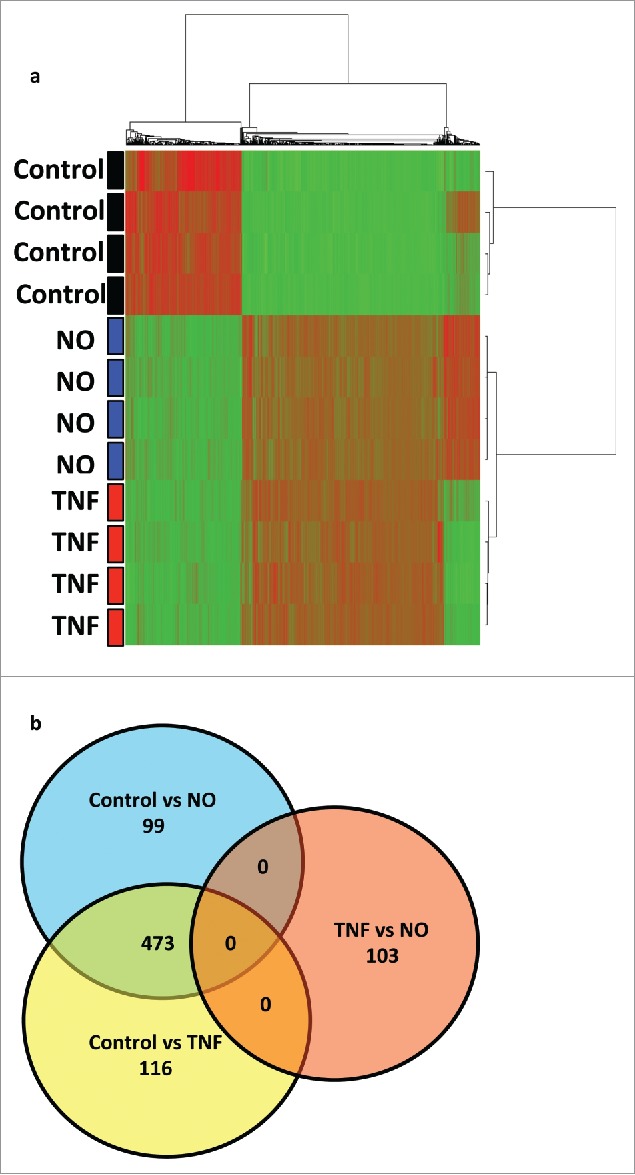
Comparative transcriptional profiling of HUVECs treated with NO donor GTN or TNF. (a) Unsupervised cluster analysis of untreated HUVECs (controls) versus HUVECs treated with low dose NO donor GTN (100 μM) or treated with TNF (10 ng/mL). (b) Venn-diagram of genes differentially expressed in control, NO, and TNF treatment (p < 0.001, fold change > 2 ). 473 genes were upregulated by both TNF and NO treatment.
Table 1.
Top upregulated genes in HUVEC after NO and TNF treatment.
| NO-treated vs. untreated |
TNF-treated vs. untreated |
||
|---|---|---|---|
| Molecule | Fold change | Molecule | Fold change |
| SELE | 72.38 | SELE | 102.3 |
| UBD | 49.68 | VCAM1 | 64.14 |
| VCAM1 | 48.1 | UBD | 49.33 |
| LTB | 44.55 | LTB | 42.26 |
| CX3CL1 | 30.57 | CX3CL1 | 41.59 |
| CCL20 | 25.56 | CCL20 | 34.34 |
| DARC | 23.53 | SLC7A2 | 27.7 |
| MMP10 | 22.4 | DARC | 26.98 |
| SLC7A2 | 19.95 | MMP10 | 26.62 |
| CXCL10 | 19.42 | CXCL10 | 25.96 |
| MX1 | 19.02 | MX1 | 25.26 |
| CXCL5 | 17.84 | CXCL5 | 22.59 |
| LAMC2 | 16.98 | CXCL6 | 21.82 |
| EBI3 | 16.59 | CXCL2 | 20.89 |
| SOD2 | 14.85 | SOD2 | 18.96 |
| CXCL2 | 14.7 | EBI3 | 18.45 |
| IL-8 | 14.49 | IL-8 | 17.97 |
Table 2.
Top activated pathways and regulator effect networks in HUVEC after NO and TNF treatment.
| NO-treated vs. untreated | TNF-treated vs. untreated | ||
|---|---|---|---|
| Top canonical pathways | Top canonical pathways | ||
| Name |
p value |
Name |
p value |
| Agranulocyte adhesion and diapedesis | 2.04 × 10−12 | Interferon signaling | 6.18 × 10−14 |
| Granulocyte adhesion and diapedesis | 3.50 × 10−12 | Granulocyte adhesion and diapedesis | 2.01 × 10−13 |
| Activation of IRF by cytosolic pattern recognition receptors | 6.05 × 10−10 | Agranulocyte adhesion and diapedesis | 6.45 × 10−12 |
| Interferon signaling | 9.37 × 10−10 | Activation of IRF by cytosolic pattern recognition receptors | 7.27 × 10−12 |
Discussion
Our results demonstrate a crucial role for macrophage-derived NO in T-cell extravasation into tumors. We show that in vivo activation of TAMs by danger signals, such as TLR ligands, induces iNOS expression. As a consequence, macrophage-derived NO, the primary product generated by iNOS, stimulates expression of adhesion molecules on tumor vessels, thereby promoting diapedesis of tumor-specific effector T cells and tumor elimination. These findings are supported by gene expression profiling studies of HUVECs activated by low concentrations of NO donor, which identified leukocyte diapedesis pathways among the major pathways induced by NO. Furthermore, the NFkB pathway emerged as one of the top signaling pathways, as indicated by the inhibition of adhesion molecule induction by NO through the NFkB inhibitor Bay-117082. TNF is a classical activator of adhesion molecules on endothelial cells, which is known to signal via NFkB.12 Interestingly, treatment of HUVECs with TNF induced a similar set of genes as treatment with NO, with leukocyte diapedesis among the top induced pathways. The precise NFkB-dependent molecular mechanism by which low doses of NO activate leukocyte extravasation pathways in endothelial cells remains to be explored.
NO can directly interact with target molecules, but it can also react with another free radical, the superoxide anion, to form the powerful oxidant peroxynitrite.23-25 It is assumed that many of the effects attributed to NO are actually indirect effects caused by peroxynitrite or other NO derivatives. Thus, it is not clear whether the functions of NO descibed here are direct or indirect effects. NO and its derivatives are important mediators of diverse biological processes including blood vessel relaxation, neuronal cell function, platelet aggregation, anti-microbial, and tumoricidal activities. In the immune system, NO was frequently, but not exclusively, found to act as an immunosuppressant.9,24,26 Nevertheless, our observation that NO exerts a stimulatory function on endothelial cells by inducing adhesion molecules was unexpected as prior publications documented an anti-adhesion effect of NO (see below). Myeloid-cell-derived NO is known to inhibit T-cell proliferation and function.8-10 On the other hand, smaller amounts of NO supported the survival and differentiation of T-cell subpopulations, notably Th1 and Th9 cells.27,28 Low quantities of NO were also reported to activate NFkB.29
Of direct relevance for the present study are the numerous reports demonstrating that NO inhibits expression of adhesion molecules on endothelia, which is in striking contrast to the results described here.11-14,22 Our observation that an exogenously added NO donor was inhibitory at high concentrations but stimulatory at low doses offers an explanation for this apparent discrepancy. Our results indicate that iNOS+ macrophages produce in vitro as well as in vivo precisely the quantities of NO that are required for induction of adhesion molecules, but not high quantities that may cause suppression. As NO is a diffusible gas with a short halflife of 1–5 sec, it is likely that for NO to exert its biological functions the location of the producing cells and target cell, timing, and concentration are important. Under the experimental conditions used here, namely T-cell transfer and modulation of tumor macrophages by CpG-ODN, we did not observe any of the potential imunosuppressive effects of macrophage-derived NO.
The combined transfer of iNOS+ macrophages with T cells induced dramatic changes in the tumor microenvironment with polarization of M2- toward M1-like macrophages, upregulation of proinflammatory cytokines, i.e., IFNγ, TNF, IL-12, T-cell-attracting chemokines such as CCL5, CXCL9, and CXCL10, and downregulation of genes encoding pro-angiogenic factors including VEGF-α, ANG2, RGS5, FGF1, and PDGF-α. Although activated iNOS deficient macrophages exhibited a very similar cytokine and chemokine expression profile as iNOS-expressing macrophages, they failed to induce such changes in the tumor, demonstrating the importance of NO as an initiator for the observed alterations.
In a pevious study, we have shown that also other danger signals such as low dose radiation of tumors can induce iNOS+ macrophages that are critical for tumor rejection.7 Thus, iNOS+ macrophages seem to play a central role in different settings of T-cell-mediated cancer immunotherapy. Importantly, our data suggest that polarization toward NO-producing macrophages by danger signals alone does not result in strong T-cell extravasation, but that assistance by the infiltrating T cells is required. As danger signals alone induce adhesion molecules only on a small fraction of endothelial cells, we envisage the following scenario that is depicted as a diagram in Fig. 7. The initial low adhesion molecule expression induced by NO will allow a small number of effector T cells to infiltrate. Effector T cells are known to secrete cytokines such as IFNγ and TNF and thereby are likely to induce more adhesion molecules so that in a self-amplifying feedback mechanism more and more T cells can infiltrate. Thus, it is possible that NO-mediated induction of adhesion molecules is mainly required for initiation of this feedback process. The effect of T-cell-derived cytokines is probably not restricted to enhancement of adhesion molecules expression, but may also drive further polarization of TAMs toward M1 macrophages30 that produce T-cell-attracting chemokines, such as CCL5, CXCL9, and CXCL10. In addtion, M1 macrophages are characterized by strongly reduced synthesis of proangiogenic factors such as VEGF, a process resulting in normalization of the tumor vasculature, decreased tumor hypoxia and enhanced T-cell infiltration and reactivity.3,21,31 Clearly, such a T-cell-driven infiltration process depends on sufficient numbers of potent tumor-specific T cells, which is in agreement with our previous observation that following treatment with radiation or CpG-ODN optimal tumor rejection is only obtained after two or more adoptive transfers of T cells.4,5,21
Figure 7.

M1 macrophage-derived NO promotes T-cell infiltration into tumors (a) initiation of extravasation by NO. 1. CpG polarises M2- toward iNOS+ M1-like macrophages. 2. M1 macrophage-derived NO induces cellular adhesion molecules (CAM) on tumor vessels. 3. T cells adhere and extravasate. (b) Feedback amplification loop. 4. Extravasating T cells produce cytokines such as IFNɣ and TNF. 5. These cytokines induce more CAMs and more iNOS+ M1-like macrophages. 6. More T cells extravasate. Not included into the diagram are additional functions of M1-like macrophages such as production of the T cell attractants CXCL9, CXCL10, tumor vessel normalization and decrease of hypoxia.
Macrophage populations in tumors are known to be heterogeneous with varying proportions of M2 and M1 macrophages. Recent clinical studies suggest a correlation between the presence of M1-skewed CD68+ HLA-DR+ iNOS+macrophages and clinical outcome in tumors such as non-small cell lung cancer, pancreatic cancer, colorectal cancer, ovarian cancer, and renal cell carcinoma32-36). NO-producing macrophages can be cytocidal in vitro against tumor cells,37,38 but formal evidence for in vivo cytoxicity against solid tumors is lacking. In our present study, transfer of activated iNOS+ M1 polarized macrophages alone had only a minor effect on tumor growth, suggesting no major cytotoxic activity in vivo. Only transfer together with T cells resulted in tumor rejection. In view of our data, it will be of interest to determine whether an increased number of M1-like macrophages in human cancers correlates with enhanced T-cell infiltration and improved response to immunotherapy. The data presented here will be valuable for the design of novel therapeutic strategies. Although tumor-associated M2 macrophages are pro-tumorigenic and interfere with T-cell-mediated immunotherapy, macrophage depletion approaches may not enhance tumor rejection as depletion would probably not only remove M2, but also the useful iNOS+ M1 macrophages. Instead, it appears more promising to use drugs or treatment modalities that polarize TAMs toward iNOS-expressing M1-like macrophages.
Materials and methods
Mice
RIP1-Tag5 (RT5) mice expressing the Tag oncogene on pancreatic β cells were generated on the C3HeBFe background and kindly provided by D. Hanahan (Swiss Institute for Experimental Cancer Research, Lausanne, Switzeland ).18 RT5 mice, TCR-8 mice expressing a Tag-specific TCR, were restricted by H-2Kk.4 C57BL/6N mice and OT-I mice expressing a TCR specific for the ovalbumin amino acids 257–264 epitope and restricted to H-2Kb were bred at the central animal facility of the German Cancer Research Center and held under specific pathogen-free conditions. iNOS-deficient mice (Nos2−/−) on a C57BL/6 background were originally obtained from the Jackson Laboratories (Bar Harbor, ME, USA; stock no. 260939) and bred under specific pathogen-free conditions at the Franz Penzoldt Preclinical Animal Research Center of FAU Erlangen. 6- to 10-week-old mice were used for experiments. Experiments were conducted according to governmental and institutional guidelines and regulations (Regierungspräsidium Karlsruhe, permit no. 35-9185.81/G205/13 and 35–9185.81/G206/12). The number of mice per group was confirmed by the Biostatistics Department of the German Cancer Research Center.
Tumor challenge
The ovalbumin-expressing B16 melanoma (B16-OVA)40 was cultured in complete DMEM medium and routinely tested for mycoplasma contamination. For tumor induction, 1×106 B16-OVA cells were injected intradermally into the right flank of C57Bl/6N mice. Tumor sizes were measured with a caliper every 2−3 d, and tumor volume was calculated according to the formula: volume = 0.5 × length × width2. Mice were killed when the tumor volume reached 2.000 mm3.
Injection of CpG oligonucleotide and adoptive transfer of CD8+ T cells
Tumor-bearing mice were injected i.v. with 25 μg of phosphothionate-stabilized CpG-ODN 1668 (Sigma Aldrich, Steinheim Germany) on days 0, 6, and 10 and with activated T cells on day 1 and 11 as described previously.4,21 Briefly, single cell suspensions were prepared from spleens and peripheral lymph nodes of naive OT-I or TCR-8 mice. Red blood cells were lysed with ACK buffer. Cells were suspended at a density of 1 × 106 cells per mL in RPMI-1640 medium supplemented with 10% FCS, 2 nM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.05 mM 2-mercaptoethanol, 10 U of recombinant IL-2 per mL. TCR-8 cells were activated with 25 nM Tag peptide 560–568 (SEFLLEKRI) and OT-I cells with 25 nM OVA peptide SIINFEKL. Three days after activation, 2.5 × 106 activated CD8+ T lymphocytes were transferred i.v. into recipient mice. For survival experiments the T-cell transfer was repeated every 10 d.
Macrophage depletion and iNOS inhibition
Macrophages were depleted from tumor-bearing mice by i.p. injection of 200 μL CLIP on days 0, 5, and 10, and then 100 μL every 5 d over 3 weeks for the duration of the experiment (Encapsula NanoSciences N. van Rooijen, VUMC). Control liposomes contained PBS. iNOS blockade was performed by continuously providing 2 mM L-N6-(1-iminoethyl)lysine9 (L-NIL) (Cayman Chemicals) in the drinking water.
Tissue digestion for population analyses
Tumors isolated from mice were placed in 3 mL of PBS containing 100 U/mL of collagenase IV and 1 mg/mL of DNase I, disrupted with forceps and were incubated at 20°C under gentle stirring with a magnet (100 rpm). After 30 min, samples were passed through a 40-μm filter. Erythrocytes were removed by incubation for 1 min with ACK buffer (Dulbecco's PBS containing 0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA). Cells were next incubated for 15 min in blocking buffer (flow cytometry buffer (Dulbecco's PBS containing 2.5% FCS) buffer with 1% normal immunoglobulin (Privigen; CSL Behring).
Antibodies, flow cytometry
Mouse cells were stained with the following fluorochrome-conjugated antibodies anti-CD3 (145-2C11), anti-CD8+ (53–6.7), anti-NK1.1 (PK136), anti-CD11c (HL3), anti-CD19 (1D3), (all from BD Biosciences); anti-iNOS (CXFNT), (all from eBiosciences); anti-CD4+ (GK1.5), anti-CD45.1 (A20), anti-CD45.2 (104) anti-CD11b (M1/70), anti-CD90.1 (OX-7), anti-I-A/I-E (M5/114.15.2), anti-Gr-1 (RB6-8C5), anti-F4/80 (BM8), CD206 (C068C2), (all from Biolegend). Human cells were stained with anti-CD14 (63D3), anti-CD68 (Y1/82A), anti-VCAM-1 (STA), anti-E-Selectin (HAE-1f), anti-CD31 (WM59), (BioLegend); anti-vWF (AHP062F), (ABD Serotec); anti-ICAM-1 (01), (SinoBiological Inc.). Propidium iodide (Sigma-Aldrich) or fixable viability dye (eBiosciences) were used as a viability dye. Labeled cells were analyzed on a FACSCanto II flow cytometer (BD Biosciences) and evaluated with FlowJo Mac software, version.8.2 (TreeStar). For analysis of macrophages purified from tumors, 1–5 × 104 cells were sorted with a FACSAria II (BD Biosciences).
Quantitative RT-PCR analysis
RNA from tumors and macrophages sorted by flow cytometry (purity >95 %) was isolated with an RNeasy Micro kit (Qiagen), followed by cDNA synthesis with an iScript cDNA synthesis kit (Bio-Rad). Gene expression was assessed by real-time PCR with SYBR green I master (Roche) and primers in a Light Cycler® 480 (Roche). Polymerase was activated at 95°C for 10 min. Samples were amplified by 40 cycles of 10 s at 95°C and 1 min at 60°C. Dissociation curves were used to confirm specificity of the PCR. Results were calculated by the change-in-cycling-threshold (ΔΔCt) method as follows (relative to the control gene Gapdh, encoding glyceraldehyde phosphate dehydrogenase): −ΔΔCt = 2 – ΔCt sample – ΔCt biggest Ct, where ΔCt = Ct target mRNA – Ct Gapdh mRNA. PCR was performed by an independent researcher unaware of sample group allocation.
Bio-Plex cytokine analyses
Tumor tissue was lysed using the Bio-Plex Cell Lysis Kit (Bio-Rad) and processed as described previously.7 Cytokines were quantified using the multiplex protein array system technology (Bio-Rad Laboratories) according to the manufacturer's protocol (R&D Systems).
Isolation of mouse and human macrophages
Bone marrow-derived macrophages were prepared from wild-type or NOS2−/− mice and cultured at 3.5 × 106 cells/mL in RPMI-1640 medium supplemented with 10% FCS, 2 nM glutamine, 100 U/mL penicilin, 100 μg/mL streptomycin, and 10 ng/mL mouse recombinant macrophage-colony stimulating factor (M-CSF) (Sigma-Aldrich, Steinheim, Germany). Five days later, culture medium was replaced by fresh medium supplemented wih M-CSF. Three days later macrophages were activated with 20 ng/mL LPS (Sigma-Aldrich, Steinheim, Germany) and 20 ng/mL mouse recombinant IFNγ (PeproTech, Hamburg, Germany) or left untreated. Eighteen hours later, macrophages were harvested using StemPro® Accutase® cell dissciation reagent (Life Technologies, Darmstadt Germany). For adoptive transfer, 5 × 106 activated and non-activated macrophages in 200 μL dPBS were injected i.v.
For generation of human macrophages, mononuclear cells were isolated from human peripheral blood by density gradient centrifugation using Ficoll-Paque™ (GE Healthcare, Freiburg, Germany. Mononuclear cells were plated at 2 × 108/175 cm2 in RPMI-1640 medium supplemented with 10% FCS, 2 nM glutamine, 100 U/mL penicilin, 100 μg/mL streptomycin and incubated for 90 min at 37°C. Non-adherent cells were discarded and adherent cells were isolated using StemPro® Accutase® cell dissociation reagent. The resulting monocytes were incubated at 2 × 106 cells with 25 ng/mL human recombinant M-CSF (PeproTech, Hamburg, Germany). After 6 d, differentiation into macrophages was confirmed by staining with CD14 and CD68 antibodies. Macrophages were left untreated (M0 macrophages) or activated in the presence of 100 ng/mL LPS and 20 ng/mL human recombinant IFNγ (PeproTech, Hamburg, Germany) for 18 h to generate M1 macrophages.
Co-culture of HUVECs and human macrophages
HUVECs from pooled donors (PromoCell, Heidelberg, Germany) were cultured in ENDOPAN 3 endothelial cell growth medium (Pan-Biotech, Aidenbach, Germany) supplemented with 3% FBS, 0.1% FGF-2, 0.1% VEGF, 0.1% R3-IGF-1, 0.1% ascorbic acid, 0.1% heparin, 0.12% gentamicin sulfate amphotericin B, 0.02% hydrocortisone. 1 × 106 M1 or M0 macrophages were added to HUVECs cultured at 1 × 105 cells/mL 3 d ago in six-well plates in the presence or absence of iNOS inhibitor L-NIL (1 mM). Eighteen hours later, HUVECs were harvested and the expression of adhesion molecules VCAM-1, ICAM-1, and E-Selectin measured by cytofluorometry. HUVECs were distinguished from macrophages by their expression of von Willebrand Factor (vWF).
Activation of HUVECs with NO donors
HUVECs were seeded at 1 × 105 cells/mL in six-well plates in ENDOPAN 3 endothelial cell growth medium. After 3 d, titrated amounts of NO donors glycerol trinitrate (GTN) (Sigma-Aldrich, Steinheim, Germany) were added to the cultures with or without 10 ng/mL TNF. After 18 h cells were harvested and the expression of adhesion molecules VCAM-1, ICAM-1 and E-Selectin measured.
Microarray analysis
Total RNA was isolated from untreated HUVECs or HUVECs treated with NO donor (GTN) or TNF, and hybridized to Human HT-12 v4 expression beadchip (Illumina, San Diego, USA) at the DKFZ Genomics and Proteomics Core Facility. Normalized gene expression intensities were compared, and genes were considered to be differentially expressed between different groups if their fold change was greater than 2.
Immunohistology
Immunohistology was performed as described previously.21 Briefly, tumors were embedded in OCT compound, snap-frozen, sectioned (Leica CM1950, Leica Biosystems), stained with hematoxylin-eosin or different antibodies. For visualization of stained sections, a ZeissAxio Observer.Z1 microscope was used. ZEN Blue software (Zeiss) was used for analysis.
Statistical analysis
Comparisons between two groups were assessed by Student's t-test. Comparisons of tumor-growth curves were assessed by analysis of variance. Survival studies were assessed by Kaplan–Meier curves and the log-rank (Mantel-Cox) test. Data were analyzed with Prism 6 software (GraphPad). p values lower than 0.05 were considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank N. Bulbuc for expert technical help.
Funding
G.J.H. was supported by the Wilhelm Sander-Stiftung, N.G. by the Deutsche Forschungsgemeinschaft (SFB 704, and C.B. by the Deutsche Forschungsgemeinschaft (SFB 643 and SFB 1181) and the Interdisciplinary Center for Clinical Research of the University Hospital Erlangen (project A61).
References
- 1.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012; 12:298-306; PMID:22419253; http://dx.doi.org/ 10.1038/nrc3245 [DOI] [PubMed] [Google Scholar]
- 2.Ryschich E, Schmidt J, Hammerling GJ, Klar E, Ganss R. Transformation of the microvascular system during multistage tumorigenesis. Int J Cancer 2002; 97:719-25; PMID:11857345; http://dx.doi.org/ 10.1002/ijc.10074 [DOI] [PubMed] [Google Scholar]
- 3.Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Grone HJ, Hammerling GJ et al.. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008; 453:410-4; PMID:18418378; http://dx.doi.org/ 10.1038/nature06868 [DOI] [PubMed] [Google Scholar]
- 4.Garbi N, Arnold B, Gordon S, Hammerling GJ, Ganss R. CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. J Immunol 2004; 172:5861-9; PMID:15128765; http://dx.doi.org/ 10.4049/jimmunol.172.10.5861 [DOI] [PubMed] [Google Scholar]
- 5.Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res 2002; 62:1462-70; PMID:11888921 [PubMed] [Google Scholar]
- 6.Cao ZA, Daniel D, Hanahan D. Sub-lethal radiation enhances anti-tumor immunotherapy in a transgenic mouse model of pancreatic cancer. BMC Cancer 2002; 2:11; PMID:12019035; http://dx.doi.org/ 10.1186/1471-2407-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L et al.. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013; 24:589-602; PMID:24209604; http://dx.doi.org/ 10.1016/j.ccr.2013.09.014 [DOI] [PubMed] [Google Scholar]
- 8.Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 2015; 36:161-78; PMID:25687683; http://dx.doi.org/ 10.1016/j.it.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 9.Liew FY. Regulation of lymphocyte functions by nitric oxide. Curr Opin Immunol 1995; 7:396-9; PMID:7546406; http://dx.doi.org/ 10.1016/0952-7915(95)80116-2 [DOI] [PubMed] [Google Scholar]
- 10.Umansky V, Schirrmacher V. Nitric oxide-induced apoptosis in tumor cells. Adv Cancer Res 2001; 82:107-31; PMID:11447761; http://dx.doi.org/ 10.1016/S0065-230X(01)82004-2 [DOI] [PubMed] [Google Scholar]
- 11.Spiecker M, Peng HB, Liao JK. Inhibition of endothelial vascular cell adhesion molecule-1 expression by nitric oxide involves the induction and nuclear translocation of IkappaBalpha. J Biol Chem 1997; 272:30969-74; PMID:9388244; http://dx.doi.org/ 10.1074/jbc.272.49.30969 [DOI] [PubMed] [Google Scholar]
- 12.Waldow T, Witt W, Weber E, Matschke K. Nitric oxide donor-induced persistent inhibition of cell adhesion protein expression and NFkappaB activation in endothelial cells. Nitric Oxide 2006; 15:103-13; PMID:16504556; http://dx.doi.org/ 10.1016/j.niox.2005.12.005 [DOI] [PubMed] [Google Scholar]
- 13.Lee SK, Kim JH, Yang WS, Kim SB, Park SK, Park JS. Exogenous nitric oxide inhibits VCAM-1 expression in human peritoneal mesothelial cells. Role of cyclic GMP and NF-kappaB. Nephron 2002; 90:447-54; PMID:11961404; http://dx.doi.org/ 10.1159/000054733 [DOI] [PubMed] [Google Scholar]
- 14.Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Pro Natl Acad Sci U S A 1996; 93:9114-9; PMID:8799163; http://dx.doi.org/ 10.1073/pnas.93.17.9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol 2007; 178:1505-11; PMID:17237399; http://dx.doi.org/ 10.4049/jimmunol.178.3.1505 [DOI] [PubMed] [Google Scholar]
- 16.Ridnour LA, Cheng RY, Weiss JM, Kaur S, Soto-Pantoja DR, Basudhar D, Heinecke JL, Stewart CA, DeGraff W, Sowers AL et al.. NOS Inhibition Modulates Immune Polarization and Improves Radiation-Induced Tumor Growth Delay. Cancer Res 2015; 75:2788-99; PMID:25990221; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q, Tomei S, Ascierto ML, De Giorgi V, Bedognetti D, Dai C, Uccellini L, Spivey T, Pos Z, Thomas J et al.. Melanoma NOS1 expression promotes dysfunctional IFN signaling. J Clin Invest 2014; 124:2147-59; PMID:24691438; http://dx.doi.org/ 10.1172/JCI69611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985; 315:115-22; PMID:2986015; http://dx.doi.org/ 10.1038/315115a0 [DOI] [PubMed] [Google Scholar]
- 19.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23:549-55; PMID:12401408; http://dx.doi.org/ 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- 20.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277-86; PMID:23518347; http://dx.doi.org/25915731 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 21.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nature Immunol 2015; 16:609-17; PMID:25915731; http://dx.doi.org/ 10.1038/ni.3159 [DOI] [PubMed] [Google Scholar]
- 22.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O'Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nature Med 2008; 14:28-36; PMID:18157142; http://dx.doi.org/ 10.1038/nm1699 [DOI] [PubMed] [Google Scholar]
- 23.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Pro Natl Acad Sci U S A 1997; 94:6954-8; PMID:9192673; http://dx.doi.org/ 10.1073/pnas.94.13.6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogdan C. Nitric oxide and the immune response. Nature Immunol 2001; 2:907-16; PMID:11577346; http://dx.doi.org/ 10.1038/ni1001-907 [DOI] [PubMed] [Google Scholar]
- 25.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007; 87:315-424; PMID:17237348; http://dx.doi.org/ 10.1152/physrev.00029.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013; 34:503-12; PMID:23354310; http://dx.doi.org/ 10.1093/carcin/bgt034 [DOI] [PubMed] [Google Scholar]
- 27.Niedbala W, Besnard AG, Nascimento DC, Donate PB, Sonego F, Yip E, Guabiraba R, Chang HD, Fukada SY, Salmond RJ et al.. Nitric oxide enhances Th9 cell differentiation and airway inflammation. Nat Commun 2014; 5:4575; PMID:25099390; http://dx.doi.org/ 10.1038/ncomms5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niedbala W, Wei XQ, Piedrafita D, Xu D, Liew FY. Effects of nitric oxide on the induction and differentiation of Th1 cells. Eur J Immunol 1999; 29:2498-505; PMID:10458764; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199908)29:08%3c2498::AID-IMMU2498%3e3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 29.Umansky V, Hehner SP, Dumont A, Hofmann TG, Schirrmacher V, Droge W, Schmitz ML. Co-stimulatory effect of nitric oxide on endothelial NF-kappaB implies a physiological self-amplifying mechanism. Eur J Immunol 1998; 28:2276-82; PMID:9710205; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199808)28:08%3c2276::AID-IMMU2276%3e3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- 30.Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E, Ponsoda S, Delneste Y, Hebbar M, Jeannin P. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer 2009; 125:367-73; PMID:19378341; http://dx.doi.org/ 10.1002/ijc.24401 [DOI] [PubMed] [Google Scholar]
- 31.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307:58-62; PMID:15637262; http://dx.doi.org/ 10.1126/science.1104819 [DOI] [PubMed] [Google Scholar]
- 32.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. The tissue microlocalisation and cellular expression of CD163, VEGF, HLA-DR, iNOS, and MRP 8/14 is correlated to clinical outcome in NSCLC. PloS One 2011; 6:e21874; PMID:21799753; http://dx.doi.org/ 10.1371/journal.pone.0021874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013; 108:914-23; PMID:23385730; http://dx.doi.org/ 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edin S, Wikberg ML, Dahlin AM, Rutegard J, Oberg A, Oldenborg PA, Palmqvist R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteri-stics of colorectal cancer. PloS One 2012; 7:e47045; PMID:23077543; http://dx.doi.org/ 10.1371/journal.pone.0047045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res 2014; 7:19; PMID:24507759; http://dx.doi.org/ 10.1186/1757-2215-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dannenmann SR, Thielicke J, Stockli M, Matter C, von Boehmer L, Cecconi V, Hermanns T, Hefermehl L, Schraml P, Moch H et al.. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunol 2013; 2:e23562; PMID:23687622; http://dx.doi.org/ 10.4161/onci.23562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med 1996; 183:1323-9; PMID:8666890; http://dx.doi.org/ 10.1084/jem.183.4.1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nowosielska EM, Wrembel-Wargocka J, Cheda A, Lisiak E, Janiak MK. Enhanced cytotoxic activity of macrophages and suppressed tumor metastases in mice irradiated with low doses of X- rays. J Radiat Res 2006; 47:229-36; PMID:16936416; http://dx.doi.org/ 10.1269/jrr.0572 [DOI] [PubMed] [Google Scholar]
- 39.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Pro Natl Acad Sci U S A 1995; 92:10688-92; PMID:7479866; http://dx.doi.org/ 10.1073/pnas.92.23.10688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Kostareli E, Suffner J, Garbi N, Hammerling GJ. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur J Immunol 2010; 40:3325-35; PMID:21072887; http://dx.doi.org/ 10.1002/eji.201041093 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.