

Abstract
Gram-negative uropathogenic Escherichia coli (UPEC) bacteria are a causative pathogen of urinary tract infections (UTIs). Previously developed antivirulence inhibitors of the type 1 pilus adhesin, FimH, demonstrated oral activity in animal models of UTI but were found to have limited compound exposure due to the metabolic instability of the O-glycosidic bond (O-mannosides). Herein, we disclose that compounds having the O-glycosidic bond replaced with carbon linkages had improved stability and inhibitory activity against FimH. We report on the design, synthesis, and in vivo evaluation of this promising new class of carbon-linked C-mannosides that show improved pharmacokinetic (PK) properties relative to O-mannosides. Interestingly, we found that FimH binding is stereospecifically modulated by hydroxyl substitution on the methylene linker, where the R-hydroxy isomer has a 60-fold increase in potency. This new class of C-mannoside antagonists have significantly increased compound exposure and, as a result, enhanced efficacy in mouse models of acute and chronic UTI.
Introduction
Since the 1970s, antibiotic resistance has been steadily increasing and now multidrug resistant pathogens are an imminent threat. Antibiotics target processes essential for bacterial replication and thus induce strong pressure to evolve resistance to these drugs. An alternate therapeutic strategy is to disarm the pathogen by inhibiting critical host–pathogen interactions necessary for persistence in a host. Herein, we describe a promising antibiotic-sparing strategy using small molecule C-mannosides that specifically block the ability of uropathogenic Escherichia coli (UPEC) to colonize the lower urinary tract by neutralizing the FimH adhesin. In mouse models, we show that C-mannosides are oral drugs that are effective in both preventing and treating urinary tract infections (UTIs). These compounds represent advanced preclinical candidates for UTI therapy.
Microbial adherence to host cells is a critical initial step in most infectious diseases.1 The ability of bacteria to bind and invade epithelial tissues allows them to establish a niche within the host and evade host immune responses.2 While antimicrobial therapy has traditionally been effective in treating bacterial infections, resistance to the most commonly used antimicrobials is on the rise,3 highlighting the need for new therapeutic strategies.4 The study of critical host–pathogen interactions during bacterial infection has revealed promising novel targets for therapeutic intervention.5 Monoclonal antibodies, vaccines, glycoconjugates, and small molecules have all been developed to disrupt bacterial adherence by competitively blocking the bacterial protein(s) involved in the recognition of various host receptors.6 This process and molecular mechanism has been most thoroughly studied in urinary tract infection (UTI) pathogenesis.
A key step in UTI pathogenesis is the initial colonization of the bladder epithelium by uropathogenic E. coli (UPEC). This binding is mediated by hair-like fibers, termed type 1 pili, that are tipped with the virulence factor FimH adhesin.7 The lectin domain of FimH binds mannosylated glycoproteins expressed on the luminal surface of human and murine bladder cells. The terminal FimH residue is a prototypical two-domain adhesin, which is joined to the distal end of the linear tip fibrillum of type 1 pili by a donor strand exchange reaction with subunit FimG.7,8 FimG then undergoes its own donor strand exchange reaction with subunit FimF, which adapts the fibrillum tip to the long pilus rod, which consists of a homopolymer of ∼1000 FimA subunits coiled into a rigid right-handed helical structure that is capable of unwinding into a linear fiber.9 The FimH lectin domain contains a deep acidic pocket that recognizes α-d-mannose with stereochemical specificity.8 FimH-mediated binding to mannosylated uroplakins10 or α1,β3 integrins11 facilitates bacterial colonization and invasion of bladder epithelial cells.12
UTIs present a significant burden for women, with nearly 20 million cases reported annually.13 Despite antibiotic treatment, 20–40% of these women will have at least one recurrence within 6 months of their initial diagnosis.14 This results in a significant economic impact, approximately two billion dollars in the U.S. alone,13 associated with these common and painful infections. The majority of UTIs (85–95%) are caused by members of the Enterobacteriaceae family; UPEC are isolated in approximately 80–85% of community-acquired UTI, and other Enterobacteriaceae account for 5–10% of infections.15 Because of the increasing prevalence of recurrent infections, as well as the increasing emergence of antibiotic resistant strains,16 including multidrug resistant UPEC such as the ESBL (extended spectrum β-lactamase) strain ST131,17 the desire for new UTI therapeutics has escalated rapidly in recent years. The requirement for FimH to cause disease has led to its classification as a promising and validated therapeutic target18 for UTI and, more recently, for Crohn’s disease.19 Inhibition of FimH function and activity circumvents bacterial bladder cell adhesion, invasion, and subsequent intracellular biofilm formation, making the bacteria unable to cause or propagate an existing infection.
We have previously developed20 small molecule glycosides based on α-d-mannose (O-mannosides), such as 1 and 4 (Figure 1), as tight-binding ligands and potent antagonists of FimH both in vitro and in animal models of UTI. Although, phenyl α-d-mannosides were first reported by Firon and Sharon in the 1980s as inhibitors of yeast agglutination by mannose-specific enterobacteria,21 the true viability of synthetic mannosides as oral therapeutics for treatment of UTI was not validated in vivo until 2010,20a,22 where the oral activity of several biphenyl α-d-mannosides in animal models of acute and chronic UTI was demonstrated. Since then, other related mannosides have also been reported23 and glycodendrimer24 FimH antagonists. X-ray crystallographic data of mannosides binding to the FimH lectin domain has aided in the rational design of higher affinity ligands, with improvement gained through interactions to the “tyrosine gate” at the periphery of the deep mannose binding pocket6c,8,20c and to a small hydrophobic pocket20b,23a,25 near the mannoside glycosidic bond. While O-mannosides have good efficacy in animal models of UTI, they have low bioavailability and half-life, which can possibly be attributed to the metabolic instability of the O-glycosidic bond20b to either hydrolysis in the stomach or intestinal tract or, alternately, from the enzymatic action of mannosidases.
Figure 1.

Examples of simple glycosidic bond replacement of early lead O-mannosides 1 and 4, with alternate linkers to the biphenyl aglycone.
In this article, we specifically address and eliminate this potential metabolic liability of O-mannosides with new C-mannosides, which have the labile O-glycosidic bond replaced with stable carbon-based linkers to the aglycone portion of the mannoside. Herein, we report on a novel, stereoselective, synthetic route to construct this new class of biaryl, C-glycoside FimH antagonists, which encompass a unique R-hydroxy methylene bond to the aglycone. We have employed X-ray crystallography and computational docking studies of mannosides to develop a pharmacophore model of stereospecific C-mannoside binding to FimH. Furthermore, we show that relative to O-mannosides, these new C-mannosides have greatly increased compound exposure, which we postulate to result from an increase in metabolic stability of the glycosidic bond. Exactingly, C-mannosides also have significantly improved in vitro FimH activity and in vivo efficacy in animal models of UTI.
Results and Discussion
N-, S-Linked Mannosides
At the onset of this work, we synthesized two compounds, one containing a nitrogen (2) and the other a sulfur atom (3) in place of the oxygen glycosidic bond (1) (Figure 1). As shown in Scheme 1A, these S- and N-mannosides were prepared using a traditional glycosylation methodology with slight modification, first coupling α-d-mannose with methyl 4′-aminobiphenyl-3-carboxylate to give N-linked mannoside 2, then glycosylating mannose-pentaacetate with 4-bromobenzenethiol to give 3a, followed by a Suzuki reaction with 3-methoxycarbonylphenyl boronic acid and deprotection to give S-linked mannoside 3. We found that both analogues had slightly lower activity (HAI = 4 μM) to that of O-mannoside 1 when tested in a hemagglutination (HA) inhibition assay using the clinical E. coli strain UTI89. To help guide our SAR, we also tested 2 and 3 in an isothermal shift melting point assay and found that they had equivalent binding affinity to FimH, relative to 1. Next, we synthesized an N-linked heterocycle, triazolomannoside, via “click” chemistry methodology. Shown in Scheme 1B, the reaction of azido mannoside 7(26) with phenylacetylene and copper sulfate, followed by sodium methoxide deacetylation, gave phenyl triazole mannoside 8 in good yield. However, mannoside 8 lost substantial potency relative to 1, only exhibiting an HAI of 32 μM.27 On the other hand, it was shown by another group that 8 still retains good FimH binding affinity (IC50 = 0.25 μM) as determined in a competitive binding assay.28 Hoping to build from these initial results with N- and S-mannosides, we aimed to construct potentially more metabolically stable, carbon-based linkers, or C-mannosides.
Scheme 1. Synthesis of N-, S-, and Triazole-Linked Biphenyl Mannosides.

Reagents and conditions: (a) methyl 4′-aminobiphenyl-3-carboxylate, EtOH, 55 °C; (b) 4-bromobenzenethiol, BF3–OEt2, DCM, 0 °C to rt; (c) 3-methoxycarbonylphenyl boronic acid, Pd(PPh3)4, Cs2CO3, dioxane/water (5/1), 80 °C; (d) NaOMe/MeOH, 0 °C to rt; (e) phenylacetylene, CuSO4, Na ascorbate, EtOH/H2O (4/1), rt.
C-Linked Amide Mannosides
Because of ease of synthesis, we first targeted a series of simple C-linked amide mannosides. Shown in Scheme 2, cyano mannoside 9(29) was obtained by reaction of mannose pentaacetate with trimethylsilyl cyanide and boron trifluoride diethyl etherate to give 9 as a 2:1 mixture of α and β isomers. Compound 9 was then hydrolyzed to carboxyl mannoside 10 by heating in 25% aq HCl for 2 days. 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) mediated coupling of 10 with methyl 4′-aminobiphenyl-3-carboxylate yielded the target C-linked amide mannoside 11. In parallel, we pursued methylene spaced amides in order to investigate the effect of length and flexibility of the linker. Thus, starting from cyano mannoside 9, we synthesized aminomethyl mannoside intermediate 12.30 Acylation with 4-bromo-benzoyl chloride gave amide mannoside 13, which was coupled to methoxycarbonylphenylboronic acid via Suzuki reaction, to give methylene-spaced amide 14 as a matched pair of 1. Testing of 11, 13, and 14 in the HA assay revealed that all analogues had reduced potency, relative to 1, with HAIs of 16 μM. We rationalized that these, and the triazole linker 8, do not allow sufficient flexibility to mimic the conformation, or key FimH binding interactions, observed for O-mannoside 1. Hence, we next pursued direct methylene (CH2) spaced C-mannosides, which provide a closer structural match to O-, N-, and S-mannosides 1–3.
Scheme 2. Synthesis of C-Linked Amide Mannosides.

Reagents and conditions: (a) 25% HCl aq, 50 °C; (b) HATU, DIPEA, 0 °C to rt, DMF; (c) 4-bromobenzoyl chloride, pyridine, rt; (d) Pd(PPh3)4, Cs2CO3, dioxane/water(5/1), 80 °C.
Direct Methylene C-Linked Mannosides
Methylene-spaced, C-mannoside analogues of O-mannoside 4 and ortho-methylated O-mannoside 23 were obtained using a newly developed, multistep synthetic procedure, starting from nitrile 15 (Scheme 3). The non-ortho substituted analogues were synthesized first. Bromide intermediates 17R and 17S were constructed in a one-pot, two-step sequence, first reducing nitrile 15 to aldehyde 16 with DIBAL, followed by the addition of an organolithium reagent (formed by lithiation of 1,4-dibromobenzene (R = H, step b). The organolithium addition yielded a mixture of alcohol isomers (17R and 17S), which were separable by silica gel chromatography. Following a palladium-mediated cross-coupling of 17R and 17S with 3-(N-methylaminocarbonyl)phenyl boronate, 18R and 18S were obtained in good yield, respectively. Subsequent benzyl deprotection produced methylene-linked C-mannosides 5, 6R and 6S. Following testing in the HA assay, we discovered that methylene-spaced analogue 5 (HAI = 1 μM) was equipotent to its matched pair O-mannoside 4 (HAI = 0.5 μM) (Figure 1). Surprisingly, we found the secondary alcohol isomers 6S and 6R showed differential potency, with a pronounced stereochemical preference for the R-hydroxyl group for FimH binding affinity. The isomer 6S had 25-fold less potency in the HA assay (HAI = 24 μM) than 5, whereas isomer 6R gained 4-fold activity (HAI = 0.25 μM).
Scheme 3. Synthesis of C-Linked Methylene and Hydroxy-Methylene Mannosides.

Reagents and conditions: (a) DIBAL, CH2Cl2, −78 °C; (b) diethyl ether, −78 °C to −20 °C; (c) Dess–Martin periodinane, pyridine, CH2Cl2, 0 °C; (d) Li(tOBu)3AlH, THF, −40 to 0 °C; (e) 3-(N-methyaminocarbonyl)phenylboronic acid pinacol ester, Pd(PPh3)4, Cs2CO3, dioxane/water (5/1), 80 °C; (f) 10% Pd/C, H2, MeOH, rt.
On the basis of these key results, we synthesized the ortho-methyl analogues 22 and 21 based on our lead O-glycoside 23 (Figure 2).20a Shown in Scheme 3, C-mannosides 21 and 22 were constructed in a similar fashion as for 5 and 6, only differing in the organolithium reagent (4-bromo-2-methyl-iodobenzene [R = Me, step b]) used for the addition to aldehyde 16. We discovered that methylene C-mannoside 22 (HAI = 2 μM) had a 30-fold decrease in activity relative to 23, which is in contrast to the results obtained from 5, which retained activity relative to its matched pair, 4. However, we did discover the same R-stereospecificity of the benzylic alcohols observed with 6R and 6S also exists in the more potent ortho-methyl series, with C-mannoside 21R having an HAI of 30 nM (relative to 62 nM for 23) and S-isomer 21S having an HAI of only 24 μM. This striking increase in the FimH activity of R-isomers 6R and 21R was attributed exclusively to a change in their relative FimH binding affinities when compared to 6S and 4 and 21S and 23, respectively (Figure 3). It should be noted that although the R and S stereochemical assignment of the hydroxymethylene linker in 6 and 21 were only speculation at this time, we later confirmed the stereochemistry through small molecule X-ray crystal structure of a derivative of a key precursor, 19R (Figure 6). From the vast potency difference seen between 21R and 21S, we can also presuppose that our potency-based assignments are correct for the less effective 6R and 6S compounds.
Figure 2.

Direct comparison of the potencies of C-linked methylene mannosides 21R, 21S, and 22 to those of similar O-mannoside analogues 23, 24, and 25.
Figure 3.

In vitro analysis of mannoside potency. The difference in the conformational stability of the FimH lectin domain in the presence of mannoside compared to that in the absence of mannoside, as determined by differential scanning fluorimetry, is presented on the left axis. Measurement of the melting temperature is described in the methods. The hemagglutination inhibition data is presented relative to d-mannose on the right axis and is described in the methods.
Figure 6.

(A) Derivatization of key building block 19R (precursor to 21R). (B) Small molecule X-ray structure of acetylated intermediate 27, confirming the R-stereochemistry of the 21R benzylic hydroxyl group.
Stereoselective Methodology for Recycling the Undesired 19S-Isomer into the 19R-Isomer
To improve upon our yield of R-hydroxybenzyl C-mannosides, we explored alternate synthetic strategies. In this study, we sought to maximize the yield of 19R in step d, depicted in Scheme 3, which generates a mixture of diastereomers, yielding 19R in only 16%, and the 19S isomer in 20%. We first investigated modified reaction conditions, including a systematic evaluation of solvents. However, these changes resulted in a lower reaction yield and also gave rise to an inseparable 4-iodo-3-methyl byproduct. Variation of other reaction parameters, such as temperature, concentration, additives, and order of addition, were also attempted, but these unproductively increased the 19S isomer ratio. As a possible solution, we envisioned a transformation to convert 19S into the desired 19R isomer. Therefore, we implemented a two-step oxidation and selective reduction protocol, allowing us to efficiently recycle the undesired 19S isomer into 19R. This stereoselective recycling method improved the R:S ratios from 1:1.2 (steps c,d) to 28:1 (steps c–f), also increasing the overall yield of 19R from 16% to 28% (calculated from compound 15). The oxidation of 19S (or unresolved mixtures of 19S and 19R) was carried out using Dess–Martin periodinane in pyridine to give the aryl ketone intermediate 19a (not shown) in 74%. Stereoselective reduction using the bulky lithium tri-tert-butoxyaluminum hydride reagent in THF gives 19R with a respectable 17:1 (R:S) diastereoselectivity and an excellent yield of 84%. Upon further investigation, we elucidated that the corresponding bulky potassium tri-sec-butylborohydride (K-selectride), the unhindered lithium aluminum hydride (LAH), and sodium borohydride (NaBH4) reagents all favored stereoselectivity for the undesired 19S isomer. This strongly suggests that the stereoselectivity for the hydride delivery, and reduction comes from a combination of both steric and electronic interactions between the chirality of the α-d-mannoside and the chosen reducing agent. A careful review of the literature reveals that this particular use of mannose, or any other sugar, as a chiral director for the stereoselective reduction of ketones has not been reported to date and could be precedent for further investigation. We surmise that this methodology could be applied to other glycoside systems to effect a large array of diastereoselective or possibly even enantioselective ketone reductions.
Prodrugs of Mannosides
In a parallel strategy to improve the oral bioavailability and half-life (compound exposure) of O-mannosides, we designed and synthesized various prodrugs31 of lead O-mannoside 23. The prodrugs include tetraacetate 23a, the 6-position phosphate 23b, and dimethylglycine acetate 23c (Scheme 4). Although 23a is the precursor in the synthesis of 23,20b it was synthesized by reacetylating mannoside 23. To selectively phosphorylate the 6-position, as in prodrug 23b, mannoside 23 was reacted with phosphoryl chloride (POCl3) in water and trimethyl phosphate. Finally, prodrug 23c was constructed via silylation of 23, followed by selective deprotection of the primary trimethylsilyl group. Next, coupling of the free 6-hydroxy group with N,N-dimethylaminoglycine was achieved using N,N-diisopropylcarbodiimide (DIC) to give prodrug 23c upon protecting group removal with TFA.
Scheme 4. Structures and Synthesis of Prodrug Analogues of O-Mannoside 23.

Reagents and conditions: (a) Ac2O, pyridine, rt; (b) POCl3 trimethylphosphate, H2O, 0 °C; (c) TMSCl, Et3N, DMF, 0 °C; (d) AcOH, acetone/MeOH, 0 °C to rt; (e) N,N-dimethylglycine hydrochloride, DMAP, DIC, CH2Cl2/DIPEA, rt; (f) TFA, CH3CN, 0 °C.
X-ray Crystallography and Computational Modeling
To better understand the molecular interactions of O-mannosides 23 and its matched disubstituted analogue 24 (Figure 2)20b with FimH, and to help us design improved mannosides based on C-mannoside 21R, we obtained co-crystal structures of 23 and 24 bound to FimH. A high-resolution 1.67 Å X-ray crystal structure of 24 (PDB 5F2F)25 and a 1.75 Å structure of 23 (PDB 5F3F) bound to the FimH lectin domain were obtained (Figure 4A) and used to develop a pharmacophore model for mannoside FimH ligands. The O-linked aglycone projects toward the “lid” of the binding pocket, which engages in favorable hydrophobic and H-bonding interactions. The ortho-methyl substituent on the biphenyl A-ring makes hydrophobic interactions in a small pocket formed by I52, Y137, and N138. The biphenyl B-ring forms favorable π–π stacking interactions with Y48, and the meta-substituted amide also makes polar contacts with FimH. For example, the carbonyl of the amide forms a predicted electrostatic or H-bonding interaction with the neighboring salt bridge, R98, and/or Y137 residues. In Figure 4B, comparison of mannosides 23 and 24 to non-ortho-substituted 1 (PDB 3MCY),20c highlights a marked difference in the biphenyl group B-ring conformation upon installation of the ortho-methyl group. On the basis of this new X-ray structure of 23 and previously reported structures,23a,25,32 it is apparent that the hydrophobic contacts of the ortho-substituents within the small pocket, and the resultant twisted B-ring conformation, improves π–π stacking in the “tyrosine gate” with Y48 and Y137.
Figure 4.

(A) X-ray structure of 23 bound to FimH (PDB 5F3F). Water-mediated H-bond of amide carbonyl to Y48. (B) Overlay of 23 with 24 (PDB 5F2F) and 1 (PBD 3MCY). Altered biphenyl ring conformation and amide carbonyl orientation of ortho-substituted mannosides 23 and 24 compared to 1.
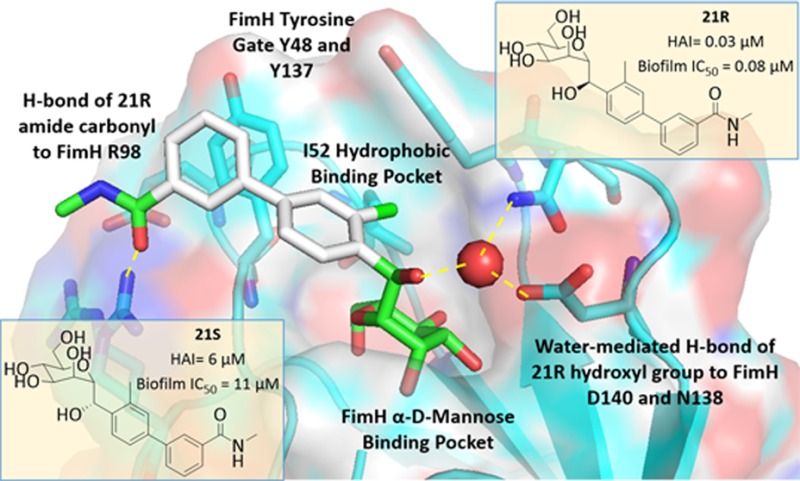
Computational Docking of C-Mannosides to FimH
Because we have not been able to obtain a co-crystal structure of C-mannoside 21R, we have formulated a computational binding model, shown in Figure 5. As seen in the structure of 23 (Figure 4A), there is a crystallized water molecule between the glycoside anomeric oxygen and the D140 and N135 FimH side chains. The location of the water suggests it is involved in a H-bonding interaction between the anomeric oxygen and FimH. Our computational docking model of the diastereomers 21S and 21R indicates that the R-isomer (21R, Figure 5B) is favored energetically over the S-isomer (21S, Figure 5A) due to a productive water-mediated H-bond in the former. This same water molecule, positioned between the hydroxyl group of the R-isomer and residue(s) D140 and N135, is not accessible to the S-isomer. This preference is also predicted from thermodynamic calculations, as the calculated ΔG of binding for the R-isomer is favored by 2.8 kcal/mol over that of the S-isomer. These observations can explain the strong stereospecific preference for FimH binding the R-isomer, relative to the S-isomer, and why methylene spaced mannoside 22 loses activity33 compared to O-mannoside 23, as the CH2 is not able to participate in H-bonding to this water molecule. In the absence of X-ray crystallography data of FimH binding, we determined the stereochemistry of the 21R benzylic hydroxyl by obtaining a small molecule X-ray crystal structure of crystalline C-mannoside aryl bromide intermediate, 27 (Figure 6). The high resolution X-ray of 27 unequivocally confirms that the benzylic hydroxyl group of intermediate 19R. By logical deduction, 19R and its final product 21R, both have the R-stereochemistry, reinforcing our computational docking model invoking a stereospecific water-mediated H-bond to FimH.
Figure 5.

Computational docking models of isomeric C-mannosides: (A) S-hydroxy (21S) and (B) R-hydroxy (21R) bound to the FimH mannose-binding pocket. The accepted model of 21R is bound to FimH through water-mediated H-bond to D140 and N135.
Pharmacokinetics (PK) of Mannosides
To further examine the therapeutic potential of the new C-mannosides and O-mannoside prodrugs, we evaluated their pharmacokinetic (PK) behavior in vivo. Mannosides were delivered orally in a suspension of 10% cyclodextrin. Previous administration utilized DMSO as a vehicle. Our decision to administer in cyclodextrin was 2-fold: (1) cyclodextrin is commonly used for increasing the aqueous solubility and bioavailability of drugs34 and (2) DMSO has been demonstrated to modulate the host immune response, obfuscating any observed results.35 The concentration of mannoside detected in the urine of mice was monitored at multiple time points following oral delivery over a period of 8 h and quantitatively measured by HPLC and mass spectrometry. As previously published, O-linked mannosides, including both 23 and 24, were found to accumulate at high concentrations in the urine as early as 1 h post dosing,20b followed by a steady almost linear decrease throughout the collection time frame (Figure 7A). Despite this continual decrease in concentration, the relative concentration of each mannoside remained well above the HAI EC90 (0.062 μM) and biofilm IC50 (0.15 μM) of 23 throughout this period. In contrast to 23 and 24, the concentrations of 21R and 23a (the acetate prodrug of 23) remained substantially higher throughout the period measured, resulting in a significantly increased AUC. Shown in Figure 7A, the concentration of both 21R and 23a in the urine 8 h following oral delivery was significantly higher than 23. Compound levels of 21R reached 50 μM at 8 h, which is 1600 times higher than its HAI potency. Acetate prodrug 23a also showed enhanced concentration, presumably from higher gut permeability, resulting in a 20-fold higher concentration (20 μM) of 23 relative to direct oral dosing of 23 (1 μM) at 8 h.
Figure 7.

(A) Mannoside levels were quantified in the urines of mice over a period of 8 h following oral gavage of drug as described in the methods. Data is presented as the mean and standard deviation from at least three independent experiments. Differences in concentration at the 8 h time point were tested for significance using Mann–Whitney U test. (* P < 0.05). (B) Measurement of mannoside in the plasma of rats following either a 10 mg/kg oral dose (dashed lines) or a 3 mg/kg intravenous dose (solid lines). Details regarding the measurement of mannoside are described in the methods.
To further expand on this result, we conducted a more thorough examination of the PK properties of 23 and 21R as well as the analogous ring-constrained isoquinolone O-mannoside 25(25) (Figure 2) in rats (Figure 7B). Recent examination of the PK behavior and efficacy of 25 in mice identified it as a leading therapeutic candidate in the O-mannosides series.25 In contrast to the previous study (Figure 7A), here we monitored the concentration of mannoside in the plasma of rats following either a 10 mg/kg oral dose (dashed lines) or a 3 mg/kg intravenous dose (solid lines) (Figure 7B). Analysis of the rat PK data revealed that, while C-mannoside 21R encompassed the highest level of compound exposure (as assessed by Cmax and AUCall) and the lowest clearance rate, at 34.9 mL/min/kg (23 clearance rate = 98.4 mL/min/kg) O-mannoside 25 shows the highest clearance rate (408 mL/min/kg) and volume of distribution (Vdss = 6.7 L) but the highest bioavailability (7%) of all compounds tested. This suggests that the biaryl B-ring isoquinolone aglycone of 25 significantly enhances permeability in the gut, while on the other hand, C-mannoside 21R and the O-mannoside prodrug 23a have a longer half-life and extended compound exposure, presumably from a combination of increased bioavailability from the prodrugs and metabolic stability of the C-mannoside. Because O-mannosides 24 and 25 were stable (t1/2 > 2h) in plasma, simulated gut fluid (pH 1.2), simulated intestinal fluid (pH 6.8), and liver microsomes, we hypothesize that the increased exposure of C-mannoside 21R is likely due to evading the enzymatic action of mannosidases. This combined data constitutes key structure–property relationships (SPR) and was useful for guiding further optimization of the C-mannosides described below.
Synthesis of Optimized C-Linked Isoquinolone Mannosides
Inspired by the enhanced potency and increased compound exposure of C-mannoside 21R, coupled with the improved oral bioavailability of 25, we synthesized its C-mannoside matched pair analogue, 28R. Suzuki cross-coupling between 27 and 1-hydroxyisoquinoline-7-boronate ester, followed by acetyl deprotection, generated biaryl isoquinolone C-mannoside 28R (Table 1). In an alternate synthesis, 19R was used in place of 27 in the Suzuki reaction, but attempts to remove the benzyl protecting groups via hydrogenation resulted in formation of the saturated dihydroisoquinolone analogue 29R. For direct comparison, we repeated this hydrogenation on O-glycoside 25 to give corresponding analogue 30. We found that both isoquinolone 28R and dihydroisoquinolone 29R have excellent potency, with HAI titers of 8 and 6 nM, respectively. This equates to a ∼4.5-fold increase in potency over the O-mannoside matched pairs, 25 and 30 (HAI = 31 and 32 nM, respectively). Following these exciting results, we evaluated fused ring systems like 25 that would still retain their H-bonding and FimH binding attributes. Using similar procedures as described above, we synthesized aminoisoquinoline 31R and isoquinoline 32R, both possessing a nitrogen atom that occupies a position equivalent to the carbonyl oxygen on the isoquinolone ring of 25. Isoquinoline 32R has a different ring fusion, being attached to the aglycone A-ring at the 5-position (versus the 7-position in 31R, 28R, and 29R). As expected, the C-mannoside 31R was 3-fold more potent than its matched O-mannoside pair 33, with an HAI of 6 nM. However, the 5-position isoquinoline C-mannoside 32R had lower potency relative to its O-mannoside pair 34(24) (HAI = 62 nM), with an HAI of 125 nM (Table 1). This unexpected result can be rationalized from docking studies (not shown), wherein we found that due to a dramatic shift in ring conformation of the C-mannosides relative to the O-mannosides (Figure 5), a steric clash exists between FimH and this 5-position ring substitution of 32R in the C-mannoside series, which does not exist in 34 of the O-mannoside series. When comparing the matched pair C- and O-mannosides shown in Table 1, it is apparent that the cLogP values are approximately 1 Log unit lower in the C-mannoside series. For example, comparing 33 to 31R, we see a drop in cLogP from 2.28 to 1.2, respectively, suggesting that 31R has respectable oral drug-like physical properties like 25 (cLogP 1.4) but with enhanced glycosidic bond stability.
Table 1. Compound Structures and Comparison of Biological Activity and Physical Properties Between O- and C-Mannoside Matched Pairs (ND = not determined).

In Vivo Efficacy of Mannosides in Acute and Chronic Cystitis
Given the improved in vivo PK behavior of both the C-mannoside 21R and the prodrug 23a, we next investigated whether these enhancements improved their potency in animal models of UTI.20a,36 The distinct pathophysiology of UTI that has been identified in these murine models has also been observed in humans, demonstrating their relevance.37 To measure efficacy in these models, we examined the ability of mannosides to prevent the initial pathogenic steps of UPEC infection in our prophylactic model of acute UTI as well as their ability to treat an established infection in our model of chronic UTI. Despite the disparity in potency and PK behavior, the efficacy of all the compounds, in either model, were nearly identical (Figure 8). When tested as a prophylaxis therapy (Figure 8A), where the mannoside is dosed 30 min prior to UPEC infection and bacterial titers enumerated 6 h postinfection, tetraacetate prodrug 23a and phosphate prodrug 23b (see Supporting Information data) each showed improved efficacy compared to 23. In contrast, the glycine acetate prodrug 23c (see Supporting Information data) did not show enhanced efficacy, perhaps due to its increased stability in conversion to drug or decreased oral absorption in the gut relative to 23a and 23b.
Figure 8.

(A) Prophylactic treatment of an acute UTI. Mannoside was dosed orally at 25 mg/kg in 10% cyclodextrin, 30 min prior to infection with 107 CFUs UPEC. Bladders were harvested 6 h postinfection and bacterial burdens enumerated. (B) Mannoside treatment of chronic UTI. Mice were treated orally with 50 mg/kg of mannoside in 10% cyclodextrin, after 14 days of chronic UPEC infection. Bladders were harvested 6 h following oral dosing and bacterial burdens enumerated. Data from at least two independent experiments are presented in each panel; bars indicate geometric means. Differences between treated groups and vehicle were tested for significance using Mann–Whitney U test (*** P = 0.0001, **** P < 0.0001).
Both the O-linked and C-linked mannosides accumulated in the urine at concentrations over 10-fold higher than their HAI EC90 and biofilm IC50 (Figure 7) up to 8 h following oral dosing. Thus, we hypothesized that 6 h post-treatment was too early to discriminate relative mannoside potency in the chronic UTI model (Figure 8B). To investigate this possibility, we first monitored the pharmacodynamics (PD) of O-linked mannoside 24 in our treatment model of chronic UTI to evaluate its therapeutic window. Diamide mannoside 24 has similar PK to monoamide mannoside 23 but was found to be 4-fold more potent in our HA assay (HAI = 16 nM), identifying it as one of our most effective O-linked mannosides in vitro. Following oral delivery of 24 in the chronic UTI model, we found a significant decrease in bladder burden from 107 CFUs to approximately 105 CFUs 2 h postdosing (Figure 9). Bladder burdens continued to decrease to 104 CFUs at 6 h but leveled off at 12 hours, when the geometric mean of the bacterial burden increased to 105. Interestingly, the 12-hour time point appears to be comprised of a bimodal distribution, consisting of mice that have either high or low bacterial burdens in the bladder (Figure 9). Taken together, this data demonstrates a rapid reduction of bladder burdens following oral delivery of O-linked mannoside 24 and an increase of burdens in a subpopulation of mice 12 hours after treatment that may represent bladder repopulation once mannoside levels fall below the HAI.
Figure 9.

Pharmacodynamics of O-linked mannoside. Chronically infected mice were treated orally with 50 mg/kg of mannoside 24. Bladders were harvested at the designated time points following oral dosing and bacterial burden enumerated. Bars indicate geometric means. Differences between the designated time point and the 0 h time point were tested for significance using Mann–Whitney U test (* P < 0.05, ** P < 0.001).
C-Mannosides Displays Significantly Improved and Prolonged Efficacy in Vivo
Given the observed bimodal distribution of bladder burdens 12 hours after treatment with mannoside 24, we next decided to investigate the influence of the improved pharmacokinetic behavior of C-mannoside 21R on its ability to treat chronic cystitis at this time point following oral delivery of the mannoside (Figure 10A). Additionally, to accentuate any differences in efficacy at this time point, we reduced the mannoside dosages by half, treating only with a 25 mg/kg dose. At 12 h post treatment, the O-linked mannoside 24 resulted in a noticeable, but not significant, decrease in bladder burdens compared to vehicle. Treatment with prodrug 23a resulted in a similar, but statistically significant, decrease in bladder burdens. In contrast, it was found that treatment with C-mannoside 21R at 25 mg/kg resulted in a dramatic and significant decrease in CFUs as compared to both vehicle and O-linked mannosides 24 and 23a. Indeed, bladder burdens of mice treated with 21R were uniformly three logs lower than untreated mice, clearly demonstrating the improved efficacy of the C-linked mannoside.
Figure 10.

Improved efficacy of C-linked mannosides. (A) Chronically infected mice were treated orally with 25 mg/kg of mannoside. Bladders were harvested 12 hours following oral dosing and bacterial burdens enumerated. (B) Mice were treated orally with 25 mg/kg of mannoside 30 min prior to infection. Bladders were harvested 6 h following infection and bacterial burdens enumerated. Data from at least two independent experiments are presented; bars indicate geometric means. Differences between treated groups and vehicle were tested for significance using Mann–Whitney U test (**P < 0.01, *** P = 0.0001, **** P < 0.0001).
Furthermore, using the newly optimized heterocyclic C-mannosides, 28R, 29R and 31R, we found significantly augmented activity relative to O-mannoside 25 when tested in the prophylactic model of UTI (Figure 10B), described above, where compound was dosed orally (25 mg/kg) 30 min before infection. The in vivo activity seen with 28R, 29R, and 31R is similar to 21R, but it can be argued that 29R and 31R perform slightly better in preventing an infection in this model of acute UTI. However, there is a clear difference in efficacy displayed between the C-mannosides and O-mannoside 25. This is nicely illustrated by direct comparison of O-mannoside 25 and its matched C-mannoside 28R, which decreases bacterial titers 2 Logs lower (100-fold). Taken together with the fact that O-mannoside 25 and C-mannoside 21R are equivalent in both FimH binding (DSF = 75.4 °C) and target inhibition (HAI = 31 nM), this result suggests that the C-mannosides have superior metabolic stability.
Conclusion
Carbohydrates, and in particular small glycosides, are typically unstable in vivo due to their inherent susceptibility to hydrolysis in the stomach or intestine and to enzymatic hydrolysis by various glycosidases present in the gut, plasma, and some tissues. This instability can critically limit the oral bioavailability, half-life. and overall therapeutic potential of glycoside-based drugs38 like FimH mannoside antagonists. The replacement of O-glycosidic bonds with sulfur, carbon, or nitrogen-based linkers is one strategy that can be utilized to increase the metabolic stability of glycosides while also theoretically helping to increase their bioavailability. However, the synthesis of unnatural glycoside linkers is challenging and the influence of these changes on activity are not always predictable or straightforward. In the current manuscript, we implemented a rational strategy to improve mannoside PK and efficacy by replacing the glycosidic bond of the O-mannosides with a number of unique carbon-based linkers that are likely stable to metabolism. To access these novel C-mannosides, we have designed a new stereoselective synthetic route for the assembly of hydroxymethyl C-mannosides. While increased stability to mannosidases is the most reasonable explanation for the improved efficacy in vivo, other properties may be contributing to this behavior. Current investigations are confirming this hypothesis through both in vitro and in vivo studies which are beyond the scope of this initial communication.
In summary, mannoside FimH antagonists represent a first-in-class antivirulence drug in the treatment of chronic UTI and the prevention of recurrent cystitis. Herein, we have rationally designed a novel C-mannoside FimH antagonist 21R, which we subsequently modified, based on O-mannoside SAR and isoquinolone derivative 25,25 to generate several optimized analogues, 28R, 29R, and 31R. These C-mannosides have become preclinical candidates for use as novel antivirulence drugs in UTI therapy.18 Along with other lead compounds, they are currently being profiled in advanced in vivo PK, efficacy, and toxicologuey studies in preparation for selecting a lead candidate drug for clinical trials. Upon successful clinical development, a mannoside would not only represent the first small molecule FimH antagonist drug for UTI but also the first antibiotic-sparing therapeutic for the treatment of Gram-negative infections. This could also pave the way for the new development of other antibiotic-sparing drugs targeting other lectins or virulence factors, which are desperately needed for other infectious diseases.
Experimental Section
General Synthesis, Purification, and Chemistry Procedures
Starting materials, reagents, and solvents were purchased from commercial vendors unless otherwise noted. In general, anhydrous solvents are used for carrying out all reactions. 1H NMR spectra were measured on a Varian 300 MHz NMR instrument or Varian 400 MHz NMR instrument equipped with an auto sampler. The chemical shifts were reported as δ ppm relative to TMS using residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the peak multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. Melting points were determined on a Kofler micro hot stage and were uncorrected. High-performance liquid chromatography (HPLC) was carried out on GILSON GX-281 using Waters C18 5 μM 4.6 mm × 50 mm and Waters Prep C18 5 μM 19 mm × 150 mm reverse phase columns, eluted with a gradient system of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05–0.1% TFA. Mass spectroscopy (MS) was performed on HPLC/MSD using a gradient system of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05–0.1% TFA on a C18 or C8 reversed phased column and electrospray ionization (ESI) for detection. All reactions were monitored by thin layer chromatography (TLC) carried out on either Merck silica gel plates (0.25 mm thick, 60F254) or Millipore Silica gel aluminum sheets (60F254) and visualized by using UV (254 nm) or dyes such as KMnO4, p-anisaldehyde, and CAM (Hannesian’s Stain). Silica gel chromatography was carried out on a Teledyne ISCO CombiFlash purification system using prepacked silica gel columns (12–330 g sizes). All compounds used for biological assays are greater than 95% purity based on NMR and HPLC by absorbance at 220 and 254 nm wavelengths.
General Procedure for the Suzuki Coupling Reactions (Amounts and Volumes Are Specified within Individual Procedures)
Cesium carbonate (Cs2CO3) was activated by adding it to a round-bottom flask, which was then heated to 250 °C under vacuum for 2 min and then allowed to cool to rt under vacuum for an additional 10 min, after which time a nitrogen atmosphere was continuously maintained. Next, the desired mannosyl bromide (or mannosyl boronate ester) derivative was dissolved into dioxane and the solution was added dropwise, followed by the addition of the desired boronate-aglycone (or bromide-aglycon if mannosyl boronate was used) and, finally a small amount of water. After allowing the reaction contents to stir for 5 min at rt, a catalytic amount of tetrakis(triphenylphosphine)palladium(0) [Pd(Ph3)4] was added and the reaction flask was evacuated under high vacuum and backfilled with N2 three times and then placed in an oil bath preheated to 80 °C and allowed to stir for the time specified (typically 1.5 h). Upon completion, the reaction was cooled to rt and solvents were evaporated under reduced pressure. The crude reaction residue was then purified and deprotected as specified.
Detailed Procedures for the Synthesis of Final Mannosides and Intermediates
4′-(α-d-Mannopyranosylamino)-[1,1′biphenyl]-3-carboxylic Acid Methyl Ester (2)
α-d-Mannose (0.360 g, 2 mmol) and methyl 4′-aminobiphenyl-3-carboxylate (0.454 g, 2 mmol) were dissolved into ethanol (5 mL), and the reaction was heated to 55 °C for 17 h. After cooling down to rt, the white precipitate that formed was collected by filtration. The precipitate was washed with ethanol (2 × 3 mL) and then dried in vacuo to afford pure 2 in 77% yield. Analytical data for 2: 1H NMR (300 MHz, acetonitrile-d3 and D2O) δ ppm 3.28–3.38 (m, 1H), 3.54–3.59 (m, 2H), 3.64–3.71 (m, 2H), 3.86 (s, 3H), 3.88–3.92 (m, 1H), 4.89 (d, J = 1.1 Hz, 1H), 6.80–6.91 (m, 2H), 7.44–7.58 (m, 3H), 7.75–7.90 (m, 2H), 8.11–8.20 (m, 1H). 13C NMR (100 MHz, methanol-d4/dimethyl sulfoxide-d6; 10/1) δ ppm 52.8, 62.8, 68.6, 72.9, 76.2, 78.9, 83.3, 115.6 (×2), 127.8, 128.1, 128.7 (×2), 130.2, 130.8, 131.7, 131.9, 142.8, 147.5, 168.4. ESI-MS found: [M + H]+, 390.1.
4′-(α-d-Mannopyranosylthio)-[1,1′biphenyl]-3-carboxylic Acid Methyl Ester (3)
Step 1: Under nitrogen atmosphere at 0 °C, boron trifluoride diethyl etherate (0.427 g, 3.0 mmol) was added dropwise into a solution of α-d-mannose pentaacetate (0.390 g, 1.0 mmol) and 4-bromobenzenethiol (0.378 g, 2.0 mmol) in 6 mL of CH2Cl2. After 5 min, the mixture was warmed to rt and allowed to stir for 48 h. The reaction was then quenched with water and extracted with CH2Cl2. The CH2Cl2 layer was collected, dried over Na2SO4, and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (ethyl acetate–hexanes gradient elution), giving rise to the intermediate 4-bromophenyl 2,3,4,6-tetra-O-acetyl-1-thio-α-d-mannopyranoside (3a) (0.40 g) in 77% yield. Analytical data for 3a: 1H NMR (300 MHz, chloroform-d) δ ppm 2.01–2.10 (m, 9H), 2.16 (s, 3H), 4.10 (dd, J = 12.1, 2.5 Hz, 1H), 4.30 (dd, J = 12.1, 6.0 Hz, 1H), 4.43–4.61 (m, 1H), 5.24–5.40 (m, 2H), 5.43–5.52 (m, 2H), 7.32–7.39 (m, 2H), 7.42–7.49 (m, 2H). ESI-MS found: [2 M + H+] 1039.1.
Step 2: Following the general Suzuki-coupling procedure, mannosyl bromide 3a (0.20 g, 0.39 mmol) from step 1, commercially available 3-methoxycarbonylphenylboronic acid (0.106 g, 0.59 mmol), cesium carbonate (0.381 g, 1.17 mmol), and tetrakis(triphenylphosphine)palladium (0.045 g, 0.04 mmol) in dioxane/water (5 mL/1 mL) were reacted under N2 at 80 °C for 1 h. Upon completion, the reaction was cooled to rt, and the mixture was filtered through a silica gel column (ethyl acetate–hexanes, 2/1 isocratic elution) to remove the metal catalyst and salts. The filtrate was concentrated then dried in vacuo. To the crude residue was added MeOH (6 mL) and a catalytic amount of sodium methoxide (0.02 M), and the mixture was stirred at rt overnight. H+ exchange resin (DOWEX 50WX4-100) was added to neutralize the mixture, the resin was filtered off, and the filtrate concentrated. The resulting residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.1% TFA)] to give 3 (0.095 g) in 63% yield. Analytical data for 3: 1H NMR (300 MHz, methanol-d4) δ ppm 3.66–3.91 (m, 4H), 3.94 (s, 3H), 4.01–4.16 (m, 2H), 5.51 (d, J = 1.4 Hz, 1H), 7.51–7.69 (m, 5H), 7.81–7.93 (m, 1H), 8.00 (dt, J = 7.8, 1.4 Hz, 1H), 8.24 (t, J = 1.7 Hz, 1H). 13C NMR (100 MHz, methanol-d4 /dimethyl sulfoxide-d6; 20/1) δ ppm 52.8, 62.6, 68.7, 73.2, 73.6, 76.0, 90.2, 128.6 (×2), 128.6, 129.5, 130.4, 132.0, 132.5, 133.2 (×2), 135.9, 140.0, 141.8, 168.1. ESI-MS found: [M + H+] 407.1.
4′-[(α-d-Mannopyranosyl)methyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (5)
The title compound was obtained from the preparation of 6S as a side product in 35% yield (0.005 g, 0.013 mmol). Analytical data for 5: 1H NMR (400 MHz, methanol-d4) δ ppm 2.93–3.00 (m, 4H), 3.04–3.13 (m, 1H), 3.62–3.88 (m, 6H), 4.09–4.17 (m, 1H), 7.39 (d, J = 8.2 Hz, 2H), 7.48–7.57 (m, 1H), 7.62 (d, J = 7.8 Hz, 2H), 7.74–7.79 (m, 2H), 8.06 (br s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 26.9, 36.1, 63.1, 69.4, 72.0, 72.7, 76.4, 80.4, 126.6, 126.9, 128.1 (×2), 130.1, 130.9 (×2), 131.0, 136.2, 139.6, 139.7, 142.6, 170.7. ESI-MS found: [M + H+] 388.2.
4′-[(α-d-Mannopyranosyl)-(R)-hydroxymethyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (6R)
A mixture of 18R (0.039 g, 0.051 mmol) and 10% wt Pd/C (0.044 g, 0.02 mmol) in MeOH (10 mL) was stirred under 1 atm of H2 for 16 h. The Pd/C was filtered off, and the filtrate was concentrated and dried in vacuo to give title compound 6R (0.020 g, 0.050 mmol) in 99% yield. Analytical data for 6R: 1H NMR (300 MHz, methanol-d4) δ ppm 2.95 (s, 3H), 3.54–3.81 (m, 4H), 3.86–4.05 (m, 2H), 4.25 (t, J = 3.0 Hz, 1H), 5.01 (d, J = 8.0 Hz, 1H), 7.46–7.61 (m, 3H), 7.67 (d, J = 8.2 Hz, 2H), 7.74–7.88 (m, 2H), 8.08 (t, J = 1.7 Hz, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 27.0, 62.8, 69.0, 69.9, 72.6, 72.9, 77.8, 82.4, 126.7, 127.0, 127.8 (×2), 128.7 (×2), 130.1, 131.0, 136.2, 140.8, 142.6, 143.5, 170.7. ESI-MS found: [M + Na+] 426.2.
4′-[(α-d-Mannopyranosyl)-(S)-hydroxymethyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (6S)
A mixture of 18S (0.028 g, 0.037 mmol) and 10% wt Pd/C (0.032 g, 0.015 mmol) in MeOH (10 mL) was stirred under 1 atm of H2 for 16 h. The Pd/C was filtered off, and the filtrate was concentrated and dried in vacuo. The resulting residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)], to give 6S (0.010 g, 0.025 mmol) in 67% yield [compound 5 was also isolated as a product in 35% yield (0.005 g, 0.013 mmol)]. Analytical data for 6S: 1H NMR (300 MHz, methanol-d4) δ ppm 2.95 (s, 3H), 3.49–3.58 (m, 1H), 3.59–3.84 (m, 4H), 3.86–4.02 (m, 2H), 5.04 (d, J = 9.0 Hz, 1H), 7.48–7.60 (m, 3H), 7.71 (d, J = 8.2 Hz, 2H), 7.76–7.84 (m, 2H), 8.08 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 27.0, 63.4, 69.1, 69.9, 71.2, 72.9, 77.1, 84.0, 126.8, 127.1, 128.3 (×2), 128.7 (×2), 130.2, 131.0, 136.2, 141.4, 142.3, 142.3, 170.6. ESI-MS found: [M + Na+] 426.2.
1-α-d-Mannopyranosyl-4-phenyl-1H-1,2,3-triazole (8)27,28
Step 1: Under a nitrogen atmosphere, ethanol/water (4 mL/1 mL) was added into the RB flask containing 2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl azide (7) (0.075 g, 0.20 mmol) (obtained from d-mannose pentaacetate27), phenyl acetylene (0.026 g, 0.24 mmol), copper(II) sulfate pentahydrate (0.01 g, 0.04 mmol), and sodium ascorbate (0.016 g, 0.08 mmol). The mixture was stirred at room temperature overnight. The solvent was removed and the residue was purified by silica gel chromatography (ethyl acetate–hexanes gradient elution), giving rise to the tetraacetate protected intermediate 1-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-4-phenyl-1H-1,2,3-triazole (8a) (0.030 g, 0.062 mmol) in 31% yield. Analytical data for 8a: 1H NMR (300 MHz, chloroform-d) δ ppm 2.05–2.12 (m, 9H), 2.20 (s, 3H), 3.89–3.99 (m, 1H), 4.08 (dd, J = 12.50, 2.61 Hz, 1H), 4.40 (dd, J = 12.50, 5.36 Hz, 1H), 5.40 (t, J = 8.79 Hz, 1H), 5.95–6.04 (m, 2H), 6.07 (d, J = 2.75 Hz, 1H), 7.34–7.53 (m, 3H), 7.79–7.92 (m, 2H), 7.96 (s, 1H). ESI-MS found: [M + Na+] 498.1.
Step 2: Tetraacetate 8a (0.028 g, 0.059 mmol) from step 1 was stirred in 6 mL of MeOH with a catalytic amount of sodium methoxide (0.02 M) at room temperature overnight. H+ exchange resin (DOWEX 50WX4-100) was added to neutralize the mixture pH. The resin was filtered off, and the filtrate was concentrated and then dried in vacuo, giving rise to the title compound (0.015 g, 0.049 mmol) in 83% yield. Analytical data for 8: 1H NMR (300 MHz, methanol-d4) δ ppm 3.34–3.44 (m, 1H), 3.70–3.92 (m, 3H), 4.12 (dd, J = 8.52, 3.57 Hz, 1H), 4.71–4.80 (m, 1H), 6.08 (d, J = 2.75 Hz, 1H), 7.30–7.41 (m, 1H), 7.41–7.51 (m, 2H), 7.77–7.90 (m, 2H), 8.51 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 62.6, 68.7, 70.1, 72.6, 78.7, 88.5, 122.1, 126.8 (×2), 129.5, 130.0 (×2), 131.5, 149.1. MS-ESI found: [M + Na+] 330.2.
4′-[(α-d-Mannopyranosylcarbonyl)amino]-[1,1′-biphenyl]-3-carboxylic Acid Methyl Ester (11)
The solution of 2,3,4,6-tetra-O-acetyl-α-d-mannosyl cyanide (9)29,30 (0.107g, 0.3 mmol) in 25% hydrochloric acid was heated at 50 °C for 48 h. The solvent was removed. Water (10 mL) and H+ exchange resin (DOWEX 50WX4-100) was added and kept stirring for 5 min. The resin was filtered off, and the filtrate was concentrated and then dried in vacuo to give crude α-d-mannopyranosyl carboxylic acid (10). To the crude product, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (0.274 g, 0.72 mmol) and anhydrous DMF (6 mL) were added at 0 °C. After stirring for 10 min, methyl 4′-aminobiphenyl-3-carboxylate (0.164 g, 0.72 mmol), and then N,N-diisopropylethylamine (0.233 g, 1.80 mmol) were added. The mixture was stirred overnight while being warmed to rt naturally. The solvent was removed and the residue was purified by silica gel chromatography (methanol–dichloromethane gradient elution) to give the title compound (0.066 g) in 52% yield. Analytical data for 11: 1H NMR (300 MHz, methanol-d4) δ ppm 3.50 (dt, J = 4.67, 2.33 Hz, 1H), 3.58–3.65 (m, 2H), 3.76 (dd, J = 11.81, 7.42 Hz, 1H), 3.86–4.03 (m, 4H), 4.52 (t, J = 2.61 Hz, 1H), 4.57 (d, J = 2.75 Hz, 1H), 7.55 (t, J = 7.69 Hz, 1H), 7.60–7.68 (m, 2H), 7.68–7.76 (m, 2H), 7.82–7.91 (m, 1H), 7.97 (dt, J = 7.69, 1.37 Hz, 1H), 8.21–8.28 (m, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 52.7, 63.3, 68.5, 70.1, 73.2, 79.6, 80.4, 121.9 (×2), 128.4 (×2), 128.6, 129.1, 130.2, 132.0, 132.4, 137.4, 138.9, 142.2, 168.5, 170.1. ESI-MS found: [M + H+] 418.1.
4-Bromo-N-[(α-d-mannopyranosyl)methyl]-benzamide (13)
To a solution of [(α-d-mannopyranosyl)methyl]amine (12) (0.58 mmol) (obtained from 2,3,4,6-tetra-O-acetyl-α-d-mannosyl cyanide 9(29,30)) in pyridine (10 mL) was added 4-bromo-benzoyl chloride (2.2 g, 10 mmol), and the reaction was stirred at RT overnight. Excess acid chloride was quenched by the addition of MeOH (2 mL). After 30 min, the reaction was concentrated in vacuo and dried. To the crude residue was added 25 mL of [0.5 M] sodium methoxide in MeOH (pH ∼ 10), and the reaction was stirred for 3 h at RT. H+ exchange resin (DOWEX 50WX4-100) was washed with MeOH and then added while stirring for 15 min. After filtration, the crude product was obtained by concentrating the filtrate and then was purified by silica gel chromatography (methanol–dichloromethane gradient elution). It was found that pyridine-HCl was a large contaminant, so the crude product was redissolved in MeOH (10 mL) and charged with 0.5 mL of 3 M NaOH and stirred for 5 min. The reaction was then neutralized with H+ exchange resin (DOWEX 50WX4-100). After filtration and concentrating the filtrate, the title product was isolated (120 mg, 0.32 mmol; 55% over 2 steps). Analytical data for 13: 1H NMR (400 MHz, methanol-d4) δ ppm 3.63–3.68 (m, 4H), 3.75–3.85 (m, 4H), 4.03–4.07 (m, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 40.2, 62.6, 70.0, 70.3, 72.7, 76.3, 77.4, 127.1, 130.3 (×2), 132.7 (×2), 134.6. ESI-MS found: [M + H+] 376.0 (100%), 378.0 (97.3%).
4′-[([(α-d-Mannopyranosyl)methyl]amino)carbonyl]-[1,1′biphenyl]-3-carboxylic Acid Methyl Ester (14)
To a stirred solution of 4-bromo-N-[(α-d-mannopyranosyl)methyl]-benzamide (13) (0.38 g, 0.1 mmol), 3-methoxycarbonylphenylboronic acid (0.027 g, 0.15 mmol), and cesium carbonate (0.098 g, 0.3 mmol) in dioxane/water (5 mL/1 mL) under nitrogen atmosphere was added tetrakis(triphenylphosphine)palladium (0.012 g, 0.01 mmol) was heated at 80 °C for 1 h. The reaction was concentrated to dryness and dissolved in DMSO and was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.1% TFA)]. After lyopholiztion of pure fractions, the title compound (24 mg, 0.056 mmol) was obtained in 55% yield. Analytical data for 14: 1H NMR (400 MHz, methanol-d4) δ ppm 3.68–3.71 (m, 4H), 3.78–3.83 (m, 3H), 3.87 (t, J = 3.2 Hz, 1H), 3.93 (s, 3H), 4.07–4.15 (m, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.73 (d, J = 7.6 Hz, 2H), 7.89 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 8.0 Hz, 2H), 8.01 (dd, J = 1.6, 8.0 Hz, 1H), 8.26 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 40.2, 52.8, 62.7, 69.9, 70.4, 72.7, 76.5, 77.5, 128.1 (×2), 129.0, 129.2 (×2), 129.9, 130.4, 132.1, 132.7, 134.8, 141.7, 144.4, 168.3, 170.0. ESI-MS found: [M + H+] 432.2.
2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl Cyanide (15)
Similar to the synthesis of compound 9, acetyl 2,3,4,6-tetra-O-benzyl-α-d-mannopyranoside (56.97 g, 97.84 mmol) was dissolved into dry acetonitrile (800 mL) under N2, and the reaction was cooled to 0 °C. Trimethylsilyl cyanide (36.86 mL, 293.53 mmol) was added, followed by the dropwise addition of boron trifluoride diethyl etherate (2.46 mL, 19.57 mmol). After 5 min, the reaction was brought to rt and stirred for an additional 30 min. Upon completion, brine (400 mL) and ethyl acetate (400 mL) were added and the reaction was stirred vigorously for 5 min. The layers were then partitioned, and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The organic portions were then combined and washed with 1 M aq HCl (2 × 200 mL) and brine (200 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude residue contained a 2:1 mixture of α- and β-anomers, which were easily separated by silica gel chromatography (ethyl acetate–hexanes gradient), the more nonpolar product being the desired α-mannoside 15, obtained in 51% yield (the β-mannoside byproduct was obtained in 27% yield). Analytical data for 15:31b1H NMR (300 MHz, chloroform-d) δ ppm 3.72–3.76 (m, 1H), 3.80–3.91 (m, 3H), 3.93–3.97 (m, 1H), 4.01–4.08 (m, 1H), 4.51–4.71 (m, 7H), 4.81 (d, J = 2.3 Hz, 1H), 4.88 (d, J = 11.0 Hz, 1H), 7.17–7.22 (m, 2H), 7.27–7.40 (m, 18H). ESI-MS found: [M + Na+] 572.2.
2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl Carbaldehyde (16)
Similarly to previously reported protocols,31b,39 at −78 °C, DIBAL/hexane (1.0 M, 11.3 mL) was added dropwise into the solution of 2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl cyanide (15) (4.97 g, 9.04 mmol) in CH2Cl2 (150 mL) under N2. The mixture was stirred for 30 min, maintaining a temperature of −78 °C. Then, the reaction was diluted with CH2Cl2 (150 mL) and then acidified with the addition of 0.2 M aq HCl (400 mL), stirring for 10 min at rt, and then filtered through Celite (to help break up emulsion) into a separatory funnel. The distinct layers were separated, and the aqueous layer was then extracted an additional time with CH2Cl2. The two organic fractions were combined and washed 2× with brine (100 mL). The organic layer was dried over Na2SO4, which also cleared up any remaining emulsion, and then concentrated to give intermediate carbaldehyde 16 as the crude product. Because of its instability, this intermediate was used without further purification, after drying 30 min to 1 h under high vacuum. Compound 16 was confirmed by ESI-MS, found [M + Na+] 575.5.
(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromophenyl)-methan-1(R/S)-ol (17R/S)
Into a flask containing 1,4-dibromobenzene (0.354 g, 1.5 mmol) in ether (5 mL) was added BuLi/hexanes (2.5 M, 0.5 mL) at −78 °C. One hour later, crude aldehyde 16 (synthesized from 0.56 mmol of carbonitrile 15) was added. The mixture was stirred at −78 °C for 1 h and then was warmed slowly to −40 °C over 1 h. Then 0.5 N aq HCl was used to quench the reaction, and ethyl acetate was use for extraction. The organic layer was collected, dried with Na2SO4, and concentrated, and the resulting residue was partially purified by silica gel chromatography (ethyl acetate–hexane gradient elution). The diastereomers, (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromophenyl)-methan-1(R)-ol (17R) and (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromophenyl)-methan-1(S)-ol (17S) were separated and collected, the more nonpolar fractions containing intermediate 17R. After separation, the impure compounds 17R (0.075 g, 0.1 mmol) and 17S (0.034 g, 0.048 mmol) were used without further purification in the synthesis of 18R and 18S, respectively. Both compounds 17R and 17S were separately confirmed by ESI-MS, found [M + Na+] 731.1.
4′-[(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(R)-hydroxymethyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (18R)
Following the general Suzuki-coupling procedure, the impure mannosyl bromide 17R (0.075 g, 0.1 mmol), commercially available N-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzamide (0.039g, 0.15 mmol), cesium carbonate (0.098 g, 0.3 mmol), and tetrakis(triphenylphosphine)palladium (0.012 g, 0.01 mmol) in dioxane/water (5 mL/1 mL) were stirred under N2, at 80 °C for 1 h. Upon completion, the solvent was removed, and the resulting residue was purified by silica gel chromatography (methanol–dichloromethane gradient elution) to give 4′-[(2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(R)-hydroxymethyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (18R) in 9% total yield (0.039 g, 0.051 mmol) (3 steps; based on 0.56 mmol carbonitrile 15). Analytical data for 18R: 1H NMR (300 MHz, acetonitrile-d3) δ ppm 2.89 (d, J = 4.7 Hz, 3H), 3.49–3.69 (m, 2H), 3.72–3.83 (m, 2H), 3.85–3.96 (m, 1H), 4.06 (dd, J = 8.0, 3.0 Hz, 2H), 4.13–4.22 (m, 1H), 4.27–4.46 (m, 2H), 4.48–4.68 (m, 5H), 4.76 (d, J = 11.3 Hz, 1H), 4.90–5.00 (m, 1H), 7.03–7.15 (m, 1H), 7.18–7.41 (m, 20H), 7.43–7.56 (m, 3H), 7.57–7.65 (m, 2H), 7.69–7.80 (m, 2H), 8.00 (t, J = 1.6 Hz, 1H). ESI-MS found: [M + Na+] 786.2.
4′-[(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(S)-hydroxymethyl]-N-methyl-[1,1′biphenyl]-3-carboxamide (18S)
The title compound was synthesized from 17S (0.034 g, 0.048 mmol) following the same procedure described for 18R. 18S was obtained in 6.5% (0.028 g, 0.037 mmol) (3 steps, based on 0.56 mmol carbonitrile 15). Analytical data for 18S: 1H NMR (300 MHz, acetonitrile-d3) δ ppm 2.90 (d, J = 4.7 Hz, 3H), 3.48 (d, J = 4.4 Hz, 1H), 3.57–3.62 (m, 1H), 3.62–3.85 (m, 3H), 3.86–3.94 (m, 2H), 3.95–4.03 (m, 1H), 4.33–4.51 (m, 6H), 4.52–4.76 (m, 2H), 4.85–4.99 (m, 1H), 6.98–7.45 (m, 22H), 7.47–7.68 (m, 4H), 7.71–7.84 (m, 2H), 8.03 (t, J = 1.7 Hz, 1H). ESI-MS found: [M + Na+] 786.2.
(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R/S)-ol (19R/S)
Into a flask containing 4-bromo-2-methyl-iodobenzene (9.04 mL, 63.29 mmol) in anhydrous Et2O (150 mL) under N2 was added BuLi/hexanes (2.5 M, 21.7 mL) dropwise at −78 °C. After 1 h, the freshly prepared crude 16 [from starting material 15 (4.97 g, 9.04 mmol)] was dissolved into Et2O (50 mL) and added via cannula over a period of 5 min. The mixture was stirred at −78 °C for 30 min and then slowly warmed to 0 °C over 1.5 h. Saturated aq NH4Cl was used to quench the reaction, and the reaction was extracted with ethyl acetate (2 × 100 mL). The organic fractions were then combined and washed with brine (100 mL), dried over Na2SO4, and concentrated in vacuo. The resulting residue was mixture of diastereomers, which were purified and separated by silica gel chromatography (1/9, v/v, ethyl acetate–hexanes, isocratic elution) to give (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol (19R) as a syrup in 16% yield (1.05g, 1.45 mmol) and (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(S)-ol (19S) as a syrup in 20% yield (1.30 g, 1.80 mmol); the fractions coming out of the column earlier containing isomer 19R. Analytical data for 19R: 1H NMR (400 MHz, chloroform-d) δ ppm 2.29 (s, 3H); 3.49 (br s, 1H), 3.70–3.83 (m, 2H), 3.89 (t, J = 5.9 Hz, 1H), 3.94–3.99 (m, 1H), 4.10 (t, J = 5.1 Hz, 1H), 4.13–4.18 (m, 1H), 4.21–4.28 (m, 1H), 4.40 (s, 2H), 4.49 (s, 2H), 4.56–4.64 (m, 3H), 4.71 (d, J = 11.7 Hz, 1H), 5.08 (d, J = 5.1 Hz, 1H), 7.13–7.18 (m, 2H), 7.28–7.41 (m, 21H). ESI-MS found: [M + Na+] 745.5 (100%), 747.5 (97.3%). Analytical data for 19S: 1H NMR (400 MHz, chloroform-d) δ ppm 2.18 (s, 3H), 3.19 (br s, 1H), 3.67–3.73 (m, 2H), 3.76–3.85 (m, 3H), 4.03–4.11 (m, 2H), 4.44–4.62 (m, 7H), 4.67–4.73 (m, 1H), 5.06 (d, J = 5.5 Hz, 1H), 7.16–7.37 (m, 23H). ESI-MS found: [M + Na+] 745.5 (100%), 747.5 (97.3%).
Two-Step Oxidation and Reduction Protocol to Convert 19S into 19R (Step 1 19S to 19a)
(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methanone (19a)
The 19S product was converted into the 19R isomer via a two-step oxidation reduction procedure. First, oxidation to the ketone intermediate (19a) was achieved by dissolving 19S (0.58 g, 0.80 mmol) in dry CH2Cl2 (50 mL) and dry pyridine (0.16 mL, 2.01 mmol) under N2 at 0 °C. Dess–Martin periodinane (DMP) (0.68 g, 1.61 mmol) was added portionwise, and the reaction mixture was kept at 0 °C for 1 h and then allowed to warm to 15 °C over an additional 1.5 h. Upon completion, the reaction flask was cooled in an ice bath and a 1:1 mixture of 10% aq Na2S2O3 (6 mL) and saturated aq NaHCO3 (6 mL) was added, and the reaction was stirred for 5 min at rt. The layers were then separated, and the aqueous layer was extracted an additional time with CH2Cl2 (5 mL). The organic fractions were combined and washed sequentially with saturated aq NaHCO3 (10 mL) and brine (10 mL), dried over Na2SO4, and concentrated in vacuo without heating. The residue was quickly purified by silica gel chromatography (ethyl acetate–hexane gradient elution), and pure compound eluent was again concentrated in vacuo in the absence of heat to afford the desired ketone intermediate (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methanone (19a) in 74% yield (0.43 g, 0.59 mmol). Analytical data for 19a: 1H NMR (400 MHz, chloroform-d) δ ppm 2.40 (s, 3H), 3.50–3.56 (m, 1H), 3.61 (d, J = 10.6 Hz, 1H), 3.67–3.77 (m, 2H), 4.05 (t, J = 9.0 Hz, 1H), 4.43 (d, J = 12.1 Hz, 1H), 4.49–4.61 (m, 3H), 4.64–4.74 (m, 2H), 4.76–4.85 (m, 2H), 4.89 (d, J = 11.0 Hz, 1H), 5.12 (d, J = 2.7 Hz, 1H), 7.19–7.24 (m, 2H), 7.31–7.45 (m, 20H), 7.60 (d, J = 8.6 Hz, 1H). ESI-MS found: [M + Na+] 743.5 (100%), 745.5 (97.3%).
Two-Step Oxidation and Reduction Protocol to Convert 19S into 19R (Step 2 19a to 19R)
(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol (19R)
Next, selective reduction to the (R)-alcohol was achieved by reacting ketone (19a) (0.31 g, 0.43 mmol) in dry THF (30 mL) under N2 at −40 °C, with the dropwise addition of lithium tri-tert-butoxyaluminum hydride (1 M in hex; 0.87 mL, 0.87 mmol). The reaction was warmed to 0 °C over 1 h and then stirred an additional 1 h at 0 °C. Upon completion, the reaction was diluted with ethyl acetate (60 mL). Saturated aq potassium sodium tartrate (30 mL) was added, and the reaction was vigorously stirred 1 h at rt. At this time, the layers were separated and the aqueous layer was additionally extracted with ethyl acetate (2 × 15 mL), using 1 M aq HCl to break up any remaining emulsion. The organic fractions were then combined, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (ethyl acetate–hexane gradient elution) to afford the desired R-isomer 19R (0.26 g, 0.36 mmol) in 84% yield (with 16% of the 19S (0.050 g, 0.069 mmol) isomer also generated). Analytical data as reported above.
4′-[(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(R/S)-hydroxymethyl]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (20R/S)
The title compound was synthesized following the general Suzuki-coupling procedure. Although separable, for this reaction the purified 19R/S (0.130 g, 0.18 mmol, generated from the reaction of aldehyde 16) was taken as a mixture of isomers and was reacted with commercially available N-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzamide (0.071g, 0.27 mmol), cesium carbonate (0.176 g, 0.54 mmol), and tetrakis(triphenylphosphine)palladium (0.021 g, 0.018 mmol) in dioxane/water (5 mL/1 mL), and the reaction was stirred under N2, at 80 °C for 1 h. Upon completion, the solvent was removed and the resulting products were separated and purified by silica gel chromatography (methanol–dichloromethane gradient elution). 20R (0.046 g, 0.059 mmol) was obtained in 33% yield as the more nonpolar compound, and the more polar 20S (0.055 g, 0.071 mmol) was obtained in 39% yield. Analytical data for 20R: 1H NMR (400 MHz, chloroform-d) δ ppm 2.38 (s, 3H), 2.88–3.17 (m, 4H), 3.65 (dd, J = 10.6, 4.3 Hz, 1H), 3.71–3.84 (m, 2H), 4.02 (d, J = 5.5 Hz, 2H), 4.09–4.21 (m, 2H), 4.30–4.44 (m, 4H), 4.51–4.68 (m, 4H), 5.17 (dd, J = 5.9, 3.1 Hz, 1H), 6.15 (d, J = 4.7 Hz, 1H), 7.08–7.39 (m, 22H), 7.43–7.54 (m, 2H), 7.62–7.67 (m, 1H), 7.68–7.73 (m, 1H), 7.87–7.90 (m, 1H). MS (ESI) found: [M + Na+] 800.6. Analytical data for 20S: 1H NMR (400 MHz, chloroform-d) δ ppm 2.27 (s, 3H), 3.03 (d, J = 4.8 Hz, 3H), 3.13 (br s, 1H), 3.61–3.89 (m, 5H), 3.98–4.10 (m, 1H), 4.12–4.20 (m, 1H), 4.33–4.61 (m, 7H), 4.69 (d, J = 11.4 Hz, 1H), 5.14 (d, J = 5.1 Hz, 1H), 6.19 (d, J = 4.3 Hz, 1H), 7.04–7.59 (m, 24H), 7.61–7.76 (m, 2H), 7.91–8.01 (m, 1H). ESI-MS found: [M + Na+] 800.6.
4′-[(α-d-Mannopyranosyl)-(R)-hydroxymethyl]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (21R)
Compound 20R (0.046 g, 0.059 mmol) and 10% wt Pd/C (0.050 g, 0.024 mmol) were stirred in MeOH (5 mL) under 1 atm of H2 for 16 h. The Pd/C was then filtered off, and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give 21R (0.020 g, 0.048 mmol) in 81% yield. [Compound 22 was also isolated as a product in 12% yield (0.003 g, 0.0072 mmol)]. 1H NMR (400 MHz, methanol-d4) δ ppm 2.51 (s, 3H), 2.95 (s, 3H), 3.57–3.78 (m, 4H), 4.00–4.07 (m, 1H), 4.10 (dd, J = 6.9, 2.4 Hz, 1H), 4.25 (t, J = 2.9 Hz, 1H), 5.24 (d, J = 6.7 Hz, 1H), 7.45–7.57 (m, 3H), 7.62 (d, J = 8.2 Hz, 1H), 7.71–7.83 (m, 2H), 8.07 (t, J = 1.6 Hz, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.6, 26.9, 63.0, 69.0, 69.9, 71.0, 73.3, 78.2, 81.6, 125.6, 126.7, 126.9, 128.9, 130.1, 130.1, 130.9, 136.1, 137.7, 140.5, 141.2, 142.6, 170.7. ESI-MS found: [M + Na+] 440.3.
4′-[(α-d-Mannopyranosyl)-(S)-hydroxymethyl]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (21S)
The title compound was synthesized from 20S (0.055 g, 0.071 mmol), following the same procedure as for 21R. Compound 21S (0.025 g, 0.062 mmol) was obtained in 88% yield. Analytical data for 21S: 1H NMR (400 MHz, methanol-d4) δ ppm 2.51 (s, 3H), 2.95 (s, 3H), 3.56 (dd, J = 1.0 Hz, 1H), 3.67 (d, J = 8.2 Hz, 1H), 3.70–3.82 (m, 3H), 3.91 (d, J = 9.4 Hz, 1H), 4.10 (dd, J = 9.0, 2.0 Hz, 1H), 5.28 (d, J = 8.6 Hz, 1H), 7.34–7.63 (m, 4H), 7.69–7.90 (m, 2H), 8.07 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.9, 27.0, 63.4, 67.5, 69.0, 69.9, 73.6, 77.3, 83.4, 126.0, 126.7, 127.0, 129.2, 130.1, 130.3, 130.9, 136.1, 137.4, 140.2, 140.9, 142.3, 170.6. ESI-MS found: [M + Na+] 440.3.
4′-[(α-d-Mannopyranosyl)methyl]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (22)
The title compound was obtained from the preparation of 21R as a side-product in 12% yield (0.003 g, 0.0072 mmol). Analytical data for 22: 1H NMR (400 MHz, methanol-d4) δ ppm 2.44 (s, 3H), 2.95 (s, 3H), 3.04 (d, J = 7.4 Hz, 2H), 3.69 (m, 3H), 3.83 (m, 2H), 3.86–3.92 (m, 1H), 4.04–4.21 (m, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.42–7.47 (m, 1H), 7.50 (m, 2H), 7.75 (m, 2H), 8.05 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.8, 26.9, 33.7, 63.1, 69.3, 72.1, 72.7, 76.5, 79.4, 125.6, 126.6, 126.8, 130.0, 130.1, 130.9, 131.7, 136.1, 137.8, 138.1, 139.8, 142.7, 170.7. ESI-MS found: [M + H+] 402.3.
4′-(2,3,4,6-Tetra-O-acetyl-α-d-mannopyranosyloxy)-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (23a)
4′-(-α-d-Mannopyranosyloxy)-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide20b (0.072 g, 0.178 mmol) was dissolved in anhydrous pyridine (1 mL) and acetic anhydride (1 mL). The solvent was removed in vacuo, and the residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.1% TFA)]. Pure fractions were combined and lyophilized to give the title compound as a white powder in 62% yield (0.063 g, 0.11 mmol). Analytical data for 23a: 1H NMR (400 MHz, dimethyl sulfoxide-d6) δ ppm 1.94 (s, 3H), 2.00 (s, 3H), 2.05 (s, 3H), 2.16 (s, 3H), 2.32 (s, 3H), 2.81 (d, J = 4.30 Hz, 3H), 3.93–4.11 (m, 2H), 4.19 (dd, J = 12.13, 5.09 Hz, 1H), 5.22 (t, J = 9.98 Hz, 1H), 5.33–5.45 (m, 2H), 5.80 (s, 1H), 7.23 (d, J = 8.61 Hz, 1H), 7.46–7.65 (m, 3H), 7.77 (d, J = 7.83 Hz, 2H), 8.07 (s, 1H), 8.54 (d, J = 4.30 Hz, 1H). 13C NMR (100 MHz, chloroform-d) δ ppm 16.4, 20.8 (×3), 21.0, 27.0, 62.3, 66.0, 69.2, 69.5, 69.6, 96.0, 114.7, 125.3, 125.7, 125.7, 128.1, 129.0, 129.7, 130.0, 135.0, 135.3, 141.1, 153.9, 168.4, 169.9, 170.1, 170.2, 170.7. ESI-MS found: [M + Na+] 594.3.
4′-(6-Dihydrogen Phosphate-α-d-mannopyranosyloxy)-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (23b)
4′-(-α-d-Mannopyranosyloxy)-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide20b (0.20 g, 0.5 mmol) was dissolved in trimethyl phosphate (5 mL) and water (9 uL, 0.5 mmol). The reaction was cooled to 0 °C, and then phosphoryl trichloride (142 μL, 1.5 mmol) was slowly added and then stirred for 3 h at 0 °C. The reaction was neutralized by adding crushed ice and then conc ammonia. The solvent was removed in vacuo, and the residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.1% TFA)]. Pure fractions were combined and lyophilized to give the title compound as a white powder in 29% yield (0.070 g, 0.145 mmol). Analytical data for 23b: 1H NMR (400 MHz, dimethyl sulfoxide-d6) δ ppm 2.26 (s, 3H), 2.81 (d, J = 4.70 Hz, 3H), 3.42–3.68 (m, 3H), 3.75 (dd, J = 9.00, 3.13 Hz, 1H), 3.86–3.97 (m, 2H), 4.03 (dd, J = 9.78, 5.87 Hz, 1H), 5.45 (d, J = 1.96 Hz, 1H), 7.24 (d, J = 8.61 Hz, 1H), 7.43–7.60 (m, 3H), 7.76 (dd, J = 7.43, 1.57 Hz, 2H), 8.06 (s, 1H), 8.56 (d, J = 4.30 Hz, 1H). 13C NMR (100 MHz, dimethyl sulfoxide-d6) δ ppm 16.1, 26.3, 66.3, 70.1, 70.6, 73.3, 73.3, 98.7, 115.1, 124.8, 125.4, 125.6, 127.3, 128.8, 128.9, 129.1, 133.0, 135.1, 139.9, 154.3, 166.6. ESI-MS found: [M + H+] 484.3
4′-[6-O-(N,N-Dimethylaminoacetyl)-α-d-mannopyranosyloxy]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (23c)
Step 1: 4′-(-α-d-Mannopyranosyloxy)-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide20b (0.202 g, 0.50 mmol) was stirred in a flask with triethylamine (0.38 mL, 2.75 mmol) and anhydrous DMF (2 mL) at under N2 at 0 °C. Trimethylsilyl chloride (0.35 mL, 2.75 mmol) was added dropwise, and the reaction was brought to rt and stirred an additional 3.5 h. The reaction was then quenched with ice water, and the reaction mixture was extracted twice with ethyl acetate. The organic extractions were then combined and concentrated under reduced pressure to give the crude 4′-[2,3,4,6-tetra-O-trimethylsilyl-α-d-mannopyranosyloxy]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide intermediate, which was then redissolved in acetone/methanol (1/1.5 mL). The reaction was cooled to 0 °C, and acetic acid (0.055 mL, 0.96 mmol) was added. The reaction was allowed to slowly warm to rt and was monitored for progress by TLC (ethyl acetate–hexanes, 1/1). After 9 h, NaHCO3 (0.16 g, 1.90 mmol) was added and the solvents were removed in vacuo. Purification by silica gel chromatography (ethyl acetate–hexanes gradient elution) gave the corresponding 4′-[2,3,4,-tri-O-trimethylsilyl-α-d-mannopyranosyloxy]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide in 61% yield (0.19 g, 0.36 mmol). Analytical data: 1H NMR (400 MHz, chloroform-d) δ ppm 0.18 (d, J = 4.3 Hz, 18H), 0.23 (s, 9H), 1.90 (dd, J = 7.0, 5.9 Hz, 1H), 2.30 (s, 3H), 3.05 (d, J = 4.7 Hz, 3H), 3.62–3.68 (m, 1H), 3.69–3.77 (m, 2H), 4.01–4.05 (m, 3H), 5.39 (d, J = 1.6 Hz, 1H), 6.20 (br s, 1H), 7.17 (d, J = 8.6 Hz, 1H), 7.36–7.44 (m, 2H), 7.45–7.50 (m, 1H), 7.67 (dt, J = 7.7, 1.8 Hz, 2H), 7.95 (t, J = 1.8 Hz, 1H). ESI-MS found: [M + H+] 489.4;
Step 2: N,N-Dimethylglycine hydrochloride (0.0154 g, 0.11 mmol) was dissolved into CH2Cl2/diisopropylethylamine (DIPEA) (5/0.2 mL). Dimethylaminopyridine (DMAP) (0.0024 g, 0.02 mmol) was added, followed by the 4′-[2,3,4,-tri-O-trimethylsilyl-α-d-mannopyranosyloxy]-N,3′-dimethyl-[1,1′biphenyl]-3-carboxamide (0.062 g, 0.10 mmol) from step 1 and finally N,N-diisopropylcarbodiimide (DIC) (0.020 mL, 0.13 mmol). The reaction was stirred for 16 h and then solvent was removed under reduced pressure and the residue was redissolved into acetonitrile (3 mL) and the reaction was cooled to 0 °C. Trifluoroacetic acid (0.08 mL) was added, and the reaction was stirred for 2 h at 0 °C. The solvent was removed in vacuo, and the residue was purified by HPLC [C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.1% TFA)]. Pure fractions were combined and lyophilized to give the title compound as a white powder (0.015 g, 0.031 mmol) in 31% yield. Analytical data for 23c: 1H NMR (400 MHz, methanol-d4) δ ppm 2.32 (s, 3H), 2.89 (s, 6H), 2.95 (s, 3H), 3.71–3.85 (m, 2H), 3.94–4.00 (m, 1H), 4.06 (d, J = 5.48 Hz, 2H), 4.11 (t, J = 2.54 Hz, 1H), 4.42 (m, 1H), 4.61 (dd, J = 11.74, 1.56 Hz, 1H), 5.57 (d, J = 1.57 Hz, 1H), 7.23 (d, J = 8.61 Hz, 1H), 7.47–7.53 (m, 3H), 7.73–7.78 (m, 2H), 8.05 (m, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 16.5, 27.0, 44.4 (×2), 57.9, 66.5, 68.3, 71.9, 72.5, 72.8, 99.9, 115.6, 126.4, 126.5, 126.5, 129.0, 130.1, 130.6, 130.6, 135.3, 136.1, 142.3, 155.5, 166.7, 170.7. ESI-MS found: [M + H+] 489.4;
(2,3,4,6-Tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol Acetate (26)
Dimethylaminopyridine (8.9 mg, 0.07 mmol) and (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol (19R) (1.06 g, 1.47 mmol) were dissolved in dry pyridine (5 mL) under N2, and the reaction was cooled to 0 °C. Acetic anhydride (0.21 mL, 2.21 mmol) was added dropwise, and after 5 min the ice bath was removed. After stirring 1 h at rt, the reaction was cooled to 0 °C and quenched with MeOH (0.5 mL) and the pyridine was removed in vacuo. The residue was redissolved in CH2Cl2, (25 mL) and washed successively with water (10 mL), 1 M aq HCl (2 × 10 mL), and water (10 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (ethyl acetate–hexane gradient elution) to afford compound 26 (1.11 g, 1.46 mmol) in 99% yield. Analytical data for 26: 1H NMR (400 MHz, chloroform-d) δ ppm 1.91 (s, 3H), 2.38 (s, 3H), 3.60–3.69 (m, 1H), 3.69–3.78 (m, 1H), 3.78–3.87 (m, 2H), 3.91–4.04 (m, 2H), 4.34 (dd, J = 6.7, 3.5 Hz, 1H), 4.39–4.46 (m, 1H), 4.50–4.69 (m, 6H), 4.81 (d, J = 11.3 Hz, 1H), 6.15 (d, J = 6.7 Hz, 1H), 7.07–7.13 (m, 1H), 7.23–7.38 (m, 22H). ESI-MS found: [M + Na+] 787.5 (100%), 789.5 (97.3%).
(2,3,4,6-Tetra-O-acetyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol Acetate (27)
Following a modified literature protocol for benzyl ether removal,40 (2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-(4-bromo-2-methylphenyl)-methan-1(R)-ol acetate (26) (1.0 g, 1.30 mmol) was dissolved in dry CH2Cl2 (50 mL) under N2, and the reaction was cooled to −78 °C. Boron trichloride (1 M in CH2Cl2; 10.4 mL, 10.4 mmol) was added dropwise, and the reaction was stirred for 30 min. Once removal of the benzyl ethers was complete, the excess reagent was quenched by the addition of MeOH (1 mL). At this time, it was observed (via TLC and LCMS) that upon quenching, a minor product corresponding to benzylic acetate cleavage was produced. The reaction mixture containing both the tetraacetate and pentaacetate intermediates was concentrated under reduced pressure and purified by silica gel chromatography (methanol–dichloromethane gradient elution). Both intermediates were collected and combined and then redissolved in dry pyridine (10 mL) under N2, and the reaction was cooled to 0 °C. Dimethylaminopyridine (7.0 mg, 0.06 mmol) was added, followed by acetic anhydride (0.77 mL, 8.16 mmol), and the reaction was stirred for 5 min at 0 °C, then brought to rt. After 1 h, the reaction was cooled to 0 °C and quenched with MeOH (0.25 mL). The pyridine was removed in vacuo, and the residue was then redissolved in CH2Cl2, (25 mL) and washed successively with water (10 mL), 1 M aq HCl (2 × 10 mL), and water (10 mL), dried over Na2SO4, and concentrated in vacuo. Purification by silica gel chromatography (ethyl acetate–hexanes gradient) gave the desired compound 27 (0.51 g, 0.88 mmol) in 68% yield. Analytical data for 27: mp 115–118 °C (diethyl ether–hexanes). 1H NMR (400 MHz, chloroform-d) δ ppm 1.90 (s, 3H), 1.97 (s, 3H), 2.01 (s, 6H), 2.08 (s, 3H), 2.37 (s, 3H), 3.84–3.90 (m, 1H), 3.94 (dd, J = 12.1, 2.0 Hz, 1H), 4.14–4.23 (m, 2H), 5.10 (t, J = 8.6 Hz, 1H), 5.30 (dd, J = 9.0, 3.1 Hz, 1H), 5.47 (t, J = 2.9 Hz, 1H), 6.12 (d, J = 7.0 Hz, 1H), 7.15–7.20 (m, 1H), 7.23–7.30 (m, 2H). ESI-MS found: [M + Na+] 595.2 (100%), 597.2 (97.3%).
7-(4-[(α-d-Mannopyranosyl)-(R)-hydroxymethyl)-3-methylphenyl]-1(2H)-isoquinolinone (28R)
Following the general Suzuki-coupling procedure, the acetylated mannosyl bromide 27 (0.10 g, 0.174 mmol), commercially available1-hydroxyisoquinoline-7-boronate ester (0.095 g, 0.35 mmol), cesium carbonate (0.17 g, 0.052 mmol), and tetrakis(triphenylphosphine)palladium (0.03 g, 0.026 mmol) in dioxane/water (5 mL/1 mL) were reacted under N2 at 80 °C for 1.5 h. Upon completion, the reaction was then cooled to rt, and solvents were evaporated under reduced pressure. The crude reaction residue was then redissolved into CH2Cl2 (typically a colorless solid byproduct remains insoluble) and partially purified by column chromatography to remove metal catalysts and salts to give the impure acetate-protected intermediate. The mannoside intermediate was then redissolved in MeOH (3 mL) and cooled to 0 °C. [1 M] Sodium methoxide in MeOH was added dropwise until a pH of 9–10 was achieved. After 5 min, the ice bath was removed and the reaction was stirred for 2 h. Upon completion, the reaction was neutralized with H+ exchange resin (DOWEX 50WX4-100). The resin was filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 28R (0.035 g, 0.082 mmol) in 47% yield. Analytical data for 28R: 1H NMR (400 MHz, methanol-d4) δ ppm 2.52 (s, 3H), 3.64–3.75 (m, 4H), 4.05 (br s, 1H), 4.09–4.15 (m, 1H), 4.26 (br s, 1H), 5.25 (d, J = 6.7 Hz, 1H), 6.70 (d, J = 7.0 Hz, 1H), 7.18 (d, J = 7.0 Hz, 1H), 7.52–7.61 (m, 2H), 7.62–7.67 (m, 1H), 7.72 (d, J = 8.2 Hz, 1H), 8.00 (d, J = 8.6 Hz, 1H), 8.54 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.6, 63.0, 69.0, 69.9, 71.0, 73.2, 78.3, 81.6, 107.7, 125.5, 125.5, 127.3, 128.2, 128.8, 129.0, 130.0, 132.6, 137.8, 138.8, 140.1, 140.7, 141.3, 165.1. ESI-MS found: [M + H+] 428.4, [M – 18 + H+] 410.3, [2M + H+] 855.6.
3,4-Dihydro-7-(4-[(α-d-mannopyranosyl)-(R)-hydroxymethyl]-3-methylphenyl)-1(2H)-isoquinolinone (29R)
Following the general Suzuki-coupling procedure, mannosyl bromide 19R (0.220 g, 0.304 mmol), commercially available1-hydroxyisoquinoline-7-boronate ester (0.165 g, 0.608 mmol), cesium carbonate (0.297 g, 0.912 mmol), and tetrakis(triphenylphosphine)palladium (0.046 g, 0.053 mmol) in dioxane/water (10 mL/2 mL) were reacted under N2, at 80 °C for 1.5 h. Upon completion, the reaction was then cooled to rt and solvents were evaporated under reduced pressure. The crude reaction residue was then redissolved into CH2Cl2 (typically a colorless solid byproduct remains insoluble) and partially purified by column chromatography to remove metal catalysts and salts to give the impure benzyl-protected intermediate. The mannoside intermediate was then redissolved in MeOH (5 mL), and 10% wt Pd/C (0.150 g, 0.14 mmol) was added. The reaction was stirred under 1 atm of H2 for 16 h. Upon completion, the Pd/C was filtered off and the filtrate was concentrated and dried in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 29R (0.06 mg, 0.140 mmol) in 46% yield. Analytical data for 29R: 1H NMR (400 MHz, methanol-d4) δ ppm 2.49 (s, 3H), 3.01 (t, J = 6.7 Hz, 2H), 3.52 (t, J = 6.7 Hz, 2H), 3.63–3.74 (m, 4H), 4.05 (br s, 1H), 4.10 (dd, J = 6.7, 2.0 Hz, 1H), 4.25 (t, J = 2.5 Hz, 1H), 5.24 (d, J = 7.0 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 7.44–7.53 (m, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.75 (dd, J = 7.8, 2.0 Hz, 1H), 8.18 (d, J = 1.6 Hz, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.6, 28.7, 40.8, 63.0, 69.0, 69.9, 71.0, 73.2, 78.2, 81.6, 125.3, 126.6, 128.9, 129.2, 129.8, 130.2, 131.7, 137.6, 139.7, 140.2, 141.0, 141.1, 168.3. ESI-MS found: [M + H+] 430.4, [M – 18 + H+] 412.4, [2M + H+] 859.6.
3,4-Dihydro-7-[4-(α-d-mannopyranosyloxy)-3-methylphenyl]-1(2H)-isoquinolinone (30)
The previously reported compound 7-[4-(α-d-mannopyranosyloxy)-3-methylphenyl]-isoquinolin-1-one20b (25) (30 mg, 0.073 mmol) was dissolved into MeOH (3 mL), and 10% wt Pd/C (30 mg) was added. The reaction was stirred under 1 atm of H2 for 16 h, after which the reaction was filtered and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 30 (0.30 mg, 0.072 mmol) in 99% yield. Analytical data for 30: 1H NMR (400 MHz, methanol-d4) δ ppm 2.30 (s, 3H), 2.96–3.03 (m, 2H), 3.49–3.55 (m, 2H), 3.57–3.65 (m, 1H), 3.72–3.81 (m, 3H), 3.97 (d, J = 9.4 Hz, 1H), 4.08 (br s, 1H), 5.55 (s, 1H), 7.31 (dd, J = 19.4, 8.0 Hz, 2H), 7.40–7.47 (m, 2H), 7.70 (d, J = 7.8 Hz, 1H), 8.14 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 16.6, 28.7, 40.9, 62.7, 68.4, 72.1, 72.7, 75.5, 99.9, 115.9, 126.3, 126.3, 128.9, 129.1, 130.2, 131.5, 135.1, 139.2, 141.0, 155.7, 168.4. ESI-MS found: [M + H+] 416.4, [2M + H+] 831.6.
7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)isoquinolin-1-amine (31a) and Isoquinolin-1-amine-7 Boronic Acid (31b)
Potassium acetate (264 mg, 2.69 mmol) was activated by adding it to a round-bottom flask, which was then heated to 250 °C under vacuum for 2 min and then allowed to cool to rt under vacuum for an additional 10 min, after which time a nitrogen atmosphere was continuously maintained. Dry DMSO (2 mL) was added, followed by the addition of commercially available 7-bromoisoquinolin-1-amine (150 mg, 0.67 mmol) and bis(pinacolato)diboron (256 mg, 1.0 mmol). Pd(dppf)Cl2 (49.2 mg, 0.067 mmol) was added, and the reaction flask was evacuated under high vacuum and then backfilled with N2 three times. The flask was then placed in an oil bath preheated to 80 °C and allowed to stir for 2.5 h. The reaction was cooled to rt, and solvents were evaporated under reduced pressure. The crude reaction residue was then redissolved into CH2Cl2 and allowed to sit for 5 min to allow for byproducts to precipitate. The precipitate was filtered off. Evaporation of the CH2Cl2 in vacuo resulted in more byproduct precipitation, and so the residue was redissolved in CH2Cl2 and the process was repeated until no further precipitation was observed. The crude brown residue was then diluted with water (1 mL) and lyophilized to removed trace DMSO, resulting in a brown solid, which was comosed of a mix of the boronate ester and the boronic acid (31a/b), as determined by LCMS. This crude mixture was used without further purification. Analytical data for 31a: ESI-MS found, [M + H+] 271.3. Analytical data for 31b: ESI-MS found, [M + H+] 189.2
7-(4-[(α-d-Mannopyranosyl)-(R)-hydroxymethyl]-3-methylphenyl)-isoquinolin-1-amine (31R)
Following the general Suzuki-coupling procedure, mannosyl bromide 27 (0.092 g, 0.160 mmol), isoquinolin-1-amine-7-boronate ester/acid 31a/b (0.086, 0.32 mmol), cesium carbonate (0.16 g, 0.48 mmol), and tetrakis(triphenylphosphine)palladium (0.028 g, 0.024 mmol) in dioxane/water (5 mL/1 mL) were reacted under N2 at 80 °C for 1.5 h. Upon completion, the solvent was removed under reduced pressure and mixture was filtered through a silica gel column (ethyl acetate–hexanes, 2/1 isocratic elution) to remove the metal catalyst and salts. The filtrate was concentrated then dried in vacuo. The crude compound was then redissolved into MeOH (3 mL) and cooled to 0 °C. Sodium methoxide [1M] in MeOH was added dropwise until a pH of 9–10 was achieved. After 5 min, the ice bath was removed and the reaction was stirred for 30 min. Upon completion, the reaction was neutralized with H+ exchange resin (DOWEX 50WX4-100). The resin was filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 31R (0.018 g, 0.042 mmol) in 26% yield. Analytical data for 31R: 1H NMR (400 MHz, methanol-d4) δ ppm 8.71 (s, 1H), 8.26 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 8.6 Hz, 1H), 7.62–7.72 (m, 3H), 7.54 (d, J = 7.0 Hz, 1H), 7.24 (d, J = 7.0 Hz, 1H), 5.27 (d, J = 7.0 Hz, 1H), 4.27 (t, J = 2.9 Hz, 1H), 4.13 (dd, J = 6.8, 2.2 Hz, 1H), 4.01–4.07 (m, 1H), 3.63–3.73 (m, 4H), 2.55 (s, 3H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.7, 63.0, 69.0, 69.9, 70.8, 73.1, 78.4, 81.5, 113.0, 119.6, 123.6, 125.8, 127.8, 129.3, 129.6, 130.3, 135.0, 137.7, 138.1, 138.9, 142.3, 143.0, 156.1. ESI-MS found: [M + H+] 427.4.
5-(4-[(α-d-Mannopyranosyl)-(R)-hydroxymethyl]-3-methylphenyl)-isoquinoline (32R)
Following the general Suzuki-coupling procedure, mannosyl bromide 27 (0.092 g, 0.160 mmol), commercially purchased 5-isoquinolinylboronic acid (0.056 g, 0.32 mmol), cesium carbonate (0.16 g, 0.48 mmol), and tetrakis(triphenylphosphine)palladium (0.028 g, 0.024 mmol) in dioxane/water (5 mL/1 mL) were reacted under N2 at 80 °C for 1.5 h. Upon completion, the solvent was removed under reduced pressure and mixture was filtered through a silica gel column (ethyl acetate–hexanes, 2/1 isocratic elution) to remove the metal catalyst and salts. The filtrate was concentrated then dried in vacuo. The crude compound was then redissolved into MeOH (5 mL) and cooled to 0 °C. Sodium methoxide [1M] in MeOH was added dropwise until a pH of 9–10 was achieved. After 5 min, the ice bath was removed and the reaction was stirred for 30 min. Upon completion, the reaction was neutralized with H+ exchange resin (DOWEX 50WX4-100). The resin was filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 32R (0.025 g, 0.060 mmol) in 38% yield. Analytical data for 32R: 1H NMR (400 MHz, methanol-d4) δ ppm 9.75 (s, 1H), 8.48 (dd, J = 15.8, 7.2 Hz, 2H), 8.28 (d, J = 6.7 Hz, 1H), 8.02–8.13 (m, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.34 (s, 1H), 5.30 (d, J = 7.0 Hz, 1H), 4.28 (br s, 1H), 4.14–4.19 (m, 1H), 4.04 (br s, 1H), 3.69 (br s, 4H), 2.54 (s, 3H). 13C NMR (100 MHz, methanol-d4) δ ppm 19.6, 63.1, 69.1, 70.1, 70.8, 73.2, 78.4, 81.4, 124.2, 128.5, 129.1, 129.6, 129.6, 130.7, 131.5, 132.9, 134.2, 137.4, 137.6, 138.1, 141.6, 142.3, 149.6. ESI-MS found: [M + H+] 412.3.
7-[4-(α-d-Mannopyranosyloxy)-3-methylphenyl]- isoquinolin-1-amine (33)
Step 1: Under nitrogen atmosphere, the mixture of 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside20b (2.869 g, 5.55 mmol), bis(pinacolato)diboron (1.690g, 6.66 mmol), potassium acetate (2.177 g, 22.18 mmol), and (1.1′-bis(diphenylphosohino)ferrocene)dichloropalladium(II) (Pd(dppf)Cl2) (0.244 g, 0.33 mmol) in DMSO (50 mL) was heated at 80 °C with stirring for 2 h. The solvent was removed, and the resulting residue was purified by silica gel chromatography (ethyl acetate–dichloromethane gradient elution) to give the intermediate boronate ester, 2-methyl-4-[4,4,5,5-tetramethyl-(1,3,2)dioxaborolan-2-yl]phenyl 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (33a),25 (2.50 g, 4.43 mmol) in 79% yield. Analytical data: 1H NMR (400 MHz, chloroform-d) δ ppm 1.34 (s, 12H), 2.05 (d, J = 2.3 Hz, 6H), 2.06 (s, 3H), 2.21 (s, 3H), 2.30 (s, 3H), 4.03–4.09 (m, 2H), 4.27–4.33 (m, 1H), 5.39 (t, J = 12.0 Hz, 1H), 5.48 (dd, J = 3.3, 1.8 Hz, 1H), 5.54–5.60 (m, 2H), 7.09 (d, J = 8.2 Hz, 1H), 7.55–7.67 (m, 2H). ESI-MS found: [M + Na+] 587.4.
Step 2: Following the general Suzuki-coupling procedure, 33a (0.150, 0.27 mmol) from step 1, 7-bromoisoquinolin-1-amine (0.060, 0.27 mmol), cesium carbonate (0.26 g, 0.80 mmol), and tetrakis(triphenylphosphine)palladium (0.031 g, 0.027 mmol) in dioxane/water (5 mL/1 mL) were reacted under N2, at 80 °C for 1.5 h. Upon completion, the solvent was removed under reduced pressure, and mixture was filtered through a silica gel column (ethyl acetate–hexanes, 2/1 isocratic elution) to remove the metal catalyst and salts. The filtrate was concentrated then dried in vacuo. The crude compound was then redissolved into MeOH (3 mL) and cooled to 0 °C. Sodium methoxide [1M] in MeOH was added dropwise until a pH of 9–10 was achieved. After 5 min, the ice bath was removed and the reaction was stirred for 2 h. Upon completion, the reaction was neutralized with H+ exchange resin (DOWEX 50WX4-100). The resin was filtered, and the filtrate was concentrated in vacuo. The resulting residue was purified by HPLC [(C18, 15 mm × 150 mm column; eluent, acetonitrile/water (0.05% TFA)] to give compound 33 (0.040 g, 0.097 mmol) in 36% yield. Analytical data for 33: 1H NMR (400 MHz, methanol-d4) δ ppm 2.34 (s, 3H), 3.57–3.64 (m, 1H), 3.72–3.83 (m, 3H), 3.98 (dd, J = 9.4, 3.1 Hz, 1H), 4.10 (br s, 1H), 5.60 (s, 1H), 7.21 (d, J = 6.7 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.52 (d, J = 7.0 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.64 (s, 1H), 7.93 (d, J = 8.6 Hz, 1H), 8.21 (d, J = 8.6 Hz, 1H), 8.64 (s, 1H). 13C NMR (100 MHz, methanol-d4) δ ppm 16.6, 62.7, 68.4, 72.1, 72.7, 75.6, 99.8, 113.0, 116.0, 119.6, 123.0, 127.0, 127.6, 129.2, 129.5, 130.6, 133.6, 134.8, 137.3, 142.9, 156.0, 156.4. ESI-MS found: [M + H+] 413.4.
Hemagglutination Assay
The hemagglutination inhibition (HAI) assay was performed with UTI89 bacteria and guinea pig red blood cells, as previously described.25
Biofilm Assay
The biofilm assay was performed with UTI89 bacteria as previously described.25
Differential Scanning Fluorimetry (DSF)
The DSF assay was performed with FimHL (10 μM) in the absence or presence of 100 μM mannoside, as previously described.25
Computational Molecular Modeling
Computer-generated docking was performed with AutoDock Vina. Ligand coordinates were generated in eLBOW within the Phenix program suite. Simple optimization of mannosides 21R and 21S, as described by its SMILES sequence, was manually inspected and modified to ensure the correct chair conformation of the mannose pucker. These pdb coordinates were then converted to pdbqt format using Babel. The X-ray coordinates of FimH from the 23 co-crystal structure was converted to its topology file using AutoDock Tools. The grid box was centered at the mannose binding pocket of FimH, and its dimensions (40 × 40 × 40 Å3) were chosen to accommodate multiple potential binding modes at or near the binding pocket. The exhaustiveness of the search was set to a value of 20. The top binding modes and scores within this grid space were generated by AutoDock Vina and visualized in PyMOL.
Animal Infections
The bacterial inoculum and infection of mice was performed as previously described.20a Briefly, the bacterial strain UTI89 with a kanamycin antibiotic cassette inserted in the chromosome (UTI89KAN) was grown under type 1-inducing conditions, 2 × 24 static growth in LB at 37 °C. Bacteria were harvested, washed, and resuspended in sterile PBS to the appropriate concentration. Six to eight week C3H/HeN female mice were purchased from Envigo (formerly Harlan Laboratories). Mice were anesthetized by inhalation of isoflurane and infected by transurethral inoculation of 50 μL of bacterial suspension at the appropriate density. At the designated time point, mice were killed by cervical dislocation under anesthesia and bladders harvested and processed by mechanical disruption. Processed bladders were serially diluted and plated on LB with 50 μg/mL of kanamycin to enumerate bacterial burden.
Treatment of Chronic Urinary Tract Infection in Mice
Mice were infected with 1–2 × 108 UTI89KAN, and the infection was allowed to continue for 2 weeks. Twelve days post inoculation, the urine from mice was collected and titered to identify chronically infected mice (urine titers ≥105). At 2 weeks post inoculation, chronically infected mice were treated orally with 100 μL of mannoside resuspended in 10% cyclodextrin to the appropriate concentration to generate the designated dosage. Bladders were harvested and titered to determine the bacterial burden at the designated time points following treatment.
Prophylactic Treatment of Acute Urinary Tract Infection in Mice
Prophylactic treatment of mice involved oral delivery of mannoside at 25 mg/kg 30 min prior to infection with UTI89KAN. Mice were infected with 1–2 × 107 UTI89KAN, and bladders were harvested and titered to determine the bacterial burden 6 h following inoculation.
Mouse Pharmacokinetic (PK) Studies
For dosing in mice, 100 μL of a solution of mannoside in 10% cyclodextrin [10 mg/mL (50 mg/kg)] was inoculated with a gavage needle into the mouse stomach. Urine was collected at 1, 3, 6, and 8 h after dosing and spiked with an internal standard. Analysis for test article levels by LC–MS/MS was performed using a C18 reversed phase column and an AB Sciex API-4000 QTrap instrument (a gradient of acetonitrile and water in 0.1% formic acid), with ion spray detection (+) ESI. Selected reaction monitoring (SRM) mode quantification was performed for the following MS/MS transitions [precursor mass/charge ratio (m/z)/product m/z]: compound 21R, 418.20/238.20 amu; compound 23, 404.20/242.20 amu (Levels of 23 were quantitated after dosing of prodrug, 23a); compound 24, 461.20/299.20 amu.
Acknowledgments
We thank Rick Stegeman (X-ray facility, Biochemistry Department) for help with X-ray crystallography studies and Scott Wildman (Biochemistry Department) for help with molecular modeling and docking of FimH ligands. We also thank Drs. Rasesh Shah and Jay Wendling at Seventh Wave for conducting the rat PK study. This work was funded in part by the NIH grants 1RC1DK086378 (Washington University) from NIDDK, and 5R43AI106112-02 (Fimbrion Therapeutics, Inc.) from NIAID. Support for the Biomedical Mass Spectrometry Resource to do the mouse urine PK bioanalytical studies is provided by the NIH/NIGMS grant no. P41GM103422. For the small molecule X-ray structure of compound 27, we acknowledge funding from the NSF (MRI, CHE-0420497) for the purchase of the ApexII diffractometer.
Glossary
Abbreviations Used
- UTI
urinary tract infection
- UPEC
uropathogenic E. coli
- CFUs
colony forming units
- SAR
structure–activity relationships
- SPR
structure–property relationships
- HA
hemagglutination
- HAI
hemagglutination Inhibition
- IBC
intracellular bacterial community
- PD
pharmacodynamics
- PK
pharmacokinetics
- Cmax
concentration maximum
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00948.
1H and 13C NMR, HPLC, and MS analytical data for new compounds and the details of the crystallography experiments, including the X-ray crystal determinants for both for the small molecule 27 and FimH co-crystal structure with 23; details of the prophylactic acute UTI mouse model with 23 and prodrugs 23a–c, as well as the rat pharmacokinetic study of 21R, 23, and 25 (PDF)
Molecular formula strings (CSV)
Author Contributions
# L.M.-M. and Z.C. contributed equally to the work
The authors declare the following competing financial interest(s): Scott Hultgren and James Janetka are co-founders and stockholders in Fimbrion Therapeutics, Inc.
Supplementary Material
References
- a Boyle E. C.; Finlay B. B. Bacterial pathogenesis: exploiting cellular adherence. Curr. Opin. Cell Biol. 2003, 15, 633–639. 10.1016/S0955-0674(03)00099-1. [DOI] [PubMed] [Google Scholar]; b Chagnot C.; Listrat A.; Astruc T.; Desvaux M. Bacterial adhesion to animal tissues: protein determinants for recognition of extracellular matrix components. Cell. Microbiol. 2012, 14, 1687–1696. 10.1111/cmi.12002. [DOI] [PubMed] [Google Scholar]
- a Ofek I.; Beachey E. H.. General concepts and principles of bacterial adherence. In Bacterial Adherence, Receptors and Recognition, Beachey E. H., Ed.; Chapman and Hall: London, 1980; Vol. 6, pp 1–29. [Google Scholar]; b Ofek I.; Doyle R. J.. Common Themes in Bacterial Adhesion. In Bacterial Adhesion to Cells and Tissues; Chapman and Hall: New York, 1994; pp 513–561. [Google Scholar]
- Aminov R. I. A brief history of the antibiotic era: lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cole S. T. Who will develop new antibacterial agents?. Philos. Trans. R. Soc., B 2014, 369, 20130430. 10.1098/rstb.2013.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Walsh C. Where will new antibiotics come from?. Nat. Rev. Microbiol. 2003, 1, 65–70. 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- a Lee Y. M.; Almqvist F.; Hultgren S. J. Targeting virulence for antimicrobial chemotherapy. Curr. Opin. Pharmacol. 2003, 3, 513–519. 10.1016/j.coph.2003.04.001. [DOI] [PubMed] [Google Scholar]; b Rasko D. A.; Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discovery 2010, 9, 117–128. 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- a Ofek I.; Hasty D. L.; Sharon N. Anti-adhesion therapy of bacterial diseases: prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. 10.1016/S0928-8244(03)00228-1. [DOI] [PubMed] [Google Scholar]; b Cozens D.; Read R. C. Anti-adhesion methods as novel therapeutics for bacterial infections. Expert Rev. Anti-Infect. Ther. 2012, 10, 1457–1468. 10.1586/eri.12.145. [DOI] [PubMed] [Google Scholar]; c Bouckaert J.; Berglund J.; Schembri M.; De Genst E.; Cools L.; Wuhrer M.; Hung C. S.; Pinkner J.; Slattegard R.; Zavialov A.; Choudhury D.; Langermann S.; Hultgren S. J.; Wyns L.; Klemm P.; Oscarson S.; Knight S. D.; De Greve H. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. 10.1111/j.1365-2958.2004.04415.x. [DOI] [PubMed] [Google Scholar]
- Jones C. H.; Pinkner J. S.; Roth R.; Heuser J.; Nicholes A. V.; Abraham S. N.; Hultgren S. J. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2081–2085. 10.1073/pnas.92.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C. S.; Bouckaert J.; Hung D.; Pinkner J.; Widberg C.; DeFusco A.; Auguste C. G.; Strouse R.; Langermann S.; Waksman G.; Hultgren S. J. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002, 44, 903–915. 10.1046/j.1365-2958.2002.02915.x. [DOI] [PubMed] [Google Scholar]
- Puorger C.; Vetsch M.; Wider G.; Glockshuber R. Structure, folding and stability of FimA, the main structural subunit of type 1 pili from uropathogenic Escherichia coli strains. J. Mol. Biol. 2011, 412, 520–535. 10.1016/j.jmb.2011.07.044. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Mo W. J.; Sebbel P.; Min G.; Neubert T. A.; Glockshuber R.; Wu X. R.; Sun T. T.; Kong X. P. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J. Cell Sci. 2001, 114, 4095–4103. [DOI] [PubMed] [Google Scholar]
- Eto D. S.; Jones T. A.; Sundsbak J. L.; Mulvey M. A. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 2007, 3, e100. 10.1371/journal.ppat.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. G.; Palermo J. J.; Schilling J. D.; Roth R.; Heuser J.; Hultgren S. J. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 2003, 301, 105–107. 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- Griebling T. L.Urinary tract infection in women. In Urologic Diseases in America; Litwin M.S., Saigal C.S., Eds.; U.S. Government Printing Office: Washington, DC, 2007; pp 587–620.
- a Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. DM, Dis.-Mon. 2003, 49, 53–70. 10.1067/mda.2003.7. [DOI] [PubMed] [Google Scholar]; b Kärkkäinen U. M.; Ikäheimo R.; Katila M. L.; Siitonen A. Recurrence of urinary tract infections in adult patients with community- acquired pyelonephritis caused by E. coli: a 1-year follow-up. Scand. J. Infect. Dis. 2000, 32, 495–499. 10.1080/003655400458767. [DOI] [PubMed] [Google Scholar]; c Ikähelmo R.; Siitonen A.; Heiskanen T.; Kärkkäinen U.; Kuosmanen P.; Lipponen P.; Mäkelä P. H. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin. Infect. Dis. 1996, 22, 91–99. 10.1093/clinids/22.1.91. [DOI] [PubMed] [Google Scholar]; d Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect. Dis. Clin. North Am. 2014, 28, 1–13. 10.1016/j.idc.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Ronald A. R.; Nicolle L. E.; Stamm E.; Krieger J.; Warren J.; Schaeffer A.; Naber K. G.; Hooton T. M.; Johnson J.; Chambers S.; Andriole V. Urinary tract infection in adults: research priorities and strategies. Int. J. Antimicrob. Agents 2001, 17, 343–348. 10.1016/S0924-8579(01)00303-X. [DOI] [PubMed] [Google Scholar]
- a Karlowsky J. A.; Hoban D. J.; Decorby M. R.; Laing N. M.; Zhanel G. G. Fluoroquinolone-resistant urinary isolates of Escherichia coli from outpatients are frequently multidrug resistant: results from the North American Urinary Tract Infection Collaborative Alliance-Quinolone Resistance study. Antimicrob. Agents Chemother. 2006, 50, 2251–2254. 10.1128/AAC.00123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b National Disease and Therapeutic Index; IMS Health, Inc.: Plymouth Meeting, PA, 2004. [Google Scholar]
- Blango M. G.; Mulvey M. A. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob. Agents Chemother. 2010, 54, 1855–1863. 10.1128/AAC.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydock-McGrane L. K.; Cusumano Z. T.; Janetka J. W. Mannose-derived FimH antagonists: a promising anti-virulence therapeutic strategy for urinary tract infections and Crohn’s disease. Expert Opin. Ther. Pat. 2016, 26, 175–197. 10.1517/13543776.2016.1131266. [DOI] [PubMed] [Google Scholar]
- a Martinez-Medina M.; Garcia-Gil L. J. Escherichia coli in chronic inflammatory bowel diseases: An update on adherent invasive Escherichia coli pathogenicity. World J. Gastrointest. Pathophysiol. 2014, 5, 213–227. 10.4291/wjgp.v5.i3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Boudeau J.; Barnich N.; Darfeuille-Michaud A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol. Microbiol. 2001, 39, 1272–1284. 10.1111/j.1365-2958.2001.02315.x. [DOI] [PubMed] [Google Scholar]
- a Cusumano C. K.; Pinkner J. S.; Han Z.; Greene S. E.; Ford B. A.; Crowley J. R.; Henderson J. P.; Janetka J. W.; Hultgren S. J. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci. Transl. Med. 2011, 3, 109ra115. 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han Z.; Pinkner J. S.; Ford B.; Chorell E.; Crowley J. M.; Cusumano C. K.; Campbell S.; Henderson J. P.; Hultgren S. J.; Janetka J. W. Lead optimization studies on FimH antagonists: discovery of potent and orally bioavailable ortho-substituted biphenyl mannosides. J. Med. Chem. 2012, 55, 3945–3959. 10.1021/jm300165m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Han Z.; Pinkner J. S.; Ford B.; Obermann R.; Nolan W.; Wildman S. A.; Hobbs D.; Ellenberger T.; Cusumano C. K.; Hultgren S. J.; Janetka J. W. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Firon N.; Ashkenazi S.; Mirelman D.; Ofek I.; Sharon N. Aromatic alpha-glycosides of mannose are powerful inhibitors of the adherence of type 1 fimbriated Escherichia coli to yeast and intestinal epithelial cells. Infect. Immun. 1987, 55, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Firon N.; Ofek I.; Sharon N. Carbohydrate-binding sites of the mannose-specific fimbrial lectins of Enterobacteria. Infect. Immun. 1984, 43, 1088–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Firon N.; Ofek I.; Sharon N. Interaction of mannose-containing oligosaccharides with the fimbrial lectin of Eshcerichia coli. Biochem. Biophys. Res. Commun. 1982, 105, 1426–1432. 10.1016/0006-291X(82)90947-0. [DOI] [PubMed] [Google Scholar]
- Klein T.; Abgottspon D.; Wittwer M.; Rabbani S.; Herold J.; Jiang X.; Kleeb S.; Lüthi C.; Scharenberg M.; Bezençon J.; Gubler E.; Pang L.; Smiesko M.; Cutting B.; Schwardt O.; Ernst B. FimH antagonists for the oral treatment of urinary tract infections: from design and synthesis to in vitro and in vivo evaluation. J. Med. Chem. 2010, 53, 8627–8641. 10.1021/jm101011y. [DOI] [PubMed] [Google Scholar]
- a Kleeb S.; Pang L.; Mayer K.; Eris D.; Sigl A.; Preston R. C.; Zihlmann P.; Sharpe T.; Jakob R. P.; Abgottspon D.; Hutter A. S.; Scharenberg M.; Jiang X.; Navarra G.; Rabbani S.; Smiesko M.; Ludin N.; Bezencon J.; Schwardt O.; Maier T.; Ernst B. FimH antagonists: bioisosteres to improve the in vitro and in vivo PK/PD profile. J. Med. Chem. 2015, 58, 2221–2239. 10.1021/jm501524q. [DOI] [PubMed] [Google Scholar]; b Sperling O.; Fuchs A.; Lindhorst T. K. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006, 4, 3913–3922. 10.1039/b610745a. [DOI] [PubMed] [Google Scholar]; c Chalopin T.; Alvarez Dorta D.; Sivignon A.; Caudan M.; Dumych T. I.; Bilyy R. O.; Deniaud D.; Barnich N.; Bouckaert J.; Gouin S. G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. 10.1039/C6OB00424E. [DOI] [PubMed] [Google Scholar]; d Alvarez Dorta D.; Sivignon A.; Chalopin T.; Dumych T. I.; Roos G.; Bilyy R. O.; Deniaud D.; Krammer E.-M.; de Ruyck J.; Lensink M. F.; Bouckaert J.; Barnich N.; Gouin S. G. The Antiadhesive Strategy in Crohn′s Disease: Orally Active Mannosides to Decolonize Pathogenic Escherichia coli from the Gut. ChemBioChem 2016, 17, 936–952. 10.1002/cbic.201600018. [DOI] [PubMed] [Google Scholar]; e Brument S.; Sivignon A.; Dumych T. I.; Moreau N.; Roos G.; Guerardel Y.; Chalopin T.; Deniaud D.; Bilyy R. O.; Darfeuille-Michaud A.; Bouckaert J.; Gouin S. G. Thiazolylaminomannosides as potent antiadhesives of type 1 piliated Escherichia coli isolated from Crohn’s disease patients. J. Med. Chem. 2013, 56, 5395–5406. 10.1021/jm400723n. [DOI] [PubMed] [Google Scholar]; f Pang L.; Kleeb S.; Lemme K.; Rabbani S.; Scharenberg M.; Zalewski A.; Schadler F.; Schwardt O.; Ernst B. FimH antagonists: structure-activity and structure-property relationships for biphenyl alpha-D-mannopyranosides. ChemMedChem 2012, 7, 1404–1422. 10.1002/cmdc.201200125. [DOI] [PubMed] [Google Scholar]; g Jiang X.; Abgottspon D.; Kleeb S.; Rabbani S.; Scharenberg M.; Wittwer M.; Haug M.; Schwardt O.; Ernst B. Antiadhesion therapy for urinary tract infections—A balanced PK/PD profile proved to be key for success. J. Med. Chem. 2012, 55, 4700–4713. 10.1021/jm300192x. [DOI] [PubMed] [Google Scholar]
- a Bouckaert J.; Li Z.; Xavier C.; Almant M.; Caveliers V.; Lahoutte T.; Weeks S. D.; Kovensky J.; Gouin S. G. Heptyl alpha-D-mannosides grafted on a beta-cyclodextrin core to interfere with Escherichia coli adhesion: an in vivo multivalent effect. Chem. - Eur. J. 2013, 19, 7847–7855. 10.1002/chem.201204015. [DOI] [PubMed] [Google Scholar]; b Gouin S. G.; Wellens A.; Bouckaert J.; Kovensky J. Synthetic Multimeric Heptyl Mannosides as Potent Antiadhesives of Uropathogenic Escherichia coli. ChemMedChem 2009, 4, 749–755. 10.1002/cmdc.200900034. [DOI] [PubMed] [Google Scholar]; c Touaibia M.; Wellens A.; Shiao T. C.; Wang Q.; Sirois S.; Bouckaert J.; Roy R. Mannosylated G(0) dendrimers with nanomolar affinities to Escherichia coli FimH. ChemMedChem 2007, 2, 1190–1201. 10.1002/cmdc.200700063. [DOI] [PubMed] [Google Scholar]; d Lindhorst T. K.; Kieburg C.; Krallmann-Wenzel U. Inhibition of the type 1 fimbriae-mediated adhesion of Escherichia coli to erythrocytes by multiantennary alpha-mannosyl clusters: the effect of multivalency. Glycoconjugate J. 1998, 15, 605–613. 10.1023/A:1006920027641. [DOI] [PubMed] [Google Scholar]
- Jarvis C.; Han Z.; Kalas V.; Klein R.; Pinkner J. S.; Ford B.; Binkley J.; Cusumano C. K.; Cusumano Z.; Mydock-McGrane L.; Hultgren S. J.; Janetka J. W. Antivirulence Isoquinolone Mannosides: Optimization of the Biaryl Aglycone for FimH Lectin Binding Affinity and Efficacy in the Treatment of Chronic UTI. ChemMedChem 2016, 11, 367–373. 10.1002/cmdc.201600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente V.; Martin J.; Jimenez-Barbero J.; Chiara J. L.; Vicent C. Hydrogen-bonding cooperativity: using an intramolecular hydrogen bond to design a carbohydrate derivative with a cooperative hydrogen-bond donor centre. Chem. - Eur. J. 2004, 10, 4240–4251. 10.1002/chem.200400042. [DOI] [PubMed] [Google Scholar]
- Janetka J. W.; Han Z.; Hultgren S.; Pinkner J. S.; Cusumano C. K.. Compounds and methods for treating bacterial infections. WO/2011/050323, 2010.
- Schwardt O.; Rabbani S.; Hartmann M.; Abgottspon D.; Wittwer M.; Kleeb S.; Zalewski A.; Smiesko M.; Cutting B.; Ernst B. Design, synthesis and biological evaluation of mannosyl triazoles as FimH antagonists. Bioorg. Med. Chem. 2011, 19, 6454–6473. 10.1016/j.bmc.2011.08.057. [DOI] [PubMed] [Google Scholar]
- Myers R. W.; Lee Y. C. Improved preparations of some per-O-acetylated aldohexopyranosyl cyanides. Carbohydr. Res. 1986, 154, 145–163. 10.1016/S0008-6215(00)90029-6. [DOI] [PubMed] [Google Scholar]
- Maity S. K.; Dutta S. K.; Banerjee A. K.; Achari B.; Singh M. Design and Synthesis of Mannose Analogs as Inhibitors of Alpha-Mannosidase. Tetrahedron 1994, 50, 6965–6974. 10.1016/S0040-4020(01)81349-1. [DOI] [Google Scholar]
- a Kleeb S.; Jiang X.; Frei P.; Sigl A.; Bezencon J.; Bamberger K.; Schwardt O.; Ernst B. FimH Antagonists: Phosphate Prodrugs Improve Oral Bioavailability. J. Med. Chem. 2016, 59, 3163–3182. 10.1021/acs.jmedchem.5b01923. [DOI] [PubMed] [Google Scholar]; b Janetka J. W.; Han Z.; Hultgren S.; Pinkner J. S.; Cusumano C. K.. Compounds and methods for treating bacterial infections. WO/2014/194270, 2014.
- Fiege B.; Rabbani S.; Preston R. C.; Jakob R. P.; Zihlmann P.; Schwardt O.; Jiang X.; Maier T.; Ernst B. The tyrosine gate of the bacterial lectin FimH: a conformational analysis by NMR spectroscopy and X-ray crystallography. ChemBioChem 2015, 16, 1235–1246. 10.1002/cbic.201402714. [DOI] [PubMed] [Google Scholar]
- de Ruyck J.; Lensink M. F.; Bouckaert J. Structures of C-mannosylated anti-adhesives bound to the type 1 fimbrial FimH adhesin. IUCrJ 2016, 3, 163–167. 10.1107/S2052252516002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari G.; Tiwari R.; Rai A. K. Cyclodextrins in delivery systems: Applications. J. Pharm. BioAllied Sci. 2010, 2, 72–79. 10.4103/0975-7406.67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Elisia I.; Nakamura H.; Lam V.; Hofs E.; Cederberg R.; Cait J.; Hughes M. R.; Lee L.; Jia W.; Adomat H. H.; Guns E. S.; McNagny K. M.; Samudio I.; Krystal G. DMSO Represses Inflammatory Cytokine Production from Human Blood Cells and Reduces Autoimmune Arthritis. PLoS One 2016, 11, e0152538. 10.1371/journal.pone.0152538. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grover S.; Srivastava A.; Lee R.; Tewari A. K.; Te A. E. Role of inflammation in bladder function and interstitial cystitis. Ther. Adv. Urol. 2011, 3, 19–33. 10.1177/1756287211398255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hung C. S.; Dodson K. W.; Hultgren S. J. A murine model of urinary tract infection. Nat. Protoc. 2009, 4, 1230–1243. 10.1038/nprot.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schwartz D. J.; Conover M. S.; Hannan T. J.; Hultgren S. J. Uropathogenic Escherichia coli superinfection enhances the severity of mouse bladder infection. PLoS Pathog. 2015, 11, e1004599. 10.1371/journal.ppat.1004599. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hannan T. J.; Mysorekar I. U.; Hung C. S.; Isaacson-Schmid M. L.; Hultgren S. J. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog. 2010, 6, e1001042. 10.1371/journal.ppat.1001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rosen D. A.; Hooton T. M.; Stamm W. E.; Humphrey P. A.; Hultgren S. J. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 2007, 4, e329. 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schlager T. A.; LeGallo R.; Innes D.; Hendley J. O.; Peters C. A. B cell infiltration and lymphonodular hyperplasia in bladder submucosa of patients with persistent bacteriuria and recurrent urinary tract infections. J. Urol. 2011, 186, 2359–2364. 10.1016/j.juro.2011.07.114. [DOI] [PubMed] [Google Scholar]; c Hansson S.; Hanson E.; Hjalmas K.; Hultengren M.; Jodal U.; Olling S.; Svanborg-Eden C. Follicular cystitis in girls with untreated asymptomatic or covert bacteriuria. J. Urol. 1990, 143, 330–332. [DOI] [PubMed] [Google Scholar]; d Hannan T. J.; Roberts P. L.; Riehl T. E.; van der Post S.; Binkley J. M.; Schwartz D. J.; Miyoshi H.; Mack M.; Schwendener R. A.; Hooton T. M.; Stappenbeck T. S.; Hansson G. C.; Stenson W. F.; Colonna M.; Stapleton A. E.; Hultgren S. J. Inhibition of Cyclooxygenase-2 Prevents Chronic and Recurrent Cystitis. EBioMedicine. 2014, 1, 46–57. 10.1016/j.ebiom.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sattin S.; Bernardi A. Glycoconjugates and Glycomimetics as Microbial Anti-Adhesives. Trends Biotechnol. 2016, 34, 483–495. 10.1016/j.tibtech.2016.01.004. [DOI] [PubMed] [Google Scholar]; b Cecioni S.; Imberty A.; Vidal S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 2015, 115, 525–561. 10.1021/cr500303t. [DOI] [PubMed] [Google Scholar]; c Sutkeviciute I.; Thepaut M.; Sattin S.; Berzi A.; McGeagh J.; Grudinin S.; Weiser J.; Le Roy A.; Reina J. J.; Rojo J.; Clerici M.; Bernardi A.; Ebel C.; Fieschi F. Unique DC-SIGN clustering activity of a small glycomimetic: A lesson for ligand design. ACS Chem. Biol. 2014, 9, 1377–1385. 10.1021/cb500054h. [DOI] [PubMed] [Google Scholar]; d Thepaut M.; Guzzi C.; Sutkeviciute I.; Sattin S.; Ribeiro-Viana R.; Varga N.; Chabrol E.; Rojo J.; Bernardi A.; Angulo J.; Nieto P. M.; Fieschi F. Structure of a glycomimetic ligand in the carbohydrate recognition domain of C-type lectin DC-SIGN. Structural requirements for selectivity and ligand design. J. Am. Chem. Soc. 2013, 135, 2518–2529. 10.1021/ja3053305. [DOI] [PubMed] [Google Scholar]; e Hauck D.; Joachim I.; Frommeyer B.; Varrot A.; Philipp B.; Möller H. M.; Imberty A.; Exner T. E.; Titz A. Discovery of two classes of potent glycomimetic inhibitors of Pseudomonas aeruginosa LecB with distinct binding modes. ACS Chem. Biol. 2013, 8, 1775–1784. 10.1021/cb400371r. [DOI] [PubMed] [Google Scholar]; f Chabre Y. M.; Giguere D.; Blanchard B.; Rodrigue J.; Rocheleau S.; Neault M.; Rauthu S.; Papadopoulos A.; Arnold A. A.; Imberty A.; Roy R. Combining glycomimetic and multivalent strategies toward designing potent bacterial lectin inhibitors. Chem. - Eur. J. 2011, 17, 6545–6562. 10.1002/chem.201003402. [DOI] [PubMed] [Google Scholar]; g Magnani J. L.; Ernst B. Glycomimetic drugs—a new source of therapeutic opportunities. Discovery Med. 2009, 8, 247–252. [PubMed] [Google Scholar]; h Ernst B.; Magnani J. L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discovery 2009, 8, 661–677. 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos S.; Jablonkai I. Preparation of 1-C-glycosyl aldehydes by reductive hydrolysis. Carbohydr. Res. 2011, 346, 1503–1510. 10.1016/j.carres.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Lin C. K.; Cheng L. W.; Li H. Y.; Yun W. Y.; Cheng W. C. Synthesis of novel polyhydroxylated pyrrolidine-triazole/-isoxazole hybrid molecules. Org. Biomol. Chem. 2015, 13, 2100–2107. 10.1039/C4OB01934B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.