

ABSTRACT
T lymphocytes, which recognize antigen peptides through specific T cell receptors (TCRs), play an important role in the human adaptive immune response. TCR diversity is closely associated with host immune response and cancer prognosis. Although tumor-infiltrating T lymphocytes have implications for tumor prognosis, few studies have performed a detailed characterization of TCR diversity in both tumor and non-tumor tissues in hepatitis B virus (HBV)-associated hepatocellular carcinoma (HCC). Here, we performed high-throughput sequencing of the TCRβ chain complementarity determining region 3 (CDR3) of liver-infiltrating T cells from 48 HBV-associated HCC patients. A significantly higher average number of CDR3 aa clonotypes (2259 vs. 1324, p < 0.001), and significantly higher TCR diversity (Gini coefficient, p < 0.001; Simpson index, p < 0.01; Shannon entropy, p < 0.001) were observed in tumor tissues compared with adjacent non-tumor tissues. The ratio of highly expanded clones (HECs) was significantly higher in non-tumor tissues than in tumor tissues when the HEC threshold was defined as 2% or greater (p < 0.05). Our analysis of the median Morisita-Horn index indicated weak TCR repertoire similarity between tumor and matched non-tumor tissues. The median number of shared clones in tumor tissue and matched non-tumor tissue from each patient was 360.5, representing 5.1–15.8% (10.6 ± 0.4%) of all clones in each patient. We observed extensive heterogeneity of T lymphocytes in tumors and higher HEC ratios in adjacent non-tumor tissues of HCC patients. The differential T cell repertoires in tumor and non-tumor tissues suggest a distinct T cell immune microenvironment in patients with HBV-associated HCC.
KEYWORDS: Diversity, hepatitis B virus, hepatocellular carcinoma, high-throughput sequencing, T cell receptor
Abbreviations
- CDR3
complementarity-determining region 3
- CHB
chronic hepatitis B
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HEC
highly expanded clone
- MHSI
Morisita-Horn similarity index
- NSDE
normalized Shannon diversity entropy
- PLC
primary liver cancer
- TCR
T cell receptor
Introduction
Primary liver cancer (PLC) is the sixth most common cancer in the world. More than 700,000 new cases occur every year worldwide, and approximately half of these cases are in China.1 Hepatocellular carcinoma (HCC) accounts for more than 80% of PLC, and more than half of HCC cases in China are caused by hepatitis B virus (HBV).2,3
Although HBV is the most prominent etiology associated with HCC development, it is not directly involved in hepatocarcinogenesis. The pathogenesis of HBV-associated HCC remains unclear. Previous studies have demonstrated that viral factors, including HBV DNA integration, HBV genotype, HBx protein transactivation, preS gene deletion, and HBV genomic mutation, may contribute to HBV-associated HCC development.4-6 Furthermore, host immune attacks caused by HBV infection may lead to hepatocyte inflammation, necrosis, and proliferation. Repeated cycles of this destructive–regenerative process may activate signaling pathways of cell proliferation, leading to oncogenic transformation. Multi-causative factors in hepatocarcinogenesis suggest that aberrant neoantigens are expressed in cancer cells, resulting in the presentation of peptides on the cells that can be bound by T cell receptors (TCRs) of T lymphocytes.
T lymphocytes play an important role in human adaptive immune responses and recognize antigen peptides via specific TCRs on the cell surface. More than 90% of TCRs consist of an α and β chain. The diversity of TCRs originates from the random combination of variable (V) and joining (J) gene segments in the α-chain and V, diversity (D) and J gene segments in the β-chain germline gene. Random insertions and deletions of non-template nucleotides at the junctions of V-(D)-J gene segments further increase the diversity of TCRs, and up to 1018 TCR types have been reported. The specificity and diversity of TCRs predominantly depend on the complementarity determining region 3 (CDR3), which is encoded by V(D)J recombination and interacts with the peptide presented by the major histocompatibility complex (MHC).7
TCR diversity is closely related to treatment effectiveness and patient prognosis in different diseases.8-11 The analysis of TCR diversity in diseases might also facilitate the elucidation of pathogenesis and the development of cellular immunotherapy approaches that target specific tumors and neoantigens.12,13 Although tumor-infiltrating T lymphocytes have implications for tumor prognosis, information on the TCR diversity of HBV-associated liver cancer remains limited.14-16 A recent investigation of HBV-associated HCC indicated that the highly expanded clone (HEC) ratio of TCRβ CDR3 (TRB CDR3) was significantly different in liver cancer compared to healthy and hepatitis B patients.17 The comparison of TRB CDR3 among three histological types of liver cancer samples revealed that the consistency between tumor tissues and adjacent non-tumor tissues was related to tumor malignancy. However, because HBV-associated HCC develops as a result of HBV infection, the immune microenvironments differ between tumor and adjacent non-tumor tissues, and the consequent TRB CDR3 diversity should be elucidated, respectively. Furthermore, there is an urgent need for data regarding whether CDR3 diversity in tumor tissues is correlated with HCC progression and prognosis. In addition, determining the heterogeneity of T lymphocytes in tumors is essential to further characterize the mechanisms of diverse neoantigens. Addressing these questions would help improve the efficacy of immunotherapy for HBV-related HCC. Consequently, TCR diversity and clinical parameters were compared between tumors and adjacent non-tumor tissues of HBV-associated HCC patients.
Results
Profiling of TRB CDR3 in HCC
Our study cohort consisted of 48 patients with HBV-associated HCC (Table S1). The total number of reads was 13,719,931, with an average of 142,916 reads per sample (Table S2). We identified 64 distinct Vβ gene segments, which could be merged into 30 Vβ gene families, and 14 distinct Jβ gene segments, which could be merged into 2 Jβ gene families from the 96 samples. The average number of distinct Vβ gene segments in each sample was 53, with a range of 47 to 59. The numbers of Vβ gene segments differed significantly between tumor and non-tumor tissues (56 vs. 53, p < 0.001, pair-wise t-test, Fig. 1). The most frequent Vβ gene segments in both tissues were TRBV20-1 (14.31%), TRBV29-1 (8.42%), TRBV5-1 (5.64%), TRBV7-2 (5.63%), TRBV28 (5.24%), and TRBV7-9 (5.12%) (Figs. S1A and B). The most frequent Jβ segments were TRBJ2-1 (22.39%), TRBJ2-7 (21.57%), TRBJ2-3 (11.12%), and TRBJ2-5 (10.07%) (Figs. S1C and D). Notably, TRBV20-1 and TRBJ2-1 were also the most frequent gene segments in other studies of tumors and healthy subjects.18,19
Figure 1.
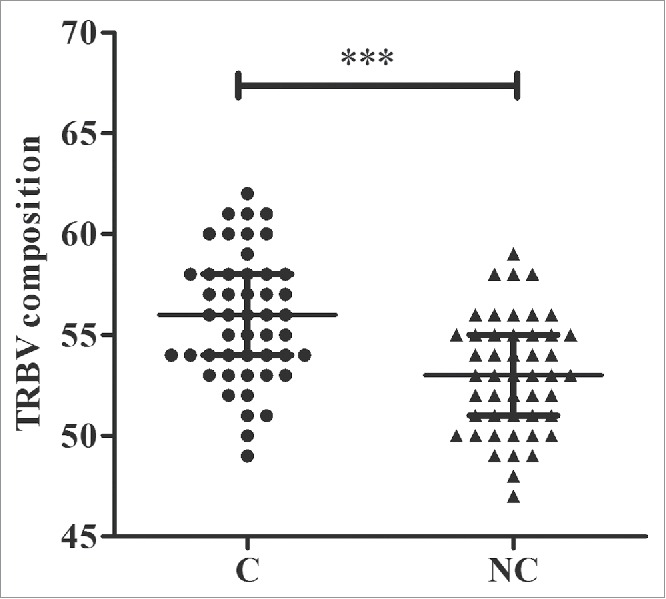
Numbers of TRBV gene segments used in tumor and adjacent non-tumor tissues. C, tumor tissues; NC, adjacent non-tumor tissues. *** p < 0.001, according to a pair-wise t-test. The error bars indicate the median with the interquartile range.
The usage patterns of Vβ and Jβ gene segments were similar between tumors and adjacent non-tumor tissues
According to the heatmap of VJ gene segments (Fig. 2), the usage patterns of Vβ and Jβ gene segments were similar between tumors and adjacent non-tumor tissues, whereas some Vβ and Jβ gene segments exhibited significantly higher frequency in non-tumor tissues, such as TRBV20-1, TRBV9, TRBV10-3, TRBV5-4, TRBV10-2, and TRBJ2-7, TRBJ2-1, and TRBJ1-5. After comparing the usage frequency, significant differences in a total of 25 Vβ gene segments were observed between tumor and non-tumor tissues (all p-values were < 0.05, Wilcoxon signed rank test, Fig. 3A). Among them, TRBV7-4, TRBV7-7, and TRBV10-3 were used more frequently in adjacent non-tumor tissues, whereas the others (22 Vβ segments, 88.8%) exhibited an increased usage frequency in tumor tissues. However, only 2 Jβ gene segments, TRBJ1-2 and TRBJ1-4, had significant usage frequency differences, and both of these segments exhibited increased frequency in tumor tissues (Fig. 3B).
Figure 2.
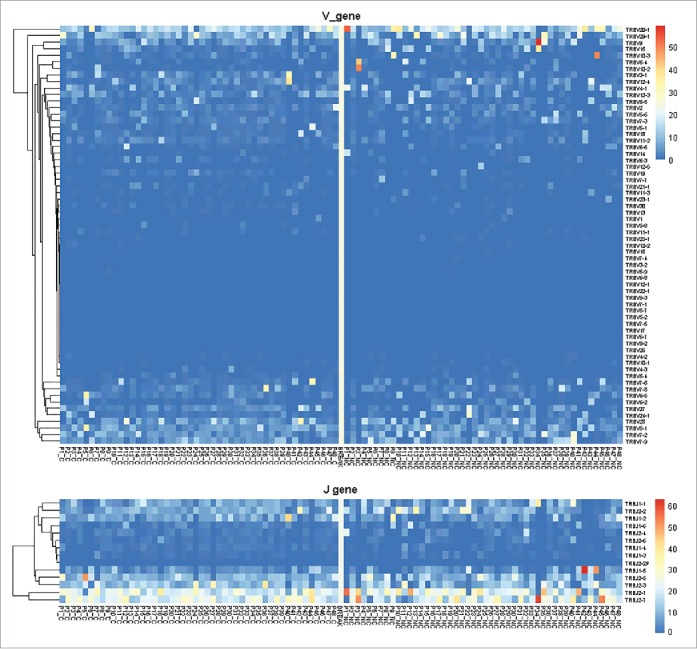
Heatmap of Vβ and Jβ gene segments usage in tumors and adjacent non-tumor tissues of HBV-associated HCC. The heatmap bar indicates the usage frequency of Vβ or Jβ gene segments in each sample.
Figure 3.

Comparison of Vβ and Jβ gene segments used in tumors and adjacent non-tumor tissues. (A) Twenty-five Vβ gene segments with different usage frequencies between tumor and non-tumor tissues (all p-values were less than 0.05, Wilcoxon signed rank test). Fold change = the mean frequency in tumor tissues divided by the mean frequency in non-tumor tissues. (B) Two Jβ gene segments with different usage frequencies between tumor (C) and non-tumor tissues (NC). *p < 0.05, **p < 0.001.
Most VJ and VDJ gene combinations were used more frequently in tumors compared with adjacent non-tumor tissues
A total of 781 VJ gene combinations and 2,100 VDJ gene combinations were identified from the 96 samples. There were 301 (38.5%) VJ gene combinations exhibited significant usage differences between tumors and adjacent non-tumor tissues (p < 0.05, Wilcoxon signed rank test; Fig. S2A, Table S3). Most of the VJ combinations (91.3%) were used more frequently in tumors than in non-tumor tissues, whereas only 26 VJ combinations (8.7%) were used more frequently in adjacent non-tumor tissues. Similarly, significant differences were observed in a total of 673 VDJ combinations (32.0%, all p-values were < 0.05) between tumors and non-tumor tissues (Fig. S2B). Only 61 VDJ gene segment combinations (9.1%) exhibited increased usage frequency in non-tumor tissues, whereas the remainder (90.9%) exhibited opposite patterns.
Higher TRB CDR3 diversity in tumors compared with adjacent non-tumor tissues
In our study, 105,895 distinct CDR3 nucleotide (nt) clonotypes and 90,228 distinct CDR3 amino acid (aa) clonotypes were identified in 96 samples. The median CDR3 nt clonotype was 1803 (range 638–9,468), and the median CDR3 aa clonotype was 1704 (range 617–8,585).
The TRB CDR3 nt lengths in most samples were 30–60 bp, with an average length of 45 ± 2 bp (mean ± SD). However, CDR3 length did not discriminate tumor from non-tumor tissues. The length distributions of TRB CDR3 were nearly identical in both tissues (Fig. S3).
To further analyze the TRB CDR3 diversity between tumors and adjacent non-tumor tissues, we compared the numbers of distinct CDR3 aa clonotypes. The number of CDR3 aa clonotypes was significantly higher in tumors compared with non-tumor tissues (2,259 vs. 1,324, p < 0.001, Fig. 4A), and identical results were obtained in different age groups (< 40 y, 40–60 y, and > 60 y, all p-values < 0.001, Fig. 4C). However, compared with patients whose ages ranged from 40 to 60 y, patients older than 60 y of age exhibited higher counts of CDR3 aa clonotypes (2,410 vs. 1,494, p = 0.020, Fig. 4B), whereas no significant differences were noted between the other 2 groups (<40 y vs.> 60 y, p = 0.745; < 40 y vs. 40–60 y, p = 0.609). No significant difference was observed in the number of CDR3 aa clonotypes between male and female subjects (p = 0.073, Fig. 4D).
Figure 4.

CDR3 aa clonotype comparisons. (A) Comparison of CDR3 aa clonotype numbers between tumors and adjacent non-tumor tissues. (B) Comparisons of CDR3 aa clonotype numbers between different age groups. (C) Comparison of CDR3 aa numbers between tumor and non-tumor tissues in different age groups. (D) Comparison of CDR3 aa numbers between the two gender groups. C, tumor tissues; NC, adjacent non-tumor tissues. *p < 0.05; ***p < 0.001; n.s, no statistical significance.
The heatmap of the Vβ and Jβ segments suggested oligo-clonal expansions in the tissues. Therefore, we first assessed the TRB diversity in samples by analyzing the proportions of the top five CDR3 aa clonotypes. The proportions of the top five CDR3 aa clonotypes were significantly higher in adjacent non-tumor compared with tumor tissues (42.0% vs. 22.0%, p < 0.001, Wilcoxon signed rank test; Fig. 5A, Fig. S4). Because the top five CDR3 aa clonotypes might include some oligo clones, we then selected HECs (defined by a CDR3 aa clonotype clonal frequency exceeding a certain threshold) from the tumor and non-tumor tissues from each patient and assessed their numbers to determine whether HECs were enriched in non-tumor tissues. We assigned this threshold across a rational range from 1% to 10% because only 50–100 mg tissue was used to amplify the TCR repertoire and perform the statistical test under each threshold. When we defined the HEC threshold as 2%, the numbers of HECs in tumors and non-tumor tissues were significantly different (6 vs. 9, p = 0.018, Fig. 5B). An identical result was obtained when we defined the threshold as 3%, 4%, 5%, or even 10% (Fig. 5C). The HEC counts were less in tumor tissues. These results suggest that the tumor tissues might have more extensive heterogeneity and, potentially, higher TRB diversity. Therefore, we used the Gini coefficient, Simpson index and normalized Shannon diversity entropy (NSDE) to compare TCR CDR3 diversity in tumors and adjacent non-tumor tissues. All results indicated that the tumor tissues exhibited significantly higher TCR CDR3 diversity compared with non-tumor tissues (all p-values were < 0.01, Fig. 6).
Figure 5.

Comparison between tumor and adjacent non-tumor tissues. (A) Comparison of the top five CDR3 aa ratios between tumor and non-tumor tissues. (B) Comparison of HEC numbers between tumor and non-tumor tissues. (C) The cutoff for HEC. C, tumor tissues; NC, adjacent non-tumor tissues. *p < 0.05; ***p < 0.001.
Figure 6.

TCR CDR3 diversity between tumor and adjacent non-tumor tissues. Comparison of TCR CDR3 diversity by the Gini coefficient (A), Simpson index (B), and normalized Shannon diversity entropy (NSDE) (C). C, tumor tissues; NC, adjacent non-tumor tissues. **p < 0.01; ***p < 0.001.
Weak similarity and overlap between tumors and adjacent non-tumor tissues
To assess the similarity between the tumor and matched adjacent non-tumor tissues from each patient, we calculated the Morisita-Horn similarity index (MHSI) of each pair of samples using the abundance or frequency of CDR3 aa sequences of each patient. The median MHSI of tumors and non-tumor tissues was 0.035 (the upper and lower quartiles were 0.119 and 0.007, respectively; Fig. S5), indicating that the similarity between the tumor and non-tumor tissue from each patient was very weak in most patients.
Because the tumor tissue and matched adjacent non-tumor tissue were obtained from the same patient, we hypothesized that certain CDR3 aa clonotypes might be shared by the tissues of the same patient. In this study, CDR3 aa clonotypes that were shared by tumor and non-tumor tissues or shared by more than half of the samples were named shared clones. The median number of shared clones in tumor and paired non-tumor tissues from each patient was 360.5, with a range of 142–967, accounting for 5.1–15.8% (10.6% ± 0.4%, mean ± std. error) of all clones in each patient. Most of the shared clones appeared with low frequency, generally less than 0.1% (Fig. S6). The percentage of shared clones was higher in adjacent non-tumor tissues than in tumor tissues (25.9% vs. 16.4%, p < 0.001, Wilcoxon signed rank test, Fig. 7A).
Figure 7.

Comparison of shared clones between different groups. (A) Comparison of percentage of shared clones in tumor and adjacent non-tumor tissues. (B) Comparison of numbers of shared clones between different frequency groups. (C) Comparison of percentages of shared clones between different frequency groups. C, tumor tissues; NC, adjacent non-tumor tissues. C > NC, the frequency in C was increased compared with NC; C<NC, the frequency in C was reduced compared with NC; C = NC, the frequencies in C and NC were identical. The error bar represents the median; **p < 0.01, ***p < 0.001, according to a non-parametric test.
Three groups of clones were divided as C > NC, C < NC, and C = NC according to their CDR3 aa clonotype frequency in tumors and non-tumor tissues (Figs. 7B and C). The number and percentage of clonotypes in group C>NC were higher compared with the other 2 groups, suggesting that most clones (including shared clones and those clones that exclusively appeared in tumor tissues) were used more frequently in tumor tissues.
We then analyzed the shared clones in 48 tumor tissues or 48 adjacent non-tumor tissues. The numbers and percentages of CDR3 aa shared clones in the 48 samples were extremely low, particularly those clones whose frequency was > 0.1%, 0.5%, or 1% (Fig. S7).
Tumor and adjacent non-tumor tissues can be well classified by multi-dimensional scaling (MDS)
To further distinguish tumors from adjacent non-tumor tissues, we compared the two groups using multi-dimensional scaling (MDS) of VDJ gene combination and CDR3 aa abundance. The abundances of the VDJ and CDR3 aa clones were scaled for normalization and used to calculate log2-fold changes of shared clones between each pair of samples. The distance between each pair of samples was the root-mean-square deviation for the log2-fold changes of shared clones. MDS of VDJ gene combination abundance distinguished tumor tissues from non-tumor tissues (Fig. 8A). Although we could not discriminate tumors from adjacent non-tumor tissues by MDS of CDR3 aa abundance (Fig. 8B), the greater dispersion of tumor tissues suggested differences between the two tissues.
Figure 8.
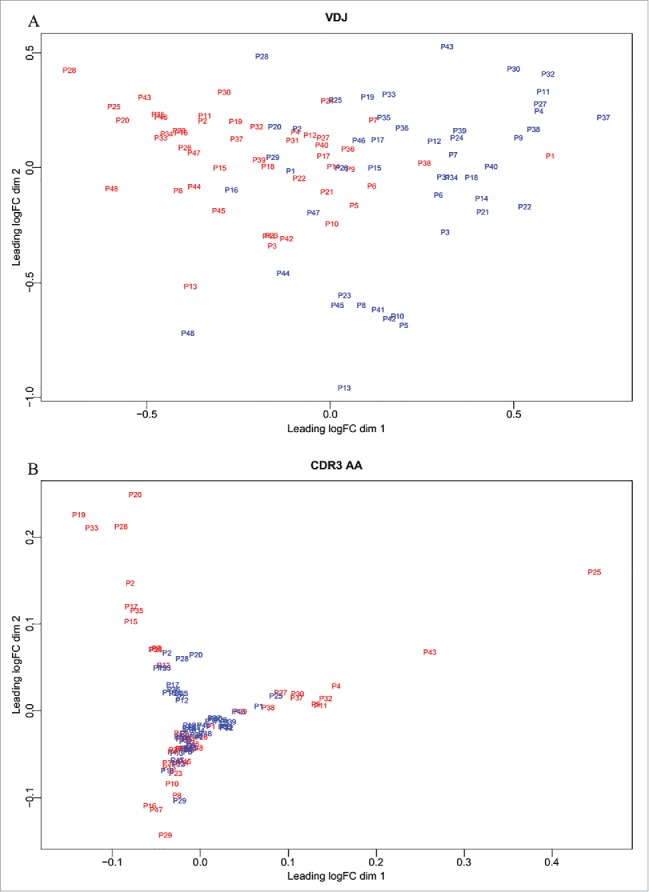
Multidimensional scaling. To distinguish tumor from adjacent non-tumor tissues, we used VDJ (A) and CDR3 aa (B) abundance to perform the MDS.
The correlation between TCR diversity and tumor characteristics was weak
To further analyze the relationship between TCR diversity and tumor characteristics, we divided the patients into two or three groups according to the tumor TNM stage, degree of differentiation and metastasis. MHSI and NSDE were compared between groups. The MHSI of stage I was higher compared with stage II (p = 0.025), but no significant differences were observed between the other two groups (Fig. 9A). There were no significant differences in MHSI between the differentiation and metastasis groups (Figs. 9B and C). Similarly, no significant differences were observed among these groups by comparing NSDE, which measures the immune diversity of the patients (Figs. 9D-F).
Figure 9.

Comparison of MHSI and NSDE in different groups. In total, 48 HCC patients were divided into three groups (stage I, stage II and stage III) according to the tumor TNM stage. In addition, patients were divided into three groups (poorly differentiated, moderate differentiated and well differentiated) according to the degree of differentiation. (A–C) comparison of MHSI; (D–F) comparison of NSDE. *p < 0.05; n.s, no statistical significance.
Discussion
The TCR diversity of chronic hepatitis B and other diseases has been previously estimated by flow cytometry, spectratyping, and Sanger sequencing.20-23 These techniques have aided the analysis of TCR diversity to some extent but are associated with disadvantages. These techniques are not suitable for large samples with millions of sequences and can only be used to analyze the dominant V gene segments, not all probable V gene segments. In addition, these techniques cannot be used to thoroughly analyze CDR3 sequences. High-throughput sequencing can simultaneously sequence millions of molecules and has been proven to be helpful to analyze the true TCR or BCR repertoires in patients.24 To reduce PCR bias as much as possible in this study, 5′ rapid amplification of cDNA ends (5′ RACE) was used as described.25
In China, most cases of HCC result from HBV infection. Because HCC is highly heterogeneous, varying immune responses are observed in different patients, and the immune microenvironment differs between tumor and adjacent non-tumor tissues. Therefore, it is meaningful to analyze the TCR diversity in tumors and non-tumor tissues of HCC patients to aid the analysis of the pathogenesis and immune response in patients and identify new biomarkers that might be related to the tumor antigens.
The Vβ and Jβ gene segments are the main components of the TCR repertoire. In this study, more diverse Vβ gene segments and CDR3 aa clonotypes were observed in tumors compared with adjacent non-tumor tissues, whereas the percentage of top five CDR3 aa clonotypes and the ratio of HECs were higher in non-tumor tissues. These results are consistent with the Gini coefficient, Simpson index, and NSDE and suggest that TCR diversity is higher in tumor tissues. In addition, some oligo-clonal expansions were observed in adjacent non-tumor tissues underlying HBV infection. Studies of other tumors, such as clear cell renal cell carcinomas and colorectal tumors, have indicated that TCR diversity in tumor tissues is much lower compared with adjacent non-invasive renal tissues or adjacent mucosal tissues.26,27 Research on esophageal squamous cell carcinoma (ESCC) has demonstrated that the tumor tissues were not significantly more oligo-clonal than adjacent normal tissues and blood samples.28 The discrepancies between the results of these studies and our study might be attributable to HBV infection, which might cause inflammation and necrosis in adjacent non-tumor tissues. In addition, years or decades are needed for hepatitis or cirrhosis to advance to HCC; thus, the immune environment in hepatitis B patients is not identical to that in other tumors.
The percentage of shared clones was 5.1–15.8% (10.6 ± 0.4%) in each HCC patient. These values are smaller than the degree of tumor and normal tissue overlap in ESCC, which accounted for approximately 25% of samples, but similar to the degree of tumor and adjacent mucosal tissue overlap in colorectal tumors, which ranged from 0.2 to 38%.29,30 This difference might be related to the different starting materials and approaches for TCR profiling. Although starting from genomic DNA and multiplexed PCR strategies is relatively easier for performing, these techniques are associated with significant bias. Thus, approaches starting with mRNA and 5′ RACE are more reliable for TCR repertoire analysis. While, all these data confirmed that the number of clones shared between tumors and non-tumor tissues is low, regardless of the tumor type. Therefore, to further elucidate the pathogenesis of cancer, it is essential to analyze the microenvironment of tumors and non-tumor tissues.
Although some shared clones were detected in our study, a majority of these shared clones were used with low frequency, generally less than 0.1%. Whether the low frequency is related to T cell exhaustion in response to tumor and HBV antigen requires further study.31 In addition, the low frequency of shared clones suggests that HCC is a heterogeneous disease and highlights neoantigen heterogeneity. McGranahan et al. recently explored the impact of neoantigen intratumor heterogeneity (ITH) on antitumor immunity in early-stage non-small cell lung cancer and determined that tumors with both a high clonal neoantigen burden and low neoantigen ITH were associated with significantly longer progression-free survival.32 Our data imply that the TCR diversity in tumor tissues is high and that the percentage of shared clones is low. This finding might help explain the poor prognosis of HCC because clonal neoantigens are rarely shared by all HCC patients.
With the exception of the shared clones, we analyzed VJ combinations in 48 HCC patients. Some fixed VJ combinations were observed in liver cancer tissues. For example, TRBV17 just assembled with J1-2, J2-1, and J2-6 in all 96 samples, and TRBV22-1 just assembled with J1-3, J2-2, J2-3, J2-4, and J2-7 (Fig. S8). We identified 60 VJ combinations that exclusively appeared in tumor tissues and 12 VJ combinations that appeared exclusively in adjacent non-tumor tissues. However, these specific VJ combinations appeared with low frequency (0.001 to 0.4%). Whether these specific VJ combinations and the shared clones are related to tumor antigens requires further study.
Recent studies have focused on adoptive cell therapies (ACT) that target specific tumors, and neoantigens have been ideal targets for cancer immunotherapy. ACT has resulted in obvious regressions in leukemia, lymphoma, melanoma, cervical cancer, bile duct cancer, and neuroblastoma.33 Tumor-specific neoantigens arise from gene mutations and could be recognized by T cells after processing and presentation on the cell surface.34 TCR sequences obtained from tumor tissues by high-throughput sequencing might allow potential neoantigens that are specific for TCR to be identified and used for TCR immunotherapy.35 Our study successfully screened out shared clones, particularly those clones that appeared exclusively in tumor tissues. Whether these clones are specific for HCC requires further study.
Some researchers have indicated that TCR diversity is related to disease prognosis. VanHeijst et al. reported that CD4+ T cell diversity was higher in DUCB (double-unit umbilical cord blood transplantation) recipients compared with TCD (T cell-depleted peripheral-blood stem cell transplantation) patients after 6 months of treatment. DUCB recipients had a lower incidence of infection, higher CD4+ T cell numbers and a reduced rate of leukemia relapse compared with TCD recipients.8 Muraro et al. reported that CD4+T and CD8+T TCR diversity in response patients was increased compared with non-response patients before and after haematopoietic stem cell transplantation.9 Jia et al. reported that high NSDE of the mucosal T cell repertoire in gastric cancer correlates with patient short-term and overall survival time.11 These results suggest that TCR diversity might be a new biomarker for the prediction of the prognosis of different diseases. In addition, comparing TCR diversity might enable the selection of a suitable therapy for a specific patient, such as chemotherapy, radiofrequency ablation, transcatheter arterial chemoembolization, and surgery resection. However, as our study is a cross-sectional study, the overall survival and relapse of HCC patients after tumor resection is unknown. Whether TCR diversity can predict HCC patient prognosis must be further analyzed in a large, longitudinal study.
In conclusion, using high-throughput sequencing technology in combination with 5′ RACE, we successfully analyzed the TCR repertoire in tumors and adjacent non-tumor tissues of HBV-associated HCC patients. Our findings suggest that TCR diversity is higher in tumors compared with adjacent non-tumor tissues. The similarity and overlap between tumor and non-tumor tissues was low. Further longitudinal studies and functional examination of specific CDR3 sequences or gene combinations are needed.
Materials and methods
Patients
A total of 48 HBV-associated HCC patients were enrolled in this study. Paired HCC tumor tissues and adjacent non-tumor tissues (at least 2 cm × 2 cm × 2 cm) were obtained from all patients who underwent liver resection. All HCC diagnoses were confirmed by pathological examination. The exclusion criteria were as follows: (1) patients with hepatitis C virus or human immunodeficiency virus co-infection; (2) tumor radiation treatment or chemotherapy; (3) previous or current use of molecular targeted anti-cancer drugs; and (4) secondary liver cancer or with other tumors. The tumor specimens were immediately frozen at −80°C or in liquid nitrogen until RNA extraction. The study was conducted in accordance with the Helsinki Declaration of 1975 and approved by the Ethics Committee of Nanfang Hospital.
RNA extraction and cDNA synthesis
Total RNA was isolated from 50–100 mg of tissue using TRIzol reagent (Invitrogen, USA) according to the manufacturer's instruction. RNA concentrations were determined using a NanoDrop 2000 spectrophotometer. Unbiased TCR cDNA libraries for high-throughput sequencing were prepared by 5′ rapid amplification of cDNA ends (RACE) using the SMARTer PCR cDNA synthesis kit (Clontech, USA) as previously described by Mamedov et al.25 Briefly, 1.5 μg of total RNA was mixed with the primer BC1R (CAGTATCTGGAGTCATTGA), which is specific for human TCR β cDNA synthesis. To denature RNA and anneal the priming oligonucleotides, RNA was incubated at 72°C for 3 min and then at 42°C for 2 min. The SMARTer IIA oligonucleotide and SMARTScribe reverse transcriptase were added for 5′-template switching and the cDNA synthesis reaction, which was performed at 42°C for 60 min.
TCR library preparation
Two-round PCR was performed for TCR library preparation as described with slight modification. Briefly, for the first round of PCR amplification, 2 μL of cDNA from the synthesis reaction was mixed with Advantage 2 polymerase mix (Clontech, USA), the universal primer Smart20 (CACTCTATCCGACAAGCAGTGGTATCAACGCAG), and the TCR β-specific primer BC2R (TGCTTCTGATGGCTCAAACAC). The reaction was performed for 18 cycles using the following temperature regimen: 95°C for 20s, 65°C for 20s, and 72°C for 50s.
For the second round of PCR amplification, the product from the first round of PCR was purified by magnetic beads (Beckman, USA), and one fifth of the purified product was used in each 50 μL PCR reaction. The primers TRB-step1 (CACTCTATCCGACAAGCAGT) corresponding to the 5′ end and Hum-bcj (ACACSTTKTTCAGGTCCTC) corresponding to the 3′ end of the libraries were used. The reaction was performed for 16–18 cycles with the following temperature regimen: 95°C for 20s, 65°C for 20s, and 72°C for 50s. PCR products were identified on 2% agarose gels, and bands centered at 500–600 bp were excised and purified using Qiaquick Gel Extraction kit (Qiagen, Germany). Illumina adaptors were ligated using NEBnext Ultra DNA Library Prep kit (New England BioLabs, USA) according to the manufacturer's protocol. Libraries were amplified using Illumina sequencing primers with different sample barcodes. Then, the purified PCR product was subjected to high-throughput sequencing using the Illumina HiSeq3000 platform with a read length of 2 × 150 bp.
Data processing and analysis
The original data obtained from high-throughput sequencing were converted to raw sequence reads by base calling, and the results were stored in FASTQ format. Low-quality sequences were discarded. TCRβ V, D, and J gene identification; CDR3 sequence extraction; and error corrections in clean reads were performed using miXCR.36
Statistical analysis
A paired t-test was used to compare matched samples between tumors and adjacent non-tumor tissues in populations assumed to be normally distributed. The Wilcoxon signed rank test was used to compare matched samples between tumor and non-tumor tissues. The Wilcoxon rank-sum test was used to compare independent samples in tumor or non-tumor tissues. The Kruskal–Wallis H test was used to compare three or more samples. The Morisita-Horn similarity index was used to measure the similarity between pairs of variables. Two-sided p-values < 0.05 were considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant number: 81371802 and 91331107).
Author contributions
ZHW and YQC conceived and designed the study. YQC, YX, and MXZ performed the experiments. YQC, YL, MXG, and CTX collected samples and clinical information. YQC, YX, and HKW analyzed the sequencing data. ZHW, YQC, and HKW interpreted the data. ZHW and YQC wrote the manuscript.
References
- 1.Stewart BW, Wild CP. World Cancer Report 2014. World Health Organization, 2014. pp.Chapter 1.1. ISBN 978-92-832-0432-9; http://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-2014 [Google Scholar]
- 2.El Serag HB, Rudolph KL. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132:2557-76; PMID:17570226; http://dx.doi.org/ 10.1053/j.gastro.2007.04.061 [DOI] [PubMed] [Google Scholar]
- 3.Sarin SK, Kumar M, Lau GK, Abbas Z, Chan HLY, Chen CJ, Chen DS, Chen HL, Chen PJ, Chien RN, et al.. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int 2016; 10:1-98; PMID:26563120; http://dx.doi.org/ 10.1007/s12072-015-9675-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arzumanyan A, Reis HMGP, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 2013; 13:123-35; PMID:23344543; http://dx.doi.org/ 10.1038/nrc3449 [DOI] [PubMed] [Google Scholar]
- 5.Bolondi L, Gramantieri L. From liver cirrhosis to HCC. Intern Emerg Med 2011; 6:93-8; PMID:22009618; http://dx.doi.org/ 10.1007/s11739-011-0682-8 [DOI] [PubMed] [Google Scholar]
- 6.McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology 2009; 49:S45-55; PMID:19399792; http://dx.doi.org/ 10.1002/hep.22898 [DOI] [PubMed] [Google Scholar]
- 7.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 2006; 24:419-66; PMID:16551255; http://dx.doi.org/ 10.1146/annurev.immunol.23.021704.115658 [DOI] [PubMed] [Google Scholar]
- 8.van Heijst JWJ, Ceberio I, Lipuma LB, Samilo DW, Wasilewski GD, Gonzales AMR, Nieves JL, van den Brink MRM, Perales MA, Pamer EG. Quantitative assessment of T cell repertoire recovery after hematopoietic stem cell transplantation. Nat Med 2013; 19:372-77; PMID:23435170; http://dx.doi.org/ 10.1038/nm.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muraro PA, Robins H, Malhotra S, Howell M, Phippard D, Desmarais C, de Paula Alves Sousa A, Griffith LM, Lim N, Nash RA, et al.. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest 2014; 124:1168-72; PMID:24531550; http://dx.doi.org/ 10.1172/JCI71691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma SW, Li YY, Zhang GW, Huang X, Sun J, Li C, Abbott WGH, Hou JL. Complementarity-Determining Region 3 Size Spectratypes of T Cell Receptor Chains in CD8+ T Cells following Antiviral Treatment of Chronic Hepatitis B. Antimicrob Agents Ch 2011; 55:888-94; PMID:21098256; http://dx.doi.org/ 10.1128/AAC.01232-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia Q, Zhou J, Chen G, Shi Y, Yu H, Guan P, Lin R, Jiang N, Yu P, Li QJ, et al.. Diversity index of mucosal resident T lymphocyte repertoire predicts clinical prognosis in gastric cancer. Oncoimmunology 2015; 4:e1001230; PMID:26137399; http://dx.doi.org/ 10.1080/2162402X.2014.1001230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Mol Oncol 2015; 9:2063-70; PMID:26404496; http://dx.doi.org/ 10.1016/j.molonc.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/ 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer 2011; 105:93-103; PMID:21629244; http://dx.doi.org/ 10.1038/bjc.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G, Peng H, Cui L, Li C. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer 2014; 110:1595-605; PMID:24504370; http://dx.doi.org/ 10.1038/bjc.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Y, Li H, Guan Y, Huang J. Immune repertoire: A potential biomarker and therapeutic for hepatocellular carcinoma. Cancer Lett 2016; 379:206-12; PMID:26188280; http://dx.doi.org/26405574 10.1016/j.canlet.2015.06.022 [DOI] [PubMed] [Google Scholar]
- 17.Han Y, Liu X, Wang Y, Wu X, Guan Y, Li H, Chen X, Zhou B, Yuan Q, Ou Y, et al.. Identification of characteristic TRB V usage in HBV-associated HCC by using differential expression profiling analysis. OncoImmunology 2015; 4:e1021537; PMID:26405574; http://dx.doi.org/ 10.1080/2162402X.2015.1021537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res 2009; 19:1817-24; PMID:19541912; http://dx.doi.org/ 10.1101/gr.092924.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai X, Zhang Q, Wu S, Zhang X, Wang M, He F, Wei T, Yang J, Lou Y, Cai Z, et al.. Characteristics of tumor infiltrating lymphocyte and circulating lymphocyte repertoires in pancreatic cancer by the sequencing of T cell receptors. Sci Rep 2015; 5:13664; PMID:26329277; http://dx.doi.org/ 10.1038/srep13664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sing GK, Li D, Chen X, Macnaughton T, Lichanska AM, Butterworth L, Ladhams A, Cooksley G. A molecular comparison of T lymphocyte populations infiltrating the liver and circulating in the blood of patients with chronic hepatitis B: evidence for antigen-driven selection of a public complementarity-determining region 3 (CDR3) motif. Hepatology 2001; 33:1288-98; PMID:11343258; http://dx.doi.org/ 10.1053/jhep.2001.24026 [DOI] [PubMed] [Google Scholar]
- 21.Yao X, Zhang GW, Ma L, Wen Q, Hou J, Meng M, Ma S, Liang M, Lin Y, Wu Z. Analysis of the CDR3 length of TCR αβ T cells in the peripheral blood of patients with chronic hepatitis B. Hepatol Res 2006; 35:10-8; PMID:16522377; http://dx.doi.org/ 10.1016/j.hepres.2006.01.006 [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Li M, Wang J, Lu H, Yao X, He J, Li L. Rapid detection of clonal expansion of T-cell receptor-beta gene in patients with HBV using the real-time PCR with DNA melting curve analysis. Hepatol Res 2010; 40:407-14; PMID:20070405; http://dx.doi.org/ 10.1111/j.1872-034X.2009.00600.x [DOI] [PubMed] [Google Scholar]
- 23.McLean-Tooke A, Barge D, Spickett GP, Gennery AR. Flow Cytometric Analysis of TCR Vβ Repertoire in Patients with 22q11.2 Deletion Syndrome. Scand J Immunol 2011; 73:577-85; PMID:21323691; http://dx.doi.org/ 10.1111/j.1365-3083.2011.02527.x [DOI] [PubMed] [Google Scholar]
- 24.Metzker ML. Sequencing technologies — the next generation. Nat Rev Genet 2009; 11:31-46; PMID:19997069; http://dx.doi.org/ 10.1038/nrg2626 [DOI] [PubMed] [Google Scholar]
- 25.Mamedov IZ, Britanova OV, Zvyagin IV, Turchaninova MA, Bolotin DA, Putintseva EV, Lebedev YB, Chudakov DM. Preparing unbiased T-Cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol 2013; 4:456; PMID:24391640; http://dx.doi.org/ 10.3389/fimmu.2013.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Jia Q, Deng T, Song B, Li L. Heterogeneous expansion of CD4+ tumor-infiltrating T-lymphocytes in clear cell renal cell carcinomas. Biochem Bioph Res Co 2015; 458:70-6; PMID:25637538; http://dx.doi.org/ 10.1016/j.bbrc.2015.01.069 [DOI] [PubMed] [Google Scholar]
- 27.Sherwood AM, Emerson RO, Scherer D, Habermann N, Buck K, Staffa J, Desmarais C, Halama N, Jaeger D, Schirmacher P, et al.. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immu 2013; 62:1453-61; PMID:23771160; http://dx.doi.org/ 10.1007/s00262-013-1446-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Zhang C, Pan Y, Xu R, Xu C, Chen Z, Lu Z, Ke Y. T cell receptor β-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J Pathol 2016; 239:450-8; ; PMID:27171315; http://dx.doi.org/24027095 10.1002/path.4742 [DOI] [PubMed] [Google Scholar]
- 29.Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, Drescher CW, Tewari M, Bielas JH, Robins HS. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol 2013; 231:433-40; PMID:24027095; http://dx.doi.org/ 10.1002/path.4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherwood AM, Emerson RO, Scherer D, Habermann N, Buck K, Staffa J, Desmarais C, Halama N, Jaeger D, Schirmacher P, et al.. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immunother 2013; 62:1453-61; PMID:23771160; http://dx.doi.org/ 10.1007/s00262-013-1446-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis 2015; 6:e1694; PMID:25789969; http://dx.doi.org/ 10.1038/cddis.2015.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al.. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463-69; PMID:26940869; http://dx.doi.org/ 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/ 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol 2016; 28:22-27; PMID:26653770; http://dx.doi.org/25924071 10.1016/j.smim.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/25924071 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 36.Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015; 12:380-81; PMID:25924071; http://dx.doi.org/ 10.1038/nmeth.3364 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.