

Abstract
cAMP-dependent protein kinase A (PKA) is a ubiquitously expressed serine/threonine kinase that regulates a variety of cellular functions. Here, we demonstrate that endothelial PKA activity is essential for vascular development, specifically regulating the transition from sprouting to stabilization of nascent vessels. Inhibition of endothelial PKA by endothelial cell-specific expression of dominant-negative PKA in mice led to perturbed vascular development, hemorrhage and embryonic lethality at mid-gestation. During perinatal retinal angiogenesis, inhibition of PKA resulted in hypersprouting as a result of increased numbers of tip cells. In zebrafish, cell autonomous PKA inhibition also increased and sustained endothelial cell motility, driving cells to become tip cells. Although these effects of PKA inhibition were highly reminiscent of Notch inhibition effects, our data demonstrate that PKA and Notch independently regulate tip and stalk cell formation and behavior.
KEY WORDS: Notch, Angiogenesis, Retina, Signal transduction, Sprouting, Vascular development
Summary: During zebrafish angiogenesis, endothelial PKA activity regulates the transition from sprouting to stabilisation of nascent vessels, preventing excessive tip cell formation and hypersprouting.
INTRODUCTION
The vascular system in vertebrates forms a vast network of blood vessels lined by endothelial cells and supporting mural cells. The architecture and branching pattern of the vasculature is highly adaptive to meet the needs of tissue growth during development or changing metabolic demands in physiology as well as during wound healing and regeneration. Maladaptation of the vasculature is a hallmark of many disease processes and is likely to result in poor tissue regeneration in compromised patients. Most of the blood vessels in the body are formed by angiogenesis, i.e. by outgrowth from pre-existing blood vessels followed by vascular remodeling. During remodeling, the nascent vascular network reorganizes by fusion, widening and pruning of blood vessels to form functioning vasculature (Eilken and Adams, 2010; Geudens and Gerhardt, 2011).
Although the main genetic and molecular players orchestrating vascular development have been identified (Benedito et al., 2012; Carmeliet and Jain, 2011; Eichmann and Thomas, 2013; Eilken and Adams, 2010; Geudens and Gerhardt, 2011; Hellström et al., 2007; Suchting et al., 2007; Wälchli et al., 2015), our understanding of the signaling program controlling this process is far from complete. One key pro-angiogenic stimulus is vascular endothelial growth factor (VEGF), which is produced by hypoxic tissues to stimulate ingrowth of blood vessels into hypoxic areas. Stimulation of endothelial cells with VEGF induces formation of tip cells, highly motile endothelial cells that explore the environment and migrate up the gradient of VEGF (Gerhardt et al., 2003; Ruhrberg et al., 2002). Tip cells are followed by stalk cells, which ensure connection to the pre-existing vasculature and formation of a new lumenized vessel (Potente et al., 2011). Since every endothelial cell has the potential to turn into a tip cell under VEGF stimulation (Jakobsson et al., 2010), a negative regulation is required to prevent all neighboring cells from becoming tip cells. This is achieved by Notch-mediated lateral inhibition. The Notch ligand DLL4, is expressed in tip cells in response to activation of VEGF receptor 2 (VEGFR2) and activates the Notch1 receptor on neighboring cells (Hayashi and Kume, 2008; Suchting et al., 2007). Activation of Notch decreases the levels of VEGFR2 and VEGFR3 in these cells, thus limiting their ability to respond to VEGF (Benedito et al., 2012; Hellström et al., 2007; Suchting et al., 2007). This mechanism drives a dynamic competition for the tip position.
To inhibit tip cell behavior, Notch signaling cooperates with other signaling pathways, with BMP signaling being the best-studied example. BMP9/BMP10-induced activation of ALK1 potentiates expression of Notch targets in a SMAD-dependent manner to promote stalk cell behavior (Larrivée et al., 2012). Importantly, our recent study has demonstrated that neuropilin-1 (NRP1), which is highly expressed in tip cells, represses ALK5- and ALK1-dependent SMAD activation. This repression is relieved in stalk cells, where activation of Notch decreases expression of Nrp1 (Aspalter et al., 2015). The results of this study challenged the concept of the tip cell being the default state of endothelial cells. Instead, it appears that tip cell formation and behavior is normally repressed by constitutive SMAD signaling, which needs to be released for tip cell activation. Moreover, in addition to the correct signaling repertoire, the tip cell requires appropriate metabolic regulation. Endothelial cell ATP production relies heavily on glycolysis and deficiency of the glycolytic activator PFKFB3 decreases sprouting and reduces the number of tip cells, even when Notch is inactive (De Bock et al., 2013).
Despite the identification of these critical regulators, the intracellular integration of multiple extracellular signaling inputs in endothelial cells in sprouting angiogenesis remains unclear. One of the major mechanisms to translate such extracellular inputs into a cellular response is protein and lipid phosphorylation. However, specific roles for regulation of phosphorylation during angiogenesis have been assigned to only a few kinases and phosphatases (Deng et al., 2013; Nakayama et al., 2013; Serra et al., 2015; Yoshioka et al., 2012).
Cyclic AMP (cAMP)-dependent protein kinase A (PKA) is a ubiquitously expressed protein kinase with a large number of potential targets (Shabb, 2001; Skroblin et al., 2010; Taylor et al., 2012). The main regulator of PKA activity is cAMP produced by adenylyl cyclases, which are both positively and negatively regulated by G protein-coupled receptors (GPCRs). Since GPCRs constitute the largest receptor family, with many of its members directly or indirectly regulating cAMP levels, PKA represents a signaling hub for a large variety of hormones, neurotransmitters and cytokines and thus regulates a plethora of physiological processes (Taskén and Aandahl, 2004). PKA has been shown to regulate different aspects of endothelial cell physiology (Aslam et al., 2010; Collins et al., 2014; Goldfinger et al., 2008; Lu et al., 2011) and to inhibit angiogenesis when activated pharmacologically (Bakre et al., 2002; Jin et al., 2010). However, a demonstration of whether endothelial PKA is required during vascular development in vivo has been missing.
Here, we describe a previously unidentified role for PKA in the regulation of angiogenesis and vascular development. Inhibition of endothelial PKA in mice results in embryonic vascular defects, hemorrhage and lethality at midgestation. Analysis of angiogenesis in mouse retina and zebrafish revealed that inhibition of endothelial PKA induces excessive formation of tip cells, increased cell migration and hypersprouting in a Notch-independent manner.
RESULTS
Endothelial PKA activity is required for embryonic vascular development
To study the role of PKA in vascular development we inhibited PKA in endothelial cells using knock-in mice carrying a single floxed dominant-negative Prkar1a allele (dnPKA; Fig. 1A) knocked into the genomic Prkar1a locus allowing tissue-specific inhibition of PKA (Willis et al., 2011). The regulatory PRKAR1A subunit of PKA is an endogenous inhibitor of PKA, which binds and keeps the catalytic subunits inactive under low cAMP levels (Kumon et al., 1970; Tao et al., 1970). With increasing cAMP levels, cAMP binds to PRKAR1A, induces its conformational change and releases the catalytic subunit (PRKAC; Fig. 1B), which is now active. A G324D point mutation introduced into PRKAR1A renders it insensitive to cAMP and thereby turns it into a constitutive inhibitor of PKA. In the absence of Cre recombinase expression, the floxed allele is silent, thus resembling a PRKARA1 knockout allele. Since PRKAR1A is essential for early embryonic development and Prkar1a−/− mice show delay in development as early as embryonic day (E)7.5 and are resorbed by E10.5 (Amieux et al., 2002), only hemizygote floxed dominant-negative Prkar1a (Prkar1aTg/+) mice are viable and were used in this study (see Fig. 1A,B for details on genetic approach and inhibition of PKA by dnPKA). Consequently, PKA in Prkar1aTg/+ cells is not completely inhibited, but only by ∼50% (Willis et al., 2011). In good agreement, we found that cAMP-induced phosphorylation of the PKA target CREB in dnPKA-expressing endothelial cells was reduced to 58±21% of levels in wild-type cells (data not shown).
Fig. 1.

Endothelial PKA is required for embryonic vascular development. (A) To inhibit PKA activity, a dominant-negative approach was used. In this approach, one of the alleles of the regulatory PKA subunit Prkar1a (a), was modified by introducing a G324D point mutation into exon 11 and inserting a floxed Neo cassette between exons 10 and 11. In the absence of Cre recombinase, the transgenic allele remains silent and the wild-type copy of Prkar1a is expressed (b). In the presence of Cre recombinase, the Neo cassette is excised and dominant-negative PKA (dnPKA) is expressed resulting in inhibition of PKA (c). (B) Schematic illustration of cAMP-dependent activation of PKA (a) and its inhibition by dnPKA transgene (b). PKA is a heterotetramer consisting of two regulatory (blue) and two catalytic (green) subunits. The regulatory subunits bind the catalytic ones and prevent their activity. Binding of cAMP to the regulatory subunits relieves their inhibitory action and leads to dissociation of the active catalytic subunit. dnPKA (red) has much lower binding affinity to cAMP and keeps the catalytic subunits bound and inactive, even at increased cAMP levels. (C) Fraction of viable embryos from the cross of Prkar1aTg/+ and Tie2-CreTg/− mice. Viability was assessed by the presence of a heartbeat during preparation of the embryos. (D) Endomucin staining of E10.5 wild-type and dnPKAEC littermates. (E,F) Appearance of the yolk sac (E) and embryo proper (F) of wild-type (left) and dnPKAEC littermates (right) at E11.5. (H) E11.5 yolk sac vasculature of wild-type (left) and dnPKAEC littermates (right) was visualized by expression of mT/mG transgene.
To achieve endothelial-specific inhibition of PKA, Prkar1aTg/+ mice were crossed with mice expressing Cre recombinase under the control of an endothelial cell-specific promoter (Tie2-Cre) (Kisanuki et al., 2001). Prkar1aTg/+;Tie2-Cre (hereafter referred to as dnPKAEC) embryos were present at a Mendelian ratio between E8.5 and E10.5; however, no dnPKAEC embryo could be recovered at E12.5 (Fig. 1C). To investigate whether inhibition of endothelial PKA results in perturbed vasculogenesis or early angiogenesis, E10.5 embryos were stained for the endothelial marker endomucin. Although dnPKAEC embryos were smaller than their wild-type littermates, endomucin staining depicted a vasculature without gross abnormalities (Fig. 1D), suggesting that vasculogenesis and early angiogenesis proceed adequately in dnPKAEC embryos.
By E11.5, only a minor fraction of dnPKAEC embryos were alive and displaying a heartbeat (Fig. 1C). All showed large hemorrhages in the abdominal area and no visible signs of blood in the yolk sac (Fig. 1E,F). Visualization of the yolk sac vessels in dnPKAEC embryos carrying the mT/mG Cre-recombination marker demonstrated that the mutants had a reduced vascular plexus density and vessels of smaller caliber, with many of them very thin and probably undergoing regression (Fig. 1H).
Inhibition of endothelial PKA results in vascular hypersprouting
To separate the primary effects of PKA inhibition on vascular development from secondary defects accompanying defective vascularization of the embryo, we studied PKA inhibition in postnatal retinal angiogenesis. An essential advantage of this model is that retinal angiogenesis takes place after a functional circulatory system has been formed. While perturbation of embryonic angiogenesis will inevitably result in malfunction of the developing cardiovascular system and may induce severe secondary defects due to hypoxia or malnutrition, the systemic postnatal circulation is much less affected by inhibition of angiogenesis. Prkar1aTg/+ mice were crossed with mice expressing tamoxifen-inducible Cre recombinase under control of endothelial-specific Cdh5 promoter (Cdh5-CreERT2; Wang et al., 2010). Recombination was induced by daily tamoxifen injection at postnatal day (P)1 to P3 and angiogenesis was analyzed in retinas from Prkar1aTg/+;Cdh5-CreERT (hereafter dnPKAiEC) and Cdh5-CreERT (wt) littermates.
Analysis of the retinal vasculature at P4 showed that inhibition of endothelial PKA resulted in formation of a hyperdense plexus front (Fig. 2A and Fig. S1A) with significantly increased endothelial cell proliferation, vessel area and number of sprouts (Fig. 2B). There was no difference in the size or weight of mutant and wild-type pups, indicating that the observed effects were not due to general developmental defects. Vascular progression assessed by radial expansion of the plexus was decreased in a fraction of mutant retinas (Fig. S1A); however, no significant difference over the whole population of analyzed pups was observed (Fig. 2B). The increased vascular density persisted at P7 (Fig. S1B,C), whereas there was no significant difference in the number of sprouts at this time point. Similar phenotypic changes were observed when expression of dnPKA was induced using a second endothelial-specific Cre line, Pdgfb-iCreERT (Claxton et al., 2008) (Fig. 2D). While the Cdh5-CreERT studies were performed on a mixed N/FVB×C57 (with at least 75% N/FVB) background, the Pdgfb-iCreERT2 studies were in the C57/Bl6 background, demonstrating that the effects of endothelial dnPKA on angiogenesis were independent of the Cre line or genetic background.
Fig. 2.

Inhibition of endothelial PKA leads to retinal hypersprouting and increased tip cell number. (A) Retinas from wild-type (Cdh5-CreERT2) and dnPKAiEC littermates were isolated at indicated time points and the vasculature was visualized using isolectin B4-Alexa Fluor 568. (B) Quantification of pup weight, retinal vascular area, incorporation of EdU, number of sprouts and vascular progression in wild-type and dnPKAiEC littermates at P4. Shown are means±s.d. of at least eight mice from at least three different litters. Data were analyzed using a Student's t-test; *P<0.05, **P<0.01. (C) Retinas from wild-type and dnPKAiEC littermates were isolated at P4 and stained with isolectin B4 for vasculature (green on overlay image) and anti-ESM-1 antibody for tip cells (red on the overlay image). (D) Retinas from PDGFB-iCre and PDGFB-iCre:dnPKA littermates were isolated at P4 and vasculature stained with isolectin B4.
After the initial formation of a relatively uniform immature plexus, the vascular network undergoes dramatic remodeling to form the hierarchically branched mature plexus (Fruttiger, 2007). Artery and vein formation occurred in dnPKAiEC mice, and a hierarchical branching pattern emerged, albeit with altered complexity, suggesting that PKA inhibition does not prevent vascular remodeling (Fig. 2A). However, we observed an increased number of arteriovenous crossovers in dnPKAiEC retinas (Fig. S1B,D). Interestingly, when comparing wild-type and dnPKAiEC retinas in 1-month-old mice, there were no obvious differences in the vascular architecture, except for the arteriovenous crossovers (Fig. S2). This indicates that inhibition of endothelial PKA specifically impairs retinal vascular development at the sprouting stage and that remodeling can ultimately correct many but not all patterning defects.
Inhibition of endothelial PKA increases tip cell numbers
The hyperdense frontal plexus and increased number of sprouts indicated an increased number of tip cells. To assess the number of tip cells, we stained retinas with the tip cell-specific marker ESM1 (del Toro et al., 2010; Xu et al., 2014). As expected, only few single cells at the vascular front were stained in wild-type retinas (Fig. 2C, top panels) confirming that ESM1 is a reliable tip cell marker. In contrast, in dnPKAiEC mice, ESM1 staining extended far into the vascular plexus (Fig. 2C, bottom panels). Combined, these data indicate that inhibition of PKA leads to hypersprouting as a result of an increased number of tip cells.
Next, we sought to understand whether the increased number of tip cells reflects an impaired regulation of angiogenesis by external factors or intrinsic properties of endothelial cells under PKA inhibition. Both pericytes and basement membrane have been shown to restrain vascular sprouting (Gaengel et al., 2009; Stenzel et al., 2011) and thus, defects in recruitment of pericytes or formation of basement membrane could be a reason for extensive sprouting in dnPKAiEC mice. To study whether defective interaction with mural cells or deposition of extracellular matrix may be the reason for the observed phenotype, retinas were stained for NG2, a marker of pericytes, and collagen IV, a component of the vascular basement membrane (Fig. S1A). However, no defects in coverage by pericytes or basement membrane could be observed in dnPKAiEC retinas, indicating that neither the recruitment of pericytes nor the ability of endothelial cells to deposit basement membrane proteins is affected by PKA inhibition.
To study whether inhibition of PKA increases the probability for an endothelial cell to gain the tip cell position cell-autonomously, we performed mosaic studies in the zebrafish model. Intersegmental vessels (ISVs) in zebrafish are formed by sprouting from the dorsal aorta. Endothelial tip/stalk cell selection, migration and proliferation establish bilateral ISVs, which upon reaching the dorsal aspect of the neural tube, form a pair of dorsal longitudinal anastomotic vessels (DLAVs). To inhibit PKA in a cell-specific manner, we injected Tg(fli1a:eGFP)y1 (Lawson and Weinstein, 2002) zebrafish eggs at the one or two cell stage with a vector expressing a specific PKA inhibitor peptide (PKI) (Murray, 2008), fused to mCherry fluorescent protein and placed under the control of the endothelial cell-specific fli1 enhancer/promoter (fli1ep; Villefranc et al., 2007). Determining the location of mCherry-positive cells in mosaic ISVs (consisting of both PKI-mCherry-positive and -negative cells), we found that PKI-expressing cells were significantly over-represented at the leading (tip cell) position compared with the stalk position (Fig. 3A). In control experiments, using a construct expressing red fluorescent protein (RFP) under the same promoter, RFP-positive control cells were equally frequent at tip and stalk position (ratio close to 1). These results suggest that PKI inhibition conveys a capacity to endothelial cells to attain the tip position. In addition, we observed that PKI-expressing cells have more filopodia than cells expressing control fluorescent protein (Fig. 3B).
Fig. 3.
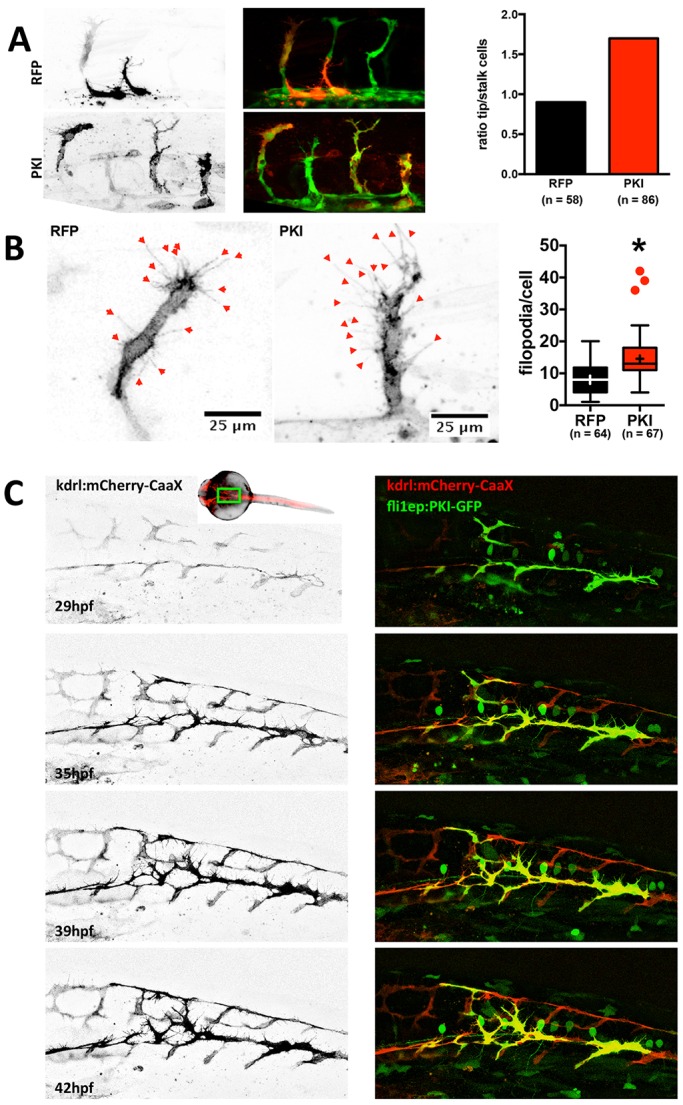
Inhibition of endothelial PKA increases the motility of endothelial cells and their probability to become a tip cell. (A) Tg(fli1a:eGFP)y1 zebrafish embryos were injected with a plasmid encoding RFP (as control) or PKI-mCherry fusion protein (left). Localization of the cells expressing red fluorescent protein was determined at 29 hpf. Mosaic ISVs (containing both transfected and untransfected cells) were used to identify whether transfected cells were in a tip or stalk cell position. RFP and PKI-mCherry signals are in black and white on the left panel and in red on the overlay image (middle); GFP signal is green on the overlay image. Ratio of tip-to-stalk cells for RFP-expressing (control) and PKI-mCherry-expressing (PKI) cells (right). For RFP and PKI, 15 and 26 embryos were analyzed, respectively. The number of analyzed cells is indicated on the figure (Chi-square test: P<0.05). (B) Filopodia in endothelial cells expressing control fluorescent protein or PKI-mCherry fusion were analyzed in fish embryos. Shown are representative images of cells expressing control fluorescent protein (RFP; left) or PKI-mCherry (PKI; middle) and quantification of filopodia number per cell (right). For RFP and PKI, 20 and 29 embryos were analyzed, respectively. The number of analyzed cells is indicated on the figure. Boxes encompass values between the 25th and 75th percentiles, centered lines represent median, mean is indicated by +, whiskers and outliers are calculated using the Tukey method (*P<0.05). (C) Tg(Kdrl:Ras-mCherry)s916 embryos were injected with plasmid expressing PKI-GFP fusion under the control of endothelial fli1ep promoter and vascular development was monitored from 29 to 42 hpf. Membrane-bound mCherry fluorescent protein (black in left panel and red on the right) depicts higher mobility and excessive filopodia formation in PKI-GFP-expressing cells (green) compared with the wild-type GFP-negative cells. The approximate area imaged is depicted on the insert showing the zebrafish embryo.
In order to gain insights into the mechanism, we assessed the dynamic behavior of PKI-expressing cells. For better visualization of the endothelial cell membrane, we used the Tg(Kdr-l:ras-Cherry)s916 line, which expresses a membrane-bound fluorescent protein (Hogan et al., 2009). To inhibit PKA, we used a PKI-GFP fusion protein. We followed the development of blood vessels in mosaic PKI-GFP-expressing fish embryos from 29 hours post fertilization (hpf) until 42 hpf (Fig. 3C and Movies 1 and 2). Compared with control cells, PKI-GFP-expressing cells formed more abundant and highly explorative filopodia (Fig. 3C and Movie 1). While control cells ceased filopodia formation after anastomosing and forming DLAVs, PKI-GFP-positive cells retained filopodial activity and kept migrating, even after anastomosis (Movie 1). This increased motility of endothelial cells correlated with an apparent instability of blood vessels, since the lumen of PKI-expressing ISVs often collapsed after perfusion had started (Movie 2). In contrast, ISVs in controls rarely display this behavior as perfusion stabilizes nascent lumen and vessel connections through shear-mediated mechanisms (Movie 2).
PKA inhibition or activation do not affect Notch signaling
Given that inhibition of endothelial PKA in dnPKAiEC mice leads to a hypersprouting phenotype that is similar to the one observed upon loss of DLL4 or inhibition of Notch signaling (Hellström et al., 2007; Suchting et al., 2007), we investigated whether inhibition of endothelial PKA affects DLL4/Notch signaling. First, we analyzed DLL4 distribution in P4 retinas. Similar to ESM1 staining (Fig. 2C), DLL4 distribution extended further into the plexus in dnPKAiEC mice (Fig. 4A) and was not visibly reduced, suggesting that the observed hypersprouting is not due to loss of DLL4 expression. In wild-type and dnPKAiEC retinas, DLL4 staining was virtually absent in the veins while present in the arteries and at the sprouting front, indicating that PKA inhibition neither reduces DLL4 nor induces generalized ectopic expression of DLL4. Instead, the wider zone of DLL4 expression at the sprouting front, similar to ESM1, is likely to be a consequence of the increased number of tip cells.
Fig. 4.

PKA and Notch independently regulate endothelial cell behavior during angiogenesis. (A) Retinas from wild-type (Cdh5-CreERT2) and dnPKAiEC littermates were isolated at P4 and stained for DLL4. For both wt and dnPKAiEC, 12 pups from three different litters were analyzed and representative images are shown. (B) Retinal endothelial cells were isolated at P6 and expression levels of the indicated genes were analyzed by qPCR. Shown are fold differences of expression in dnPKAiEC over wt cells. Data are means±s.d. of four independent experiments. (C) HUVECs were transfected with the luciferase reporter for PKA (CRE-Luc) or Notch (TP1-Luc) and seeded onto dishes coated with DLL4 (DLL4+) or BSA (DLL4−). To activate PKA, 50 µM CPT-cAMP (a cell-permeable cAMP analog) was added where indicated. Data were normalized to the activity in the presence of DLL4 (for TP1-Luc) and cAMP (for CRE-Luc). Shown are means±s.d. for three independent experiments, each performed in triplicate. (D) Wild-type (Cdh5-CreERT2) and dnPKAiEC littermates were injected with tamoxifen at P1-P3 and with 100 mg/kg DAPT or vehicle only at P2 and P3, retinas were dissected at P4 and the vasculature was visualized using isolectin B4-Alexa Fluor 568. Four litters were analyzed and representative images are shown; number of pups analyzed for each condition is indicated on the figure.
To further investigate whether Notch signaling is affected by PKA inhibition, we analyzed the expression of genes regulated by Notch in endothelial cells isolated from retinas of wild-type and dnPKAiEC mice. QPCR analysis demonstrated that out of all Notch targets studied (Dll4, Hes1, Hes2, Hey1, Vegfr1, Vegfr2, Esm1 and Nrp1), only Esm1 showed an increase in expression (about 2-fold) in dnPKAiEC mice compared with the wild type (Fig. 4B). Since most of these markers were not affected, we concluded that inhibition of PKA had only a limited, if any, effect on Notch signaling. We speculate that the increase of Esm1 expression reflects the increased number of tip cells rather than a direct induction due to inhibition of Notch. Indeed, it has been demonstrated that Esm1 expression is increased 11-fold under inhibition of Notch (del Toro et al., 2010), whereas we find only a 2-fold increase under inhibition of PKA. We also tested expression of another gene, Cxcl2, which has been shown to be upregulated more than 13-fold under inhibition of Notch (del Toro et al., 2010). Again, we detected only a modest 1.4-fold increase of mRNA in endothelial cells from dnPKAiEC retinas (data not shown). Altogether, these data indicate that PKA does not operate upstream of Notch signaling in regulating endothelial tip/stalk specification.
An alternative explanation for hypersprouting in dnPKAiEC mice is that Notch acts upstream of PKA, with PKA mediating the anti-angiogenic effect of Notch. To test this hypothesis, we transfected HUVECs with a luciferase-based reporter for PKA (CRE-Luc, firefly luciferase under control of the cAMP response element) or Notch [TP1-Luc, firefly luciferase under the control of the Epstein Barr Virus terminal protein 1 (TP1) gene, which contains two Rbp-Jκ binding sites (Grossman et al., 1994; Henkel et al., 1994)]. To activate Notch, cells were seeded on plates coated with DLL4; for PKA activation, cells were treated with a membrane-permeable cAMP analog, CPT-cAMP. DLL4 strongly increased the signal of the TP1-Luc reporter, but cAMP had no significant effect on the Notch reporter, either in the presence or in the absence of DLL4 (Fig. 4C). Furthermore, DLL4 did not affect the activity of the PKA reporter and did not enhance its activation induced by the cAMP analog (Fig. 4C), suggesting that the cAMP/PKA-dependent pathway and Notch signaling are not intertwined in endothelial cells.
Inhibition of PKA and Notch has an additive effect on retinal angiogenesis
To directly test in vivo whether PKA inhibition exerts its effects on retinal angiogenesis in parallel to and independent of Notch signaling, we inhibited both PKA and Notch and analyzed retinal angiogenesis. Notch signaling was inhibited by DAPT {N-[(3,5-Difluorophenyl)acetyl]-L-alanyl-2-phenylglycine-1,1-dimethylethyl ester} an inhibitor of γ-secretase, which is required for Notch processing and activation. Similar to PKA inhibition and as published before (Benedito et al., 2012; Hellström et al., 2007; Suchting et al., 2007), DAPT treatment led to increased sprouting at the vascular front and increased vessel density (Fig. 4D). Interestingly, inhibition of Notch in dnPKAiEC mice resulted in even stronger hypersprouting and higher number of tip cells than in DAPT-treated wild-type mice or untreated dnPKAiEC mice (Fig. 4D). Combined, these data demonstrate that although inhibition of PKA and Notch has very similar phenotypic effects on retinal angiogenesis, mechanistically, the two pathways operate largely independently.
DISCUSSION
During the past decade, several signaling pathways regulating angiogenesis have been discovered. As both deficient and excessive angiogenesis may be deleterious, a tight balance between positive and negative regulators is required to ensure that the formation of functional vessels meeting the needs of the organism. Here, we show that the signaling hub PKA has crucial anti-angiogenic activity during vascular development. Inhibition of endothelial PKA in mice caused severe vascular abnormalities, vascular leakage, hemorrhage and ultimately, embryonic lethality during development. Postnatal inhibition of PKA in the retinal angiogenesis model revealed profound hypersprouting and increased tip cell numbers.
It has previously been demonstrated that pharmacological activation of PKA in chick chorioallantoic membrane or zebrafish embryo reduced angiogenesis (Bakre et al., 2002; Jin et al., 2010). However, whether PKA is activated endogenously as a part of a signaling network regulating angiogenesis has remained unanswered. Earlier studies that applied the PKA inhibitor H89 to zebrafish embryos noted the formation of ectopic sprouts (Jin et al., 2010). However, considering the global inhibition of PKA by the inhibitor and its potential to inhibit multiple protein kinases (Bain et al., 2007), it has been unclear to what extent endothelial PKA was responsible for the observed effects. Using genetic inhibition of PKA in endothelial cells in mouse, we demonstrate that endothelial PKA is cell-autonomously essential for normal vascular development.
In vivo studies in mouse and zebrafish allowed us to unravel a specific role for PKA during vascular development. While diminished PKA activity does not prevent vasculogenesis and early angiogenesis, it leads to excessive formation of tip cells and hypersprouting.
Since the effects of PKA inhibition are reminiscent of the effects of Notch inhibition, one possible explanation would be that both pathways converge in endothelial cells. PKA has been shown to activate Notch signaling in monocytes (Larabee et al., 2013) and to increase the level of the Notch ligand Jagged1 in osteoblasts (Weber et al., 2006). Moreover, both PKA and Notch synergistically promote self-renewal of glioma-initiating cells (Wang et al., 2014). However, the results of our study do not support an interaction between PKA and Notch in endothelial cells. First, activation of PKA in HUVECs did not activate Notch; neither did activation of Notch affect PKA activity. Second, in endothelial cells isolated from dnPKAiEC retinas, Notch signaling seems to operate normally, since the expression of Notch targets (Hes1, Hey1, Hey2, Vegfr1, Vegfr2, Nrp1 and Cxcl2) was similar to that in endothelial cells from wild-type retinas. The only exception was Esm1, which was significantly increased in the total population of isolated retinal dnPKAiEC-expressing endothelial cells. Esm1 has previously been shown to be increased in retinal endothelial cells under Notch inhibition (del Toro et al., 2010). However, while Esm1 was increased ∼2-fold in cells from dnPKAiEC retinas, it increased by more than 11-fold under inhibition of Notch (del Toro et al., 2010). Also, by immunostaining, we found that DLL4 ligand expression levels and pattern were similar to wild-type retinas, although with a wider distribution in the hypersprouting plexus at the front, corresponding to the region of increased tip cell formation. This would suggest that Notch itself is not affected by PKA inhibition. Instead, we hypothesize that the quantitative increase in Esm1 mRNA is related to the increased number of tip cells in dnPKAiEC retinas. Indeed, immunostaining identified that the number of ESM1-expressing cells is much higher in dnPKAiEC retinas compared with wild-type retinas. Taken together, these data led us to the conclusion that the role of endothelial PKA during sprouting angiogenesis is to restrain formation of tip cells. Future studies will need to clarify whether PKA is (transiently) activated in tip cells and facilitates their transition to stalk cells, or whether it is selectively active in stalk cells and prevents them from becoming tip cells. Furthermore, the molecular mechanism behind PKA regulation of tip/stalk activity will need to be elucidated. One possibility would be that the increase in ESM1 observed under PKA inhibition is directly causing increased hypersprouting. ESM1 plays an active role during retinal angiogenesis since its deletion leads to a delay in vascular outgrowth and a decreased number of endothelial cell filopodia during retinal angiogenesis (Rocha et al., 2014). Therefore one might speculate that the elevated ESM1 in dnPKAiEC mechanistically drives hypersprouting.
Another important question will be what is the upstream activator of endothelial PKA during vascular development. Usually, PKA is activated in response to stimulation of GPCRs and consecutive elevation of cAMP. Several GPCRs and their ligands have been shown to regulate cAMP level or PKA activity in endothelial cells (Aslam et al., 2012; Bakre et al., 2002; Dormond et al., 2002), some of which have been proposed to activate PKA during vascular development (Jin et al., 2010). However, it remains unclear as to which GPCRs indeed regulate endothelial PKA during vascular development. Interestingly, in endothelial cells, PKA can be also activated by shear stress (Dixit et al., 2005; Funk et al., 2010), tension (Collins et al., 2014) or VEGF (Lu et al., 2011). PKA mediates alignment of endothelial cells in response to flow (Goldfinger et al., 2008), adjustment of endothelial cell stiffness in response to pulsatile force (Collins et al., 2014) and VEGF-induced activation of nitric oxide synthase (Lu et al., 2011). Importantly, in vivo data suggest that activation of PKA in response to flow may be mediated through a well-known GPCR axis. Siekmann and colleagues have demonstrated that vessel perfusion in zebrafish and the subsequent decrease in filopodia number is accompanied by decreased expression of CXCR4 in endothelial cells (Bussmann et al., 2011). Since CXCR4 is a GPCR that activates Gαi protein (Klein et al., 2001) and thus inhibits adenylyl cyclase-mediated cAMP production, downregulation of CXCR4 may result in activation of PKA endothelial cells in response to flow. One may speculate that such activation of PKA might function to switch off exploratory behavior of endothelial cells after perfusion has been established. While the upstream regulator of endothelial PKA remains to be identified, our data demonstrating sustained exploratory behavior of endothelial cells under PKA inhibition are a strong indication that PKA is indeed required for a flow-mediated switch in endothelial cell behavior.
In summary, our data demonstrate that PKA is an essential regulator of angiogenesis and vascular development, which controls tip/stalk cell behavior independent of Notch signaling. It remains to be revealed whether PKA and Notch operate simultaneously to reinforce the anti-angiogenic effect of each other or whether they regulate different cellular events during tip cell specification and tip-to-stalk cell transition.
MATERIALS AND METHODS
Animal procedures
All experimental animal procedures were approved by the Institutional Animal Care and Research Advisory Committee of the University of Leuven and performed according to European guidelines. The following mouse strains were used: Prkar1atm2Gsm (Willis et al., 2011), Tg(Cdh5-cre/ERT2)1Rha (Wang et al., 2010), Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (Muzumdar et al., 2007), Tg(Pdgfb-icre/ERT2)1Frut (Claxton et al., 2008) and Tg(Tek-cre)1Ywa/J (Kisanuki et al., 2001). All animals used in the experiments were mixed N/FVB×C57/Bl6 mice with at least 75% N/FVB. All comparisons were done between littermates only. For staging the embryos, the morning when the vaginal plug was detected was considered to be E0.5. For retinal angiogenesis assay, 50 µg tamoxifen per pup was applied by intraperitoneal injection daily on P1-P3 (Pitulescu et al., 2010). Eyes were harvested at the indicated time point, fixed in 4% paraformaldehyde at room temperature for 30 min and retinas were dissected.
Retinal angiogenesis assay
Isolation and staining of the retinas was performed according to a published protocol (Pitulescu et al., 2010). The vasculature was visualized with isolectin B4 coupled to Alexa Fluor 568. The following primary antibodies were used for staining: ESM1 (R&D Systems, AF1999; 1:100), NG2 (Millipore, AB5320; 1:100), collagen IV (AbD Serotec, 2150-1470; 1:200). Alexa Fluor 647-coupled secondary antibodies (dilution 1:500) were used (Thermo Fisher Scientific). After mounting, images were recorded using a Leica SP8 system.
Live imaging in zebrafish
Tg(fli1a:eGFP)y1 (Lawson and Weinstein, 2002) and Tg(Kdr-l:ras-Cherry)s916 (Hogan et al., 2009) zebrafish lines were maintained according to EU regulations on laboratory animals. Embryos were injected at the one- to two-cell stage with 50 pg plasmid and 50 pg Tol2 transposase RNA. Capped mRNAs were transcribed with the SP6 mMESSAGE mMACHINE Kit (Ambion). For real-time imaging, embryos were mounted in 0.8% low-melting agarose containing 0.01% Tricaine (Sigma) and bathed in Danieau's buffer and imaged every 25 min on a confocal Leica SP8 microscope at 28°C.
Retinal endothelial cell isolation
Mouse retinal endothelial cells were isolated according to a published protocol (del Toro et al., 2010). Briefly, pups were genotyped and retinas of the same genotype were pooled together (4-10 retinas depending on the size of the litter and the mutant to wild type proportion) for endothelial cell isolation. Retinas were incubated in 5 ml Dulbecco's modified Eagle's medium (4.5 g/l glucose with L-glutamine) containing 50 mg/ml collagenase (Invitrogen, 17018-029) in 15 ml tube for 10-15 min at 37°C. Another 5 ml DMEM containing 10% FBS, 50 U/ml penicillin and 50 µg/ml streptomycin was added to the incubation tube and the cells were passed through 40 µm Nylon cell strainer (BD Falcon), centrifuged at 180 g for 5 min, resuspended in cold washing buffer (PBS supplemented with 0.1% BSA and 0.5 mM EDTA) and rotated with Dynabeads sheep anti-rat IgG (Invitrogen, 11035) pre-incubated with purified rat anti-mouse CD31 (BD Pharmingen, 553370) for 20 min at 4°C. The beads were separated and washed with cold washing buffer using a magnetic particle concentrator (Dynal MPC-S; Invitrogen) and stored at −80°C. The cells not bound to the beads were saved as the non-endothelial cell fraction and used as reference to validate enrichment of vascular markers in the endothelial cell preparation.
Quantitative PCR
Total RNA was purified from retinal endothelial cells using Qiagen RNeasy Mini kit following the manufacturer's protocol. Samples were quality controlled using a Nanodrop 2000 (Thermo Fisher Scientific). Total RNA (300 ng) was reverse transcribed into cDNA using a High Capacity RNA-to-cDNA Kit (Applied Biosystems). Quantitative PCR (QPCR) was performed using Fast SYBR Green PCR Master Mix (Applied Biosystems) in a 7500 fast Real-Time PCR system (Applied Biosystems) with specific primers as listed in supplementary Materials and Methods. For each condition, four independent experiments were analyzed and fold changes were calculated using the comparative −ΔΔCT method using Gapdh as a housekeeping gene. Then, fold changes were normalized to Pecam1 level to account for possible differences in the number of endothelial cells.
Luciferase assay
Human umbilical vein endothelial cells (HUVECs) were purchased from Promocell (Heidelberg, Germany). To detect PKA and Notch activity in endothelial cells, 70% confluent HUVECs growing on a 10 cm dish were transfected with a plasmid encoding firefly luciferase reporter either using CRE-Luc (Promega) or TP1-Luc. Transfection was performed using X-tremeGENE HP DNA transfection reagent (Roche). Cells were split 6 h after transfection and plated on 24-well dishes coated with 1 µg/ml BSA in 0.1% gelatin (control) or 1 µg human recombinant Dll4 (R&D Systems) in 0.1% gelatin (DLL4 treatment). Cells were treated with the cell-permeable cAMP analog CPT-cAMP (BIOLOG Life Science Institute)18 h after plating. After 6 h, cells were lysed and luciferase activity was measured.
Statistical analysis
Statistical analyses were performed using Prism 6 (GraphPad). The Student's t-test was used to compare two different groups (Fig. 2B and Fig. 3B, Fig. S1C) and one-way ANOVA to compare more than two experimental groups (Fig. 4B). One-tailed Fisher's test was used to compare tip/stalk cells ratio for Fig. 3A.
Acknowledgements
We would like to thank our colleagues Li-Kun Phng and Ilse Geudens for their help with zebrafish experiments; E. Yu, Marly Balcer and Bing Yan for excellent technical assistance; Ann Zovein, Stephen Wilson and Shaun Coughlin for discussions and comments; Petya Georgieva for critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Designed experiments: P.I.N., R.J.M., K.E.M. and H.G. Performed experiments: P.I.N., X.Z., T.M., I.M.A. and F.S. Analyzed the data: P.I.N., F.S., R.J.M. and H.G. Wrote the paper: P.I.N. and H.G.
Funding
This study was supported by grants from the Fonds voor Wetenschappelijk Onderzoek (FWO) [G.0742.15N to H.G]; the Else-Kröner-Fresenius-Stiftung [2014_A26 to H.G and P.I.N]; the European Research Council [ERC consolidator grant RESHAPE 311719 to H.G.]; Herculesstichting [AKUL/11/33 to H.G]; the Stichting tegen Kanker [2012-181 to H.G.]; and the National Institutes of Health [5R01 DK074398 and 5R01 DK091530 to K.E.M]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.134767.supplemental
Reference
- Amieux P. S., Howe D. G., Knickerbocker H., Lee D. C., Su T., Laszlo G. S., Idzerda R. L. and McKnight G. S. (2002). Increased basal cAMP-dependent protein kinase activity inhibits the formation of mesoderm-derived structures in the developing mouse embryo. J. Biol. Chem. 277, 27294-27304. 10.1074/jbc.M200302200 [DOI] [PubMed] [Google Scholar]
- Aslam M., Härtel F. V., Arshad M., Gündüz D., Abdallah Y., Sauer H., Piper H. M. and Noll T. (2010). cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: role of CPI-17. Cardiovasc. Res. 87, 375-384. 10.1093/cvr/cvq065 [DOI] [PubMed] [Google Scholar]
- Aslam M., Pfeil U., Gündüz D., Rafiq A., Kummer W., Piper H. M. and Noll T. (2012). Intermedin (adrenomedullin2) stabilizes the endothelial barrier and antagonizes thrombin-induced barrier failure in endothelial cell monolayers. Br. J. Pharmacol. 165, 208-222. 10.1111/j.1476-5381.2011.01540.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspalter I. M., Gordon E., Dubrac A., Ragab A., Narloch J., Vizán P., Geudens I., Collins R. T., Franco C. A., Abrahams C. L. et al. (2015). Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat. Commun. 6, 7264 10.1038/ncomms8264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S. C., Alessi D. R. and Cohen P. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297-315. 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakre M. M., Zhu Y., Yin H., Burton D. W., Terkeltaub R., Deftos L. J. and Varner J. A. (2002). Parathyroid hormone-related peptide is a naturally occurring, protein kinase A-dependent angiogenesis inhibitor. Nat. Med. 8, 995-1003. 10.1038/nm753 [DOI] [PubMed] [Google Scholar]
- Benedito R., Rocha S. F., Woeste M., Zamykal M., Radtke F., Casanovas O., Duarte A., Pytowski B. and Adams R. H. (2012). Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 484, 110-114. 10.1038/nature10908 [DOI] [PubMed] [Google Scholar]
- Bussmann J., Wolfe S. A. and Siekmann A. F. (2011). Arterial-venous network formation during brain vascularization involves hemodynamic regulation of chemokine signaling. Development 138, 1717-1726. 10.1242/dev.059881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. and Jain R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298-307. 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K. and Fruttiger M. (2008). Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis 46, 74-80. 10.1002/dvg.20367 [DOI] [PubMed] [Google Scholar]
- Collins C., Osborne L. D., Guilluy C., Chen Z., O'Brien E. T. III, Reader J. S., Burridge K., Superfine R. and Tzima E. (2014). Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat. Commun. 5, 3984 10.1038/ncomms4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B. W., Cantelmo A. R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G. et al. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651-663. 10.1016/j.cell.2013.06.037 [DOI] [PubMed] [Google Scholar]
- del Toro R., Prahst C., Mathivet T., Siegfried G., Kaminker J. S., Larrivée B., Bréant C., Duarte A., Takakura N., Fukamizu A. et al. (2010). Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116, 4025-4033. 10.1182/blood-2010-02-270819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., Larrivée B., Zhuang Z. W., Atri D., Moraes F., Prahst C., Eichmann A. and Simons M. (2013). Endothelial RAF1/ERK activation regulates arterial morphogenesis. Blood 121, 3988-3996. 10.1182/blood-2012-12-474601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit M., Loot A. E., Mohamed A., Fisslthaler B., Boulanger C. M., Ceacareanu B., Hassid A., Busse R. and Fleming I. (2005). Gab1, SHP2, and protein kinase A are crucial for the activation of the endothelial NO synthase by fluid shear stress. Circ. Res. 97, 1236-1244. 10.1161/01.RES.0000195611.59811.ab [DOI] [PubMed] [Google Scholar]
- Dormond O., Bezzi M., Mariotti A. and Rüegg C. (2002). Prostaglandin E2 promotes integrin alpha Vbeta 3-dependent endothelial cell adhesion, rac-activation, and spreading through cAMP/PKA-dependent signaling. J. Biol. Chem. 277, 45838-45846. 10.1074/jbc.M209213200 [DOI] [PubMed] [Google Scholar]
- Eichmann A. and Thomas J.-L. (2013). Molecular parallels between neural and vascular development. Cold Spring Harb. Perspect. Med. 3, a006551 10.1101/cshperspect.a006551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilken H. M. and Adams R. H. (2010). Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 22, 617-625. 10.1016/j.ceb.2010.08.010 [DOI] [PubMed] [Google Scholar]
- Fruttiger M. (2007). Development of the retinal vasculature. Angiogenesis 10, 77-88. 10.1007/s10456-007-9065-1 [DOI] [PubMed] [Google Scholar]
- Funk S. D., Yurdagul A., Green J. M., Jhaveri K. A., Schwartz M. A. and Orr A. W. (2010). Matrix-specific protein kinase A signaling regulates p21-activated kinase activation by flow in endothelial cells. Circ. Res. 106, 1394-1403. 10.1161/CIRCRESAHA.109.210286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K., Genove G., Armulik A. and Betsholtz C. (2009). Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 29, 630-638. 10.1161/ATVBAHA.107.161521 [DOI] [PubMed] [Google Scholar]
- Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D. et al. (2003). VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161, 1163-1177. 10.1083/jcb.200302047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geudens I. and Gerhardt H. (2011). Coordinating cell behaviour during blood vessel formation. Development 138, 4569-4583. 10.1242/dev.062323 [DOI] [PubMed] [Google Scholar]
- Goldfinger L. E., Tzima E., Stockton R., Kiosses W. B., Kinbara K., Tkachenko E., Gutierrez E., Groisman A., Nguyen P., Chien S. et al. (2008). Localized alpha4 integrin phosphorylation directs shear stress-induced endothelial cell alignment. Circ. Res. 103, 177-185. 10.1161/CIRCRESAHA.108.176354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman S. R., Johannsen E., Tong X., Yalamanchili R. and Kieff E. (1994). The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 91, 7568-7572. 10.1073/pnas.91.16.7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H. and Kume T. (2008). Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS ONE 3, e2401 10.1371/journal.pone.0002401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström M., Phng L.-K., Hofmann J. J., Wallgard E., Coultas L., Lindblom P., Alva J., Nilsson A.-K., Karlsson L., Gaiano N. et al. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776-780. 10.1038/nature05571 [DOI] [PubMed] [Google Scholar]
- Henkel T., Ling P. D., Hayward S. D. and Peterson M. G. (1994). Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265, 92-95. 10.1126/science.8016657 [DOI] [PubMed] [Google Scholar]
- Hogan B. M., Bos F. L., Bussmann J., Witte M., Chi N. C., Duckers H. J. and Schulte-Merker S. (2009). Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat. Genet. 41, 396-398. 10.1038/ng.321 [DOI] [PubMed] [Google Scholar]
- Jakobsson L., Franco C. A., Bentley K., Collins R. T., Ponsioen B., Aspalter I. M., Rosewell I., Busse M., Thurston G., Medvinsky A. et al. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943-953. 10.1038/ncb2103 [DOI] [PubMed] [Google Scholar]
- Jin H., Garmy-Susini B., Avraamides C. J., Stoletov K., Klemke R. L. and Varner J. A. (2010). A PKA-Csk-pp60Src signaling pathway regulates the switch between endothelial cell invasion and cell-cell adhesion during vascular sprouting. Blood 116, 5773-5783. 10.1182/blood-2010-07-296210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki Y. Y., Hammer R. E., Miyazaki J.-I., Williams S. C., Richardson J. A. and Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230-242. 10.1006/dbio.2000.0106 [DOI] [PubMed] [Google Scholar]
- Klein R. S., Rubin J. B., Gibson H. D., DeHaan E. N., Alvarez-Hernandez X., Segal R. A. and Luster A. D. (2001). SDF-1α induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development 128, 1971-1981. [DOI] [PubMed] [Google Scholar]
- Kumon A., Yamamura H. and Nishizuka Y. (1970). Mode of action of adenosine 3′,5′-Cyclic phosphate on protein kinase from rat liver. Biochem. Biophys. Res. Commun. 41, 1290-1297. 10.1016/0006-291X(70)90228-7 [DOI] [PubMed] [Google Scholar]
- Larabee J. L., Shakir S. M., Barua S. and Ballard J. D. (2013). Increased cAMP in monocytes augments Notch signaling mechanisms by elevating RBP-J and transducin-like enhancer of split (TLE). J. Biol. Chem. 288, 21526-21536. 10.1074/jbc.M113.465120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivée B., Prahst C., Gordon E., del Toro R., Mathivet T., Duarte A., Simons M. and Eichmann A. (2012). ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 22, 489-500. 10.1016/j.devcel.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N. D. and Weinstein B. M. (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318. 10.1006/dbio.2002.0711 [DOI] [PubMed] [Google Scholar]
- Lu Y., Xiong Y., Huo Y., Han J., Yang X., Zhang R., Zhu D.-S., Klein-Hessling S., Li J., Zhang X. et al. (2011). Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc. Natl. Acad. Sci. USA 108, 2957-2962. 10.1073/pnas.1009395108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A. J. (2008). Pharmacological PKA inhibition: all may not be what it seems. Sci. Signal. 1, re4-re4 10.1126/scisignal.122re4 [DOI] [PubMed] [Google Scholar]
- Muzumdar M. D., Tasic B., Miyamichi K., Li L. and Luo L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593-605. 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Nakayama M., Nakayama A., van Lessen M., Yamamoto H., Hoffmann S., Drexler H. C. A., Itoh N., Hirose T., Breier G., Vestweber D. et al. (2013). Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat. Cell Biol. 15, 249-260. 10.1038/ncb2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu M. E., Schmidt I., Benedito R. and Adams R. H. (2010). Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 5, 1518-1534. 10.1038/nprot.2010.113 [DOI] [PubMed] [Google Scholar]
- Potente M., Gerhardt H. and Carmeliet P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873-887. 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- Rocha S. F., Schiller M., Jing D., Li H., Butz S., Vestweber D., Biljes D., Drexler H. C. A., Nieminen-Kelhä M., Vajkoczy P. et al. (2014). Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ. Res. 115, 581-590. 10.1161/CIRCRESAHA.115.304718 [DOI] [PubMed] [Google Scholar]
- Ruhrberg C., Gerhardt H., Golding M., Watson R., Ioannidou S., Fujisawa H., Betsholtz C. and Shima D. T. (2002). Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16, 2684-2698. 10.1101/gad.242002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra H., Chivite I., Angulo-Urarte A., Soler A., Sutherland J. D., Arruabarrena-Aristorena A., Ragab A., Lim R., Malumbres M., Fruttiger M. et al. (2015). PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 6, 7935 10.1038/ncomms8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabb J. B. (2001). Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381-2412. 10.1021/cr000236l [DOI] [PubMed] [Google Scholar]
- Skroblin P., Grossmann S., Schäfer G., Rosenthal W. and Klussmann E. (2010). Mechanisms of protein kinase a anchoring. Int. Rev. Cell Mol. Biol. 283C, 235-330. 10.1016/S1937-6448(10)83005-9 [DOI] [PubMed] [Google Scholar]
- Stenzel D., Franco C. A., Estrach S., Mettouchi A., Sauvaget D., Rosewell I., Schertel A., Armer H., Domogatskaya A., Rodin S. et al. (2011). Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 12, 1135-1143. 10.1038/embor.2011.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchting S., Freitas C., le Noble F., Benedito R., Bréant C., Duarte A. and Eichmann A. (2007). The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 104, 3225-3230. 10.1073/pnas.0611177104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao M., Salas M. L. and Lipmann F. (1970). Mechanism of activation by adenosine 3′:5′-Cyclic monophosphate of a protein phosphokinase from rabbit reticulocytes. Proc. Natl. Acad. Sci. USA 67, 408-414. 10.1073/pnas.67.1.408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskén K. and Aandahl E. M. (2004). Localized effects of cAMP mediated by distinct routes of protein kinase a. Physiol. Rev. 84, 137-167. 10.1152/physrev.00021.2003 [DOI] [PubMed] [Google Scholar]
- Taylor S. S., Ilouz R., Zhang P. and Kornev A. P. (2012). Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646-658. 10.1038/nrm3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villefranc J. A., Amigo J. and Lawson N. D. (2007). Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn. 236, 3077-3087. 10.1002/dvdy.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälchli T., Wacker A., Frei K., Regli L., Schwab M. E., Hoerstrup S. P., Gerhardt H. and Engelhardt B. (2015). Wiring the vascular network with neural cues: a CNS perspective. Neuron 87, 271-296. 10.1016/j.neuron.2015.06.038 [DOI] [PubMed] [Google Scholar]
- Wang Y., Nakayama M., Pitulescu M. E., Schmidt T. S., Bochenek M. L., Sakakibara A., Adams S., Davy A., Deutsch U., Lüthi U. et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483-486. 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- Wang H., Sun T., Hu J., Zhang R., Rao Y., Wang S., Chen R., McLendon R. E., Friedman A. H., Keir S. T. et al. (2014). miR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J. Clin. Invest. 124, 4489-4502. 10.1172/JCI75284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. M., Forsythe S. R., Christianson C. A., Frisch B. J., Gigliotti B. J., Jordan C. T., Milner L. A., Guzman M. L. and Calvi L. M. (2006). Parathyroid hormone stimulates expression of the Notch ligand Jagged1 in osteoblastic cells. Bone 39, 485-493. 10.1016/j.bone.2006.03.002 [DOI] [PubMed] [Google Scholar]
- Willis B. S., Niswender C. M., Su T., Amieux P. S. and McKnight G. S. (2011). Cell-type specific expression of a dominant negative PKA mutation in mice. PLoS ONE 6, e18772 10.1371/journal.pone.0018772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Hasan S. S., Schmidt I., Rocha S. F., Pitulescu M. E., Bussmann J., Meyen D., Raz E., Adams R. H. and Siekmann A. F. (2014). Arteries are formed by vein-derived endothelial tip cells. Nat. Commun. 5, 5758 10.1038/ncomms6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K., Yoshida K., Cui H., Wakayama T., Takuwa N., Okamoto Y., Du W., Qi X., Asanuma K., Sugihara K. et al. (2012). Endothelial PI3K-C2α, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat. Cell Biol. 18, 1560-1569. 10.1038/nm.2928 [DOI] [PubMed] [Google Scholar]