

Abstract
Vascular endothelial growth factor a (Vegfa) is essential for blood vessel formation and can induce activation of numerous signaling effectors in endothelial cells. However, it is unclear how and where these function in developmental contexts during vascular morphogenesis. To address this issue, we have visualized activation of presumptive Vegfa effectors at single-cell resolution in zebrafish blood vessels. From these studies, we find that phosphorylation of the serine/threonine kinase ERK (pERK) preferentially occurs in endothelial cells undergoing angiogenesis, but not in committed arterial endothelial cells. pERK in endothelial cells was ectopically induced by Vegfa and lost in Vegfa signaling mutants. Both chemical and endothelial autonomous inhibition of ERK prevented endothelial sprouting, but did not prevent initial artery differentiation. Timed chemical inhibition during angiogenesis caused a loss of genes implicated in coordinating tip/stalk cell behaviors, including flt4 and, at later stages, dll4. ERK inhibition also blocked excessive angiogenesis and ectopic flt4 expression in Notch-deficient blood vessels. Together, these studies implicate ERK as a specific effector of Vegfa signaling in the induction of angiogenic genes during sprouting.
KEY WORDS: ERK, Vegfa, Angiogenesis, Artery differentiation, Zebrafish
Summary: ERK acts as a specific effector of Vegfa signaling to induce angiogenic genes during vascular sprouting in zebrafish.
INTRODUCTION
Vascular development requires coordinated molecular outputs coupled to distinct cellular behaviors. During initial stages of vertebrate vascular development, progenitor cells that will line the blood vessels emerge within the lateral mesoderm (Cleaver and Krieg, 2010). By mid-somitogenesis stages, endothelial gene expression is evident in these progenitors and they commit to arterial and venous cell fates. Subsequently, endothelial progenitors coalesce to form major trunk and head vessels through the process of vasculogenesis. Thereafter, new blood vessels sprout from the pre-existing vascular network by angiogenesis. Together, these processes give rise to the stereotypical vascular pattern of the circulatory system in vertebrate embryos.
The Vascular endothelial growth factor (Vegf) family of proteins plays major roles in blood vessel development (Koch and Claesson-Welsh, 2012). In particular, Vegfa is essential for vascular development and can induce differentiation, proliferation and survival of endothelial cells. The vegfa gene encodes multiple secreted protein isoforms that bind and activate Vegf receptor-2 (Vegfr2, also referred to as Kdr or Flk1), an endothelial-specific receptor tyrosine kinase (RTK; Koch and Claesson-Welsh, 2012). Mice lacking Vegfa or Vegfr2 exhibit defects in blood vessel formation and endothelial differentiation (Carmeliet et al., 1996; Shalaby et al., 1997; Stalmans et al., 2002). In endothelial cell lines, Vegfa binding to Vegfr2 activates downstream effectors common to RTK signaling, including lipid kinases and lipases, such as phosphoinositide-3-kinase (PI3K) and phospholipase C gamma 1 (Plcg1), as well as serine/threonine kinases including ERK (MAPK), AKT, protein kinases A and C, and RAF (Koch and Claesson-Welsh, 2012). Signaling through these molecules induces common responses in cell culture, e.g. proliferation and migration, although their in vivo roles are sometimes distinct. For example, PI3K and AKT are essential for Vegfa-mediated endothelial cell survival in culture (Gerber et al., 1998), but in developmental contexts PI3K is required for migration during angiogenesis (Graupera et al., 2008). Thus, although it is known that many signaling effectors can act downstream of Vegfa to elicit particular endothelial cell behaviors, how these function in vivo is less clear.
The zebrafish is a valuable model for investigating Vegf signaling during angiogenesis. Zebrafish embryos possess a simple trunk vascular network comprising a single posterior cardinal vein and dorsal aorta, from which intersomitic vessels (ISVs) sprout by angiogenesis (Isogai et al., 2003). Embryos mutant for kdrl, which encodes one of two zebrafish Vegfr2 orthologs (Bussmann et al., 2008; Covassin et al., 2006), exhibit multiple defects in trunk vessels, loss of artery differentiation, delayed ISV sprouting, and arteriovenous shunting (Covassin et al., 2009, 2006). Similar defects are associated with knockdown of the Vegfaa ligand, or mutations in plcg1 (Lawson et al., 2003, 2002). Thus, Vegfa signaling through kdrl and plcg1 is essential for artery differentiation and ISV angiogenesis in zebrafish trunk vessels. Plcg1 is similarly important for Vegfa signaling in mammals as well. Signaling through tyrosine 1175 in human VEGFR2 is essential for phosphorylation of PLCG1 (Takahashi et al., 2001) and mouse embryos bearing a point mutation in this residue exhibit vascular defects comparable to those associated with a vegfr2 null allele (Sakurai et al., 2005). Thus, zebrafish serves as an excellent model to define conserved effectors of Vegfa signaling.
Although Vegfa signaling is required for both artery differentiation and angiogenesis, these are likely to be governed through distinct context-dependent effectors. Notably, the Vegfa and Notch pathways show opposing genetic interactions in these contexts. Although Notch or Vegfa deficiency leads to similar defects in artery differentiation, they cause opposite effects on angiogenesis (Covassin et al., 2006; Lawson et al., 2001, 2002; Leslie et al., 2007; Siekmann and Lawson, 2007). Notch activation blocks ISV sprouting, similar to a ‘loss-of-Vegfa’ phenotype, but rescues artery differentiation in Vegfa-deficient embryos (Lawson et al., 2002; Siekmann and Lawson, 2007). Thus, Notch appears to act as a switch between distinct Vegfa signaling outputs, suggesting that distinct downstream effectors act specifically to drive either process.
Most RTK pathway components are kinases and regulation occurs through phosphorylation at serine, threonine or tyrosine residues on these proteins or their substrates. These residues are often conserved and can be recognized by available phospho-specific antibodies. Here, we take advantage of this fact by using optimized immunostaining protocols to identify context-dependent effectors of Vegfa signaling in zebrafish trunk vessels. Our results suggest that ERK is activated specifically in sprouting endothelial cells during vascular development. Furthermore, functional perturbation of ERK reveals an essential and specific role in angiogenesis, but not in initial arterial endothelial differentiation.
RESULTS
ERK is activated by Vegfa specifically in sprouting ISV endothelial cells and their precursors
To identify effectors of Vegfa that may play a specific role in arterial endothelial differentiation or angiogenesis, we optimized a whole-mount immunostaining protocol to assess phosphorylation of candidate kinases, or their substrates, in zebrafish trunk vessels at 24 hours post-fertilization (hpf). At this stage, the trunk vasculature comprises a single dorsal aorta (DA) and posterior cardinal vein (PCV), as well as actively sprouting ISVs (Isogai et al., 2003). Immunostaining of Tg(fli1a:egfp)y1 embryos with phospho-specific antibodies revealed that presumptive Vegfa signaling effectors were activated in endothelial cells. Notably, we observed Plcg1 phosphorylation at Y771 and 783 in endothelial cells lining the DA and ISV, but not the PCV (Fig. S1A,B), consistent with the requirement for Plcg1 during artery differentiation and ISV angiogenesis (Lawson et al., 2003). We also observed phospho-specific epitopes indicative of protein kinase A and C activation in endothelial cells lining all blood vessels (Fig. S1C,D), whereas phosphorylation of the p38 MAP kinase (Mapk14) was not apparent in developing blood vessels (Fig. S1E). Thus, unlike Plcg1, the patterns of PKA, PKC and p38 did not appear in a pattern that would implicate them specifically in either artery differentiation or angiogenesis.
The serine/threonine kinase ERK is thought to be important for initiation of arterial endothelial cell identity (Hong et al., 2006; Wythe et al., 2013). Therefore, we expected that its pattern of phosphorylation would resemble that of Plcg1. However, most phosphorylated ERK (pERK)-positive endothelial cells were observed in ISVs and cells at the base of the ISV, whereas adjacent cells and most DA endothelial cells were pERK negative (Fig. 1A-A″; Fig. S2A). At earlier stages (10 somite stage, ss), pERK was apparent in developing somites, but only rarely in endothelial progenitor cells (Fig. 1B,C-D″). This pattern was different from that of Notch activation, which marks the numerous DA-committed endothelial progenitors in the lateral mesoderm at this stage (Quillien et al., 2014). By 18 ss, we detected robust pERK in selected endothelial cells in the nascent DA, whereas endothelial progenitors still in the lateral mesoderm remained negative (Fig. 1E,E′). Soon thereafter, pERK was restricted to individual ISV cells beginning to sprout from the DA, but surrounding cells remained negative (Fig. 1F). These observations suggested that ERK activation specifically marks endothelial cells that are angiogenic, rather than those committed to an arterial fate.
Fig. 1.
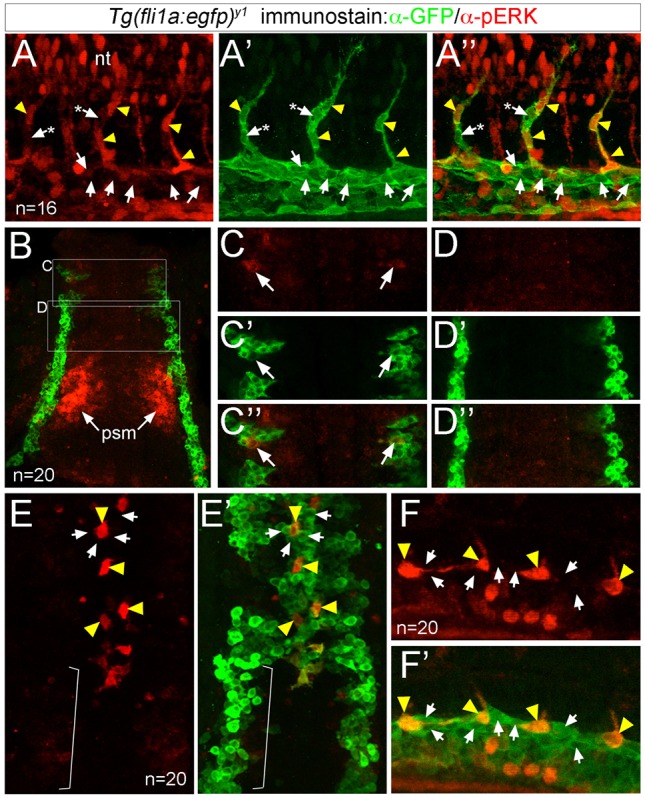
ERK is phosphorylated in sprouting ISV endothelial cells. (A-F′) Confocal images of endothelial cells in Tg(fli1a:egfp)y1 embryos immunostained to detect pERK (red) and GFP (green). Yellow arrowheads indicate pERK-positive endothelial cells, and arrows denote pERK-negative endothelial cells. In A-A″, asterisks denote pERK-negative endothelial cells in the ISV adjacent to pERK-positive cells. (A-A″) Embryo at 24 hpf showing pERK immunostaining (A), GFP immunostaining (A′), or an overlay of pERK and EGFP (A″). (B-D″) Embryo at 10 ss (dorsal view, anterior is up) showing pERK immunostaining (C,D), GFP immunostaining (C′,D′) and the overlay (B,C″,D″). Boxed regions in B indicate areas of higher magnification in C-D″. ERK staining in the presomitic (psm) mesoderm is indicated. (E,E′) Embryo at 18 ss (dorsal view, anterior is up) showing pERK immunostaining (E), and an overlay of pERK and GFP immunostaining (E′). Bracket indicates endothelial cells that have not yet migrated to the midline and are pERK negative. (F,F′) Embryo at 20 ss (lateral view, anterior to the left, dorsal is up) showing pERK immunostaining (F) and an overlay of EGFP expression and pERK immunostaining (F′). Number of embryos analyzed is indicated.
Vegfa and the related ligand Vegfc can induce ERK activation in endothelial cell culture (Pajusola et al., 1994; Takahashi et al., 2001). Accordingly, embryos injected with mRNA encoding the 121 amino acid isoform of Vegfa (Vegfa121), or Vegfc, exhibited ectopic pERK throughout the trunk vessels, although the effect of Vegfc was mostly limited to the PCV, where its receptor, Flt4, is expressed (Fig. 2A-C′). Conversely, embryos bearing a kinase dead (y17) or a null allele (um19) of kdrl had significantly reduced numbers of pERK-positive DA endothelial cells compared with wild type (Fig. 2D,E,H). By contrast, embryos bearing a null allele in flt4 (flt4um131) display normal numbers of pERK-positive DA endothelial cells at 20 hpf (Fig. 2F,H). The remaining pERK-positive cells in kdrl mutants were unlikely to be a result of residual signaling through the paralogous Kdr receptor (Covassin et al., 2006) as treatment with the pan-Vegfr inhibitor SU5416 caused a similar phenotype (Fig. 2H). Rather, remaining cells may be non-endothelial fli1a:egfp-positive cells (e.g. hematopoietic cells) that retain pERK. We further find that embryos bearing a mutation in the conserved lipase domain of plcg1 exhibit a loss of DA pERK-positive cells (Fig. 2G), similar to kdrl mutants. Thus, Vegfa signaling through Plcg1 is essential for ERK activation in sprouting ISV endothelial cells.
Fig. 2.

ERK phosphorylation in sprouting ISVs requires Vegfa signaling. (A-G) Confocal images of Tg(fli1a:egfp)y1 embryos at 21 hpf (A-C′) and 20 hpf (D-G) immunostained for GFP (green) and pERK (red). Lateral views, anterior to the left, dorsal is up. Number of analyzed embryos is indicated, representative images are shown. Embryos were injected with 100 pg mcherry mRNA (A,A′), 25 pg vegf121 mRNA (B,B′) or 100 pg vegfc mRNA (C,C′). (D) Wild-type embryo. (E) kdrly17 mutant embryo. (F) flt4um131 mutant embryo. (G) plcg1y13 mutant embryo. Arrows indicate pERK-positive cells, or lack thereof. (H) Graph showing number of pERK+ endothelial cells per embryo at 20 hpf in a defined segment of the DA in kdrl and flt4 mutant embryos, or in embryos treated with 10 µM SU5416. Number of embryos for each genotype or treated with compound is indicated on the x-axis. Error bars represent s.d.; ***P<0.001; ns, not statistically significant.
ERK activation is required for ISV angiogenesis
The pERK pattern in trunk blood vessels suggested a role during ISV angiogenesis. To determine if this was the case, we treated zebrafish embryos with MEK inhibitors to block ERK phosphorylation. A number of studies have applied 60 µM SL327 to investigate the effects of ERK inhibition on vascular development (e.g. Hong et al., 2006). However, we find that treatment at this dose beginning at 10 hpf leads to developmental delay and the appearance of tissue necrosis in the intermediate cell mass and tail (Fig. 3A,B). Treatment with 15 µM SL327 did not cause similar toxicity, yet was sufficient to potently block ERK phosphorylation (Fig. 3C,D). By contrast, 40 µM of U0126 failed to reduce pERK (Fig. 3D). Therefore, we used 15 µM SL327 from 20 to 30 hpf to reduce possible indirect effects on ISV formation. Under these conditions, pERK was largely extinguished within 30 min of SL327 treatment, but no overt defects were detected by 30 hpf (Fig. 3E,F; Fig. S2B). Importantly, SL327 caused a significant decrease in ISV length compared with embryos treated with DMSO (Fig. 3G-I). This defect was associated with a slight, but significant, decrease in the number of endothelial cells per ISV sprout (Fig. 3J). Reduced ISV numbers were not likely to be due to proliferation defects as 5-bromo-2′-deoxyuridine (BrdU) incorporation was unchanged in SL327-treated embryos, but was clearly blocked by exposure to hydroxyurea and aphidicolin (Fig. 3K).
Fig. 3.
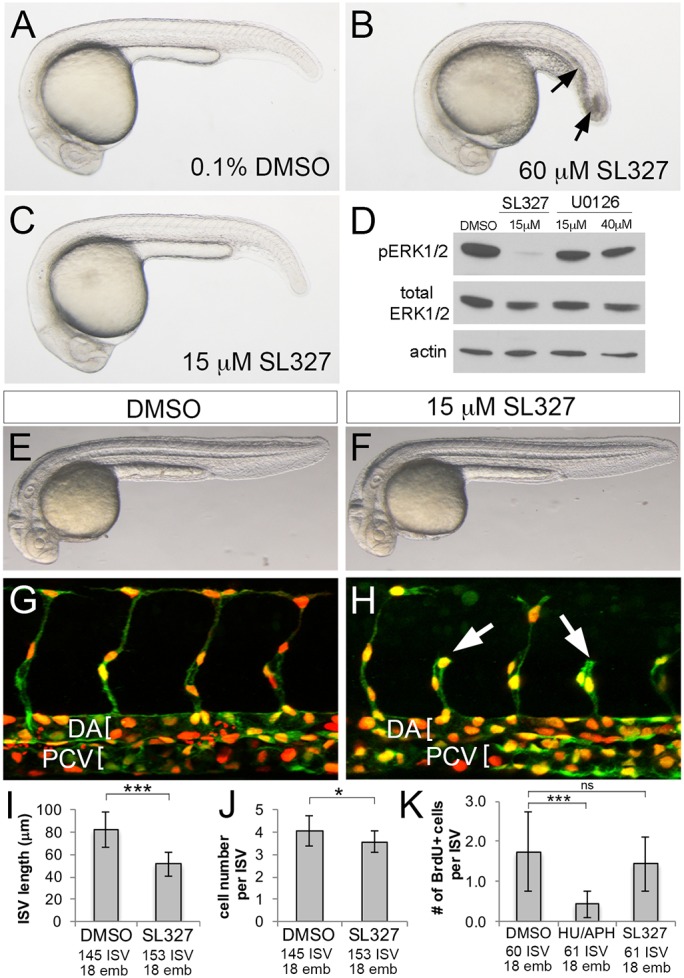
Chemical inhibition of ERK blocks ISV sprouting. (A-C) Transmitted light images of embryos treated with the indicated compound and concentration from 10 hpf to 24 hpf. Arrows in B indicate necrosis. (D) Western blot analysis of pERK, total ERK and actin in embryos treated from 24 to 25 hpf as indicated. (E-H) Transmitted light images (E,F) and confocal images (G,H) of Tg(cdh5:gal4ff)mu101;(uas:nTom)um152;(uas:egfp)nkuasgfpa1 embryos at 32 hpf. Embryos were treated with 0.1% DMSO (E,G) or 15 µM SL327 (F,H) from 20 to 32 hpf. DA and PCV are denoted by brackets in G,H and shorter ISVs are indicated by arrows in H. (I-K) Quantification of ISV length (I), cell number per ISV (J) and BrdU-positive cells per ISV (K) following treatment with the indicated compound. *P<0.05, ***P<0.001; ns, not statistically significant; numbers of ISVs and embryos counted across three experiments are indicated on the x-axis. Error bars represent s.d.
To confirm that ISV defects were autonomous to endothelial cells, we generated transgenic zebrafish in which the UAS promoter drives a stabilized form of dual specificity phosphatase 6 (referred to as dusp6dm), an ERK-specific phosphatase (Marchetti et al., 2005; Muda et al., 1996) along with myc-tagged nuclear-localized tdTomato (nTom) separated by a viral 2A sequence [Tg(uas:ntom2a-dusp6dm)um151]. In parallel, we utilized UAS-driven nTom as a control [Tg(uas:ntom)um152]. In both cases, we found that GAL4-driven expression in stable lines is mosaic, similar to other UAS transgenes in zebrafish (Goll et al., 2009). Mosaic nTom expression was observed much more frequently in pERK-positive endothelial cells than was dusp6dm, consistent with the ability of Dusp6 to inhibit ERK phosphorylation (Fig. S2C,D). We next assessed the ability of transgene-expressing endothelial cells to contribute to different blood vessels in a mosaic trunk vasculature, similar to using cell transplantation to assess angiogenic potential (Siekmann and Lawson, 2007). In control Tg(cdh5:gal4ff)mu101;(uas:egfp)nkuasgfp1a;(uas:nTom)um152 embryos, approximately half of nTom-expressing endothelial cells contributed to the dorsal aorta, while 30% and 15% of the remaining cells were in the ISV and dorsal longitudinal anastomotic vessel (DLAV), respectively (Fig. 4A,C). By contrast, significantly fewer dusp6dm-expressing cells were observed in the ISV and DLAV; most cells were found instead within the DA (Fig. 4B,C). Conversely, to determine whether ERK activation could drive ISV sprouting, we expressed an activated form of zebrafish Mek2b (actMek2b; Map2k2b), an upstream kinase for ERK. In this case, we injected plasmids containing Tol1 direct repeats flanking a UAS driving expression of nTom or myc-tagged Mek2b, along with mRNA encoding the Tol1 transposase, into Tg(cdh5:gal4ff)mu101 embryos. As above, this approach results in mosaic expression of the injected transgene in endothelial cells, which could be visualized by co-immunostaining for Myc and Fli1b. Following injection, most nTom-expressing endothelial cells contributed to the DA at 23 hpf, but 20% of cells were found in ISVs (Fig. 4D,F). Endothelial cells expressing actMek2b were observed at a significantly higher rate in ISVs compared with the DA (Fig. 4E,F). Together, these results demonstrate that endothelial autonomous ERK activation is important to drive ISV sprouting.
Fig. 4.
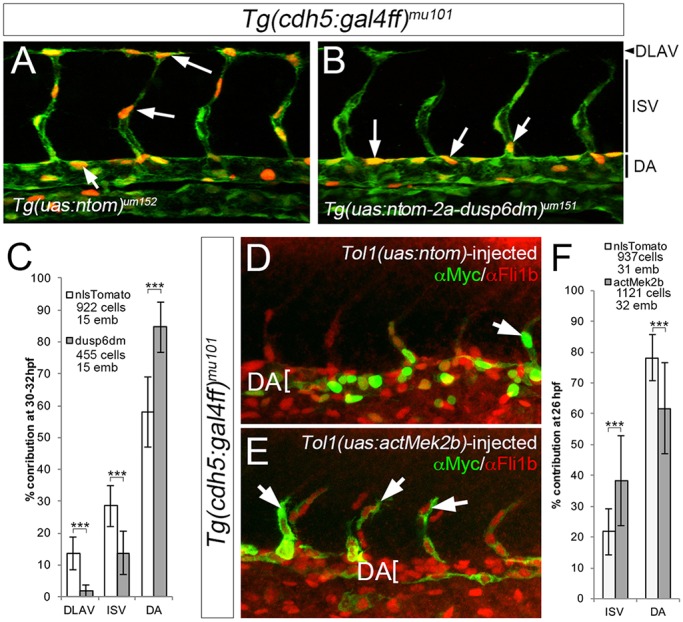
Endothelial cell-autonomous inhibition of ERK blocks ISV sprouting. (A,B) Confocal images of Tg(cdh5:gal4ff)mu101;(uas:egfp)nkuasgfpa1 embryos bearing the indicated transgenes. uas:egfp is green, nTom is red; selected nTom-positive cells indicated by arrows. Vessel positions shown on the right. (C) Contribution of nlsTom- or dusp6dm-expressing cells to the indicated vessel between 30 and 32 hpf. ***P<0.001. (D,E) Confocal images of Tg(cdh5:gal4ff)mu101 embryos injected with Tol1 transgenes expressing nTom (D) or actMek2b (E) immunostained for Myc (green) and Fli1b (red). Arrows denote transgene-expressing ISV cells. (F) Quantification of nTom- and actMek2b-expressing endothelial cells in the indicated vessel at 23 hpf. (C,F) Numbers of cells and embryos used for quantification are shown. Error bars represent s.d.
ERK is required for ISV endothelial cell gene expression
The ISV pattern of pERK is similar to that of flt4 mRNA, which is specific to ISVs and absent from DA endothelial cells (Siekmann and Lawson, 2007). Indeed, double staining for flt4 and pERK phosphorylation shows colocalization in sprouting ISV cells at 24 hpf (Fig. 5A-A″), raising the possibility that flt4 and ERK act in a common pathway during ISV sprouting. Because flt4 itself is dispensable for ERK phosphorylation (Fig. 2F,H), we hypothesized that ERK activation induces flt4 expression in ISVs. Therefore, we assessed flt4 expression in SL327-treated embryos. We again timed the SL327 exposure to avoid confounding effects on ISV sprout growth. As controls, we assessed expression of kdrl and the Notch ligand dll4, a presumed target of Vegfa signaling through ERK (Wythe et al., 2013). In DMSO-treated embryos, flt4 is expressed in ISV cells and trailing cells as they sprout from the DA, but is absent from adjacent DA endothelial cells at 23 hpf and 28 hpf (Fig. 5B,H). At the same time points, kdrl and dll4 are expressed in sprouting ISV endothelial cells, but also throughout neighboring DA endothelial cells (Fig. 5C,D,I,J; plane is focused on ISVs in these panels). Treatment with 15 µM SL327 from 16 to 23 hpf, or from 24 to 28 hpf, severely reduced flt4 expression in ISV and trailing cells within the DA, whereas PCV flt4 expression remained (Fig. 5E,K,N). By contrast, kdrl was unaffected (Fig. 5F,L,N). Interestingly, SL327 blocked dll4 in both ISV and DA endothelial cells only at the later stage (Fig. 5G,M,N). Consistent with a role for Vegfa upstream of ERK in the induction of flt4 and dll4, expanded expression of these transcripts in response to exogenous Vegfa121 is also blocked by SL327 (Fig. S3). Together, these results suggest that ERK activation is important for flt4 and dll4 expression in sprouting ISV endothelial cells.
Fig. 5.

ERK activation is essential for flt4 and dll4 expression in sprouting ISVs. (A-A″) Confocal images of embryo co-stained for flt4 transcript (green) and pERK (red). Yellow arrows denote ISVs, white arrowheads indicate non-expressing cells in the dorsal aorta. (B-M) Whole-mount in situ hybridization with riboprobes against the indicated transcripts (flt4, kdrl or dll4). Unless otherwise noted, arrows indicate ISVs in all panels; in B and E, tip and trailing positions are noted by arrows. In B-D, arrowheads denote expression (or lack thereof) in intervening DA endothelial cells. In B-E, blue brackets indicate PCV. Compound used for treatment indicated at left, interval of treatment and analysis stage indicated at right. (N) Quantification of marker expression; treatment stages and concentrations are indicated, as are numbers of embryos used for analysis. ***P<0.001; ns, not statistically significant.
ERK activation is dispensable for initial arterial endothelial differentiation
Despite the presumed role for ERK in initiating artery identity (Hong et al., 2006; Wythe et al., 2013), SL327 treatment or dusp6dm expression did not block expression of ephrin-B2a (efnb2a; Fig. S4A,B). Previous studies used an earlier starting point for treatment (10 hpf) and a higher SL327 dose, although we find this dose clearly causes delay and necrosis. Nonetheless, we treated wild-type embryos with 15 µM or 60 µM SL327 beginning at 10 hpf and assessed multiple endothelial markers at 24 hpf. As above, we noted flt4 downregulation in ISV cells with 15 µM treatment starting at 10 hpf, but no effect on kdrl or dll4 (Fig. 6A; Fig. S4C-E). efnb2a, hey2 and notch1b were also unaffected by 15 µM SL327 in this time frame (Fig. 6A-D), similar to the Notch-responsive tp1:egfp transgene (Fig. 6A; Fig. S4F). After treatment with 60 µM SL327, we noted a significant reduction in the number of embryos expressing every marker except hey2 (Fig. 6A-D; Fig. S4C-F). By comparison, treatment with the pan-Vegfr inhibitor SU5416 led to a decrease in all endothelial markers (Fig. 6A-D; Fig. S4A,C-J). Later SL327 treatment caused potent downregulation of dll4 and flt4 in a significant majority of embryos. Interestingly, both notch1b and the tp1:egfp transgene were reduced with later treatment with 15 µM SL327, whereas efnb2a and hey2 were not affected (Fig. 6E; Fig. S4G-J). Even after treatment with a high dose of SL327 from 24 to 28 hpf, efnb2a remained unaffected, although there was a reduction in the proportion of hey2-expressing embryos (Fig. 6E; Fig. S4G-I). Given the severe effects of a high dose of SL327 and the ability of 15 µM SL327 to potently repress ERK phosphorylation, our observations suggest that ERK activation is not likely to be an early signaling event during artery differentiation. Instead, ERK induces and maintains expression of genes involved in ISV sprouting.
Fig. 6.

ERK inhibition does not affect initial artery differentiation. (A,E) Quantification of marker expression following treatment with the indicated compounds from 10 to 24 hpf (A) or 24 to 28 hpf (E). Numbers of embryos used for analysis and probes are indicated. *P<0.05, ***P<0.001; ns, not statistically significant. (B-D) Whole-mount in situ hybridization on 24 hpf embryos treated as indicated from 10 hpf using probes against efnb2a (B), hey2 (C) or notch1b (D). Arrows denote staining in the DA.
Ectopic ERK activation in the absence of Notch contributes to the ‘hyper-angiogenic’ phenotype
In zebrafish ISVs, Notch signaling limits angiogenic cell behaviors (Leslie et al., 2007; Siekmann and Lawson, 2007). Furthermore, flt4 expression is ectopically expressed in the DA and contributes to excessive angiogenesis in Notch-deficient embryos (Hogan et al., 2009; Siekmann and Lawson, 2007). Because ERK is required for flt4 ISV expression, we investigated ERK activation and function in Notch-deficient embryos. As above, wild-type sibling embryos at 21 hpf exhibit pERK in both sprouting and trailing ISV endothelial cells, but not in adjacent DA cells (Fig. 7A). By contrast, pERK appears to be expanded throughout all DA endothelial cells in mindbomb mutant embryos (mibta52b; Fig. 7B), which are deficient for Notch signaling (Itoh et al., 2003). Likewise, treatment with DAPT, but not DMSO, caused a significant increase in the number of pERK-positive endothelial cells in the DA (Fig. 7C-E). We next investigated whether increased ERK activation in Notch-deficient embryos contributed to ectopic flt4 expression. In wild-type siblings, flt4 expression is detectable in endothelial cells within the ISV and PCV, but not the DA at 29 hpf (Fig. 7F), whereas kdrl is observed in both the ISV and the DA (Fig. 7G). In mibta52b mutant embryos, flt4 expression is induced in the ISVs and is ectopically expressed in the DA, whereas kdrl expression is largely unchanged (Fig. 7H,I). Treatment of mibta52b mutant embryos with 30 µM SL327 from 25 to 29 hpf blocked flt4 expression in both ISV and DA endothelial cells, whereas PCV expression is retained (Fig. 7J; PCV is out of the focal plane in this panel; Fig. S5A). By contrast, SL327 treatment does not affect kdrl expression (Fig. 7K; Fig. S5A). Therefore, ectopic expression of flt4 caused by Notch deficiency is likely to be a result of expanded ERK activation.
Fig. 7.

Ectopic ERK activation in Notch-deficient embryos induces flt4 and the hyper-angiogenic phenotype. (A,B) Embryos of the indicated genotype at 21 hpf immunostained for pERK (red). (C,D) Tg(fli1a:egfp)y1 embryos immunostained for GFP (green) and pERK (red) at 20 hpf following treatment with the indicated compound from 14 hpf. In A-D, DA is indicated by bracket; arrows denote pERK-positive cells; arrowheads indicate pERK-negative DA cells. (E) Quantification of pERK-positive DA endothelial cells following treatment with DMSO or 100 µM DAPT. (F-K) Whole-mount in situ hybridization with riboprobes against the indicated transcript (top). Arrows denote ISVs; arrowhead shows ectopic DA expression in H. DA and PCV are indicated by brackets. Genotype is noted on the left, chemical treatments are indicated in respective panels. Embryos in J,K were treated with 30 µM SL327 from 25 to 29 hpf. (L,M) Quantification of endothelial cell number per ISV (L) and ISV length (M) in embryos of the indicated genotype and after chemical treatment as indicated. Embryos were treated with 15 µM SL327 from 20 to 34 hpf. (E,L,M) Numbers of cells and embryos used for analysis are shown. Error bars are ±s.d.; *P<0.05, **P<0.01, ***P<0.001.
As increased angiogenic behaviors in Notch-deficient embryos can be partially reversed by reducing flt4 signaling (Hogan et al., 2009; Siekmann and Lawson, 2007; Villefranc et al., 2013) and ERK is important for flt4 expression, we reasoned that SL327 should block ectopic angiogenesis in the absence of Notch. Therefore, we treated wild-type and mibta52b mutant sibling embryos with SL327 from 20 to 34 hpf and assessed endothelial cell number and ISV length. At 34 hpf, mibta52b mutant embryos display an increased number of cells per ISV compared with wild type, whereas 15 µM SL327 treatment slightly, but significantly, reduced this number, similar to the modest effect on wild-type siblings (Fig. 7L). We observed a more severe effect of ERK inhibition on sprout growth as SL327 treatment caused a significant shortening of ISV length in both wild-type and mibta52b mutant embryos (Fig. 7M). Moreover, SL327 treatment blocked the ectopic ISV branching associated with dll4 deficiency (Fig. S5B,C). Taken together, these results suggest that the hyper-angiogenesis phenotype observed in Notch-deficient embryos is driven, in part, through ectopic ERK activation.
DISCUSSION
Vegfa induces a broad range of endothelial cell behaviors through a number of downstream signaling effectors (Koch and Claesson-Welsh, 2012). However, it is not clear if Vegfa utilizes distinct effectors to coordinate the diverse cellular behaviors required for vascular morphogenesis in vivo. In this study, we took advantage of the zebrafish as a model to identify context-dependent regulators of Vegfa signaling. By developing an optimized immunostaining protocol, we were able to visualize activation of presumptive Vegfa effectors at single-cell resolution in a basic vascular network in vivo. We subsequently applied chemical and genetic approaches to demonstrate that ERK acts as a specific regulator of angiogenic sprouting downstream of Vegfa in the zebrafish.
Previous studies suggested a model in which ERK activation defines artery identity within endothelial progenitors (Hong et al., 2006). This work noted DA-specific immunostaining of pERK and found that SL327 treatment led to a reduction in artery expression of efnb2a. However, image analysis of pERK-immunostained embryos was not performed to single-cell resolution in this case. As the DA comprises a mixture of endothelial cells, only some of which contribute to the ISVs, ISV markers such as flt4 and pERK may initially appear to be ‘arterial’ (i.e. in the DA) when observed at low temporal or spatial resolution. Our studies using confocal microscopy at single-cell resolution and at multiple stages clearly demonstrate that pERK is found within endothelial cells that contribute to sprouting ISVs, but not within arterial cells remaining in the aorta. This pattern is distinct from that seen for Notch activation, which we have previously found to mark committed DA progenitors in the lateral mesoderm (Quillien et al., 2014), further underscoring a role for ERK in ISV sprouting, but not initial artery differentiation. Earlier work also applied a much higher SL327 dose (60 µM) and overall effects of this treatment on development were not reported. We find that 60 µM SL327 causes significant developmental delay and necrosis, suggesting that effects on artery differentiation noted in previous studies were likely to be secondary to these severe defects. The lower SL327 doses that potently block ERK phosphorylation in our study had no global effect on artery differentiation. Furthermore, endothelial autonomous inhibition of ERK (also lacking in previous studies) did not affect commitment to the DA or Efnb2 expression. Although we believe our results likely rule out a role for ERK in artery differentiation, there are some caveats. In particular, it remains possible that low level pERK retained in SL327-treated embryos or dusp6dm-expressing endothelial cells is sufficient for artery differentiation. Addressing this issue will entail genetic ablation of ERK1 and 2 to demonstrate their roles definitively. However, we would note that artery gene expression is normal in endothelial cells from mouse embryos lacking both ERK1 and ERK2, although severe defects in vascular morphogenesis are apparent (Srinivasan et al., 2009). Taken together with our studies, these results suggest that activation of ERK downstream of Vegfa does not play a major role in initiating artery identity during development.
Although ERK inhibition did not have a general effect on early artery marker gene expression, we observed a specific reduction in dll4, a known artery-specific gene, at later stages. Furthermore, the loss of dll4 expression following ERK inhibition was accompanied by a concomitant loss of Notch activation. These observations are consistent with our previous studies showing that loss of dll4 or notch1b leads to loss of Notch activation in the dorsal aorta, but only at later stages (Quillien et al., 2014). In zebrafish, deficiency in either of these genes causes excessive angiogenic behaviors in ISVs, but does not affect initial artery differentiation (Leslie et al., 2007; Quillien et al., 2014; Siekmann and Lawson, 2007). This is likely to be due to the redundant function of other artery-expressed Notch ligands (deltac) and receptors (notch1a and 3), whereas in mouse, dll4 is essential for both artery differentiation and reducing angiogenesis (Duarte et al., 2004; Hellstrom et al., 2007). The importance of ERK in regulating dll4 expression has also been noted in recent studies focused on a conserved artery-specific enhancer in the dll4 gene (Wythe et al., 2013). This element bears Ets binding sites essential for reporter activity in response to Vegfa signaling in zebrafish embryos. Parallel endothelial cell culture manipulations demonstrated that ERK activation was important for Vegfa-mediated induction of DLL4, although other artery genes could also be induced in this context (Wythe et al., 2013). This led to a model, based in part on studies from Hong et al. (2006), in which Vegfa induces ERK activation to initiate artery differentiation during development. However, timed developmental studies of ERK inhibition to support this model were lacking in this previous study. Although our work supports an important role for ERK in regulating dll4 expression, we do not observe a more global effect on artery marker gene expression. Moreover, our results suggest that ERK is acting at later stages, but not earlier, to induce dll4 expression, maintain Notch activation and reduce angiogenic potential in the zebrafish ISVs.
Studies over the past decade have established a general model describing the cellular behaviors and signaling pathways important for angiogenesis (Potente et al., 2011). In this model, sprout initiation requires a selection step in which cells within a patent vessel exhibit ‘tip’ cell characteristics that promote their migration towards a source of Vegfa. At the same time, the angiogenic response of neighboring cells is tempered, in part through Notch activation, to prevent all surrounding cells from migrating en masse, which would compromise the structure of the patent vessel. In this case, Notch activation switches Vegfa signaling from a sprouting output to an artery differentiation output, but how downstream effectors act in this context has not been clear. We find that Vegfa signaling through Plcg1 induces preferential ERK activation in emerging tip cells, as well as trailing cells entering the ISVs. Interestingly, phosphorylation of Plcg1 is observed throughout arterial and ISV endothelial cells, yet ERK phosphorylation occurs only in sprouting cells. This suggests that all DA and ISV cells are experiencing Vegfa activation and that Notch switches Vegfa output in part by regulating ERK activation (Fig. 8). An important effect of this switch is the induction of angiogenic genes, such as flt4, which are dependent on ERK activity for expression in ISV endothelial cells. However, our studies clearly show that loss of ERK activation leads to a more severe ISV sprouting defect than that of flt4 deficiency. Likewise, ERK inhibition can potently block ISV sprouting even in the absence of Notch signaling, but flt4 deficiency simply normalizes angiogenesis in this context (Hogan et al., 2009; Siekmann and Lawson, 2007), suggesting that flt4 is one of numerous genes regulated by ERK activation. Indeed, we observe a dynamic requirement of ERK for expression of dll4, which is also essential for coordinating tip and trailing cell behaviors by activating Notch in trailing endothelial cells (Fig. 8). Thus, Vegfa signaling through ERK is acting to induce dynamically genes, such as flt4, that play a pro-angiogenic role in sprouting, and dll4, which is important to reduce angiogenic behaviors in neighboring cells.
Fig. 8.
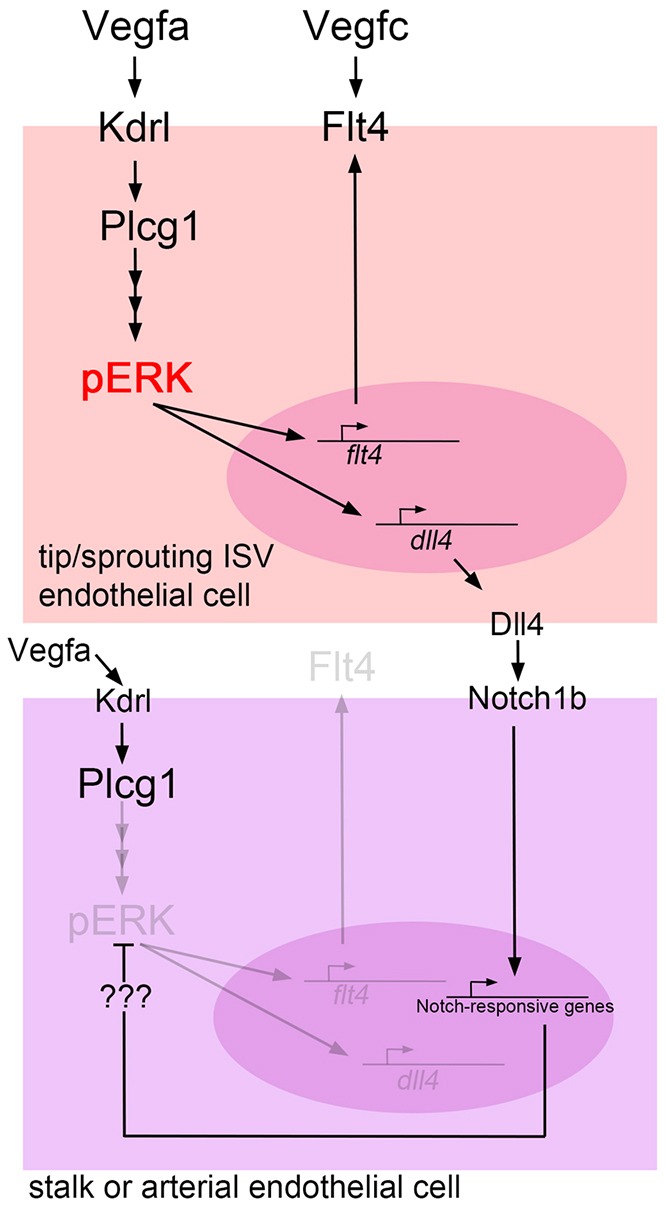
Model depicting the role of ERK in angiogenesis. In sprouting cells, Vegfa activates ERK through Plcg1, which leads to increased flt4 and dll4 expression. Subsequent activation of Flt4 by its ligand Vegfc promotes sprouting, while Dll4-mediated activation of Notch signaling in adjacent cells leads to reduction of ERK activation and reduced sprouting.
MATERIALS AND METHODS
Zebrafish and maintenance
Zebrafish were handled according to approved University of Massachusetts Medical School Institutional Animal Care and Use Committee protocols. The Tg(fli1a:egfp)y1, Tg(kdrl:egfp)la116, Tg(cdh5:gal4ff)mu101, Tg(uas:egfp)nkuasgfp1a, kdrly17, kdrlum19, flt4um131, plcg1y13 and mibta52b lines have been described elsewhere (Anderson et al., 2008; Asakawa and Kawakami, 2008; Bussmann et al., 2011; Covassin et al., 2009, 2006; Itoh et al., 2003; Lawson et al., 2003; Lawson and Weinstein, 2002; Shin et al., 2016). Additional transgenic lines are described below.
Plasmid construction
Gateway-compatible UAS vectors were generated by modifying pBS-2X-MAR-Notch:intra (Scheer and Campos-Ortega, 1999). Tol2 elements were cloned up- and downstream of uas:Notch1intra, along with a crystallin:CFP cassette (referred to as ‘CC’). The Notch1intra sequence was replaced by either an attR1-attR2-polyA or attR1-attR3 –polyA cassette from pCSDest or pCSDest2, respectively (Villefranc et al., 2007), to give pmTol-ins-UAS-R1R2-CC and pmTol-ins-UAS-R1R3-CC, respectively. 6x myc-tagged tdTomato-2A with a nuclear localization signal (MPKKKRKV) was cloned by recombination into pDONR221 to give pME-myc-nlstdTomato-2A. Full-length dusp6 cDNA was used as a template to mutate serines 159 and 196 to alanines to prevent proteasome degradation stimulated by ERK phosphorylation (Marchetti et al., 2005). dusp6dm was cloned into pDONRP2P3 to give p3E-dusp6dm. For generation of UAS lines, pME-myc-nlstdTomato-2a was cloned into pDest insUAS R1R2 CC using LR clonase II (Thermo Fisher Scientific), to give pinsUAS:nlstdTomato, whereas pME-myc-nlstdTomato-2a and p3E-dusp6dm were cloned into pDest insUAS R1R3 CC, resulting in pinsUAS:nlstdTomato-2a-dusp6dm-CC. Protocols for construction and application of Tol1 actMek2b plasmids can be found elsewhere (Shin et al., 2016). Plasmids are available at Addgene.
Microinjections and generation of transgenic lines
pinsUAS:nlstdTomato-CC and pinsUAS:nlstdTomato-2a-dusp6dm-CC were injected with ISceI (NEB) into one-cell-stage embryos and those with lens CFP expression were grown to adulthood. Tg(uas:nlstdTomato)um152 and Tg(uas:nlstdTomato2A-dusp6dm)um151 lines were identified by lens CFP expression in progeny and UAS-driven expression was confirmed by outcrossing with Tg(cdh5:gal4ff)mu101. Both lines exhibit mosaic transgene expression. mcherry, vegf121 and vegfc-2a-mcherry mRNAs were prepared and injected at the indicated amount as previously described (Lawson et al., 2002). Tol1 constructs and mRNA were prepared and injected as described elsewhere (Shin et al., 2016). dll4 MO2 (15 ng per embryo) was injected as previously described (Siekmann and Lawson, 2007). 5′-CGAATgTTAgCTAgAGcTAcATCCG-3′ (5mis-MO) was used as a control.
Inhibitor treatments
SL327, SU5416, DAPT and aphidicolin (EMD Millipore) stocks were dissolved in DMSO and diluted in egg water at indicated concentrations. Hydroxyurea (Sigma) was dissolved in water. For DAPT and aphidicolin/hydroxyurea, embryos were dechorionated and incubated on agarose-coated dishes. When required, embryos were incubated with 1× phenylthiourea to prevent pigmentation.
Western blotting
Dechorionated embryos at 25 hpf treated with DMSO, 15 µM SL327, and 20 µM or 40 µM U0126 (Cell Signaling) for 30 min were de-yolked in cold calcium free 0.5× Ginzburg buffer (55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3) with trituration on ice and lysed in Laemmli buffer. Lysate equivalent to five embryos was separated by SDS-PAGE and subjected to western blotting. Blots were incubated with anti-phospho-p44/42 MAPK (Thr202/Tyr204) XP rabbit monoclonal antibody (1:2000; #4370, Cell Signaling), anti-p44/42 MAPK antibody (1:10,000; #9102, Cell Signaling) and anti-αActin antibody (1:20,000; A-5060, Sigma).
BrdU incorporation
BrdU (2-4 nl of 10 µg/µl) in PBS was injected into the yolk of dechorinated Tg(fli1a:egfp)y1 embryos at 24 hpf lined on a 2% agar injection plate. Embryos were immediately transferred to egg water containing 4% DMSO and 1× 1-phenyl-2-thiourea (PTU), or 15 µM SL327 or 150 µM aphidicolin/20 mM hydroxyurea, on a 1% agar-coated Petri dish, incubated for 6 h and fixed with 8% paraformaldehyde/phosphate buffered saline (PFA/PBS) followed by BrdU and GFP immunostaining (see below).
In situ hybridization and immunostaining
Fluorescence and standard in situ hybridization was carried out as previously described (Quillien et al., 2014). To detect pERK and GFP, embryos were fixed with 8% PFA/PBS overnight at 4°C, quenched with 3% H2O2 in methanol and stored in methanol at −20°C until use. Embryos were washed with cold Tris-buffered saline/0.1% Tween20/0.1% Triton X-100 (TBSTwT) and permeabilized with TBSTw/1% Triton X-100 for 30 min on ice. After blocking with TBSTwT/1% bovine serum albumin (BSA)/10% goat serum (GS) (hereafter termed ‘blocking solution’) for a few hours on ice, embryos were incubated with 1:250 monoclonal mouse anti-GFP antibody (A-11120, Thermo Fisher Scientific) in blocking solution overnight at 4°C followed by washing three times with TBSTwT for 30 min at 4°C. Embryos were then incubated with 1:500 goat anti-mouse IgG-Alexa488 (A-11029, Thermo Fisher Scientific) overnight at 4°C. Embryos were washed three times with TBSTwT for 30 min at 4°C, washed twice with cold citrate buffer (0.1 M trisodium citrate dehydrate/0.05% Tween20, pH6.0) and boiled in citrate buffer for 20 min at 98°C for antigen retrieval. After blocking for 3 h on ice, embryos were incubated with 1:250 phospho-p44/42 MAPK (Thr202/Tyr204) XP rabbit monoclonal antibody overnight at 4°C. After briefly washing with TBSTwT at room temperature (RT), embryos were rinsed with maleic buffer (150 mM maleic acid/100 mM NaCl/0.05% Tween20, pH7.4), blocked with maleic blocking buffer (maleic buffer/2% blocking reagent; Roche) for 3 h at RT and incubated with 1:1000 goat anti-rabbit IgG-HRP (#81-1620, Thermo Fisher Scientific) overnight 4°C. Embryos were washed five times with maleic buffer for 30 min at RT, then PBS, and incubated with 1:50 TSA Plus tyramide-Cy3 in 1× amplification diluent (Perkin Elmer) for detection. Embryos were washed with TBSTwT overnight and kept in 4% PFA/PBS at 4°C until imaging. Immunostaining was performed as above for Plcg1, PKC, PKA and p38 using 1:100 phospho-PLCg1 (Tyr771) antibody, 1:100 phospho-PLCg1 (Tyr783) antibody, 1:1000 phospho-(Ser) PKC Substrate antibody, 1:1000 phospho-(Ser/Thr) PKA Substrate antibody, 1:1000 phospho-p38 MAPK (Thr180/Tyr182) XP rabbit monoclonal antibody (#2824, #2821, #2261, #9621, #4511, respectively; Cell Signaling). Fli1b polyclonal antibody (1:5000; Moore et al., 2013) was used for detecting endothelial cell nuclei. To detect EphrinB2, embryos were fixed with 4% PFA/PBS for 1 h at RT, quenched and stored in methanol. After washing with PBS/0.1% Tween20 (PBSTw) and permeabilization with PBSTw/1% Triton X-100, embryos were blocked with PBSTw/2% blocking reagent (Roche) and incubated with 1:100 goat anti-zEphrinB2 antibody (AF-1088, R&D Systems) at 4°C. Embryos were washed with PBSTw and incubated with 1:5000 ZyMAX rabbit anti-goat IgG(H+L)-HRP (#81-1620, Thermo Fisher Scientific) overnight at 4°C and reacted with TSA Plus tyramide-FITC (NEL741001KT, Perkin Elmer) after washing with PBSTw/0.1% Triton X-100. The embryos stained with zEphrinB2 antibody were incubated in citrate buffer for 20 min at 98°C to denature the primary and secondary antibodies. After blocking with PBSTw/1% BSA/10% GS, embryos were incubated with 1:250 monoclonal Myc antibody (9E10; Sigma) and stained with 1:500 goat anti-mouse IgG-Alexa568 (A-11004, Thermo Fisher Scientific). To detect BrdU and GFP, fixed embryos were incubated with 1 N HCl for 1 h at RT, washed with PBSTw and incubated with 1:500 polyclonal anti-BrdU antibody (600-401-C29, Rockland) and 1:500 monoclonal anti-GFP Ab (sc-9996, Santa Cruz Biotechnology) overnight at 4°C after blocking with PBSTw/0.5% Triton X-100/1% DMSO/1% BSA/0.1% GS. After washing, embryos were incubated with 1:1000 goat anti-rabbit IgG(H+L)-HRP and 1:500 goat anti-mouse IgG (H+L)-Alexa488 overnight at 4°C. BrdU was detected with tyramide-Cy3. To detect flt4 mRNA and pERK, we subjected embryos to fluorescence in situ hybridization using digoxygenin (DIG)-labeled flt4 antisense riboprobe as described elsewhere (Quillien et al. 2014), sheep anti-DIG-POD antibody (1:500; #11207733910, Roche) for the secondary antibody and TSA Plus tyramide-FITC, and quenching to remove residual POD activity. The embryos were stained for pERK with tyramide-Cy3.
Imaging and processing
Embryos were placed in 1.5% low melt agarose on a glass-bottomed culture dish filled with egg water. For live embryos, 1× tricaine was included. Confocal images were acquired using a Zeiss LSM710 equipped with W Plan-APOCHROMAT 20×/1.0 DIC D=0.17 M27 70 mm objective lens. GFP or Alexa488, and tdTomato, Alexa568 or Cy3 were simultaneously excited at 488 nm and 561 nm of confocal lasers, respectively. Alternatively, embryos were imaged by two-photon microscopy using a Chameleon Ti:Sapphire pulse laser (Coherent) at 900 nm through a 500-550 nm band path filter into a BiG detector and acquired using LSM710 or Zeiss LSM7 equipped with W Plan-APOCHROMAT 20×/1.0 DIC (UV) VIS-IR objective lens. Image stacks were assembled and quantified using ImageJ with Fiji or Imaris (Bitplane).
Measurements and statistics
pERK+GFP+ cells in the DA and ISVs on both sides between somites 8 and 11 were counted in two-photon or confocal images derived from three clutches of embryos for each experiment. ISV cell numbers between somites 9 and 16 were scored based on nlstdTomato transgene expression or Fli1b antibody immunostaining in nuclei of endothelial cells in two-photon or confocal images from three clutches of embryos. Cell contribution to the DLAV, ISV and DA between somites 9 and 16 was counted by location of nlstdTomato transgene expression of Tg(uas:nlstdTomato2A-dusp6dm)um151 in two-photon images from three clutches of embryos. The vertical length of each ISV recognized by transgene expression or immunostaining of GFP in endothelial cells was measured at the same time as when cell number was scored. Similarly, the number of BrdU+ cells in each ISV between somites 9 and 16 was scored in confocal images from three clutches of embryos. The number of branch points per ISV in morpholino-injected Tg(kdrl:egfp)la116 at 3 dpf were manually scored in 3D stacks visualized using Imaris. Student's t-test was used to calculate significance for quantification of cell numbers, cell contribution and measurements. Fisher's Exact test with Bonferroni adjustment was used to calculate P-values for in situ quantification.
Acknowledgements
We are grateful to Arndt Siekmann for providing the cdh5:gal4ff line. We thank members of the Lawson Lab for technical and intellectual input and the UMass Med Aquatics staff for excellent fish care.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
M.S. designed and performed experiments for all aspects of this work and contributed to the editing of the manuscript. T.B. performed in situ hybridization experiments and contributed to maintenance of zebrafish lines described in this study. A.Q. designed and generated the insUAS constructs. I.M. contributed to identification and maintenance of zebrafish lines described in this study. L.J.Z. performed statistical analyses. N.D.L. designed experiments and wrote the manuscript.
Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (NHLBI) [R01HL093467 and R01HL122599 to N.D.L.]; by the Uehara Memorial Foundation (M.S.); and by the Japan Society for the Promotion of Science (JSPS). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.137919.supplemental
References
- Anderson M. J., Pham V. N., Vogel A. M., Weinstein B. M. and Roman B. L. (2008). Loss of unc45a precipitates arteriovenous shunting in the aortic arches. Dev. Biol. 318, 258-267. 10.1016/j.ydbio.2008.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa K. and Kawakami K. (2008). Targeted gene expression by the Gal4-UAS system in zebrafish. Dev. Growth Differ. 50, 391-399. 10.1111/j.1440-169X.2008.01044.x [DOI] [PubMed] [Google Scholar]
- Bussmann J., Lawson N., Zon L., Schulte-Merker S. and Zebrafish Nomenclature Committee. (2008). Zebrafish VEGF receptors: a guideline to nomenclature. PLoS Genet. 4, e1000064 10.1371/journal.pgen.1000064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann J., Wolfe S. A. and Siekmann A. F. (2011). Arterial-venous network formation during brain vascularization involves hemodynamic regulation of chemokine signaling. Development 138, 1717-1726. 10.1242/dev.059881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C. et al. (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435-439. 10.1038/380435a0 [DOI] [PubMed] [Google Scholar]
- Cleaver O. and Krieg P. A. (2010). Vascular development. In Heart Development and Regeneration (ed. Rosenthal N. and Harvey R. P.), pp. 487-528. London, UK: Elsevier Inc. [Google Scholar]
- Covassin L. D., Villefranc J. A., Kacergis M. C., Weinstein B. M. and Lawson N. D. (2006). Distinct genetic interactions between multiple Vegf receptors are required for development of different blood vessel types in zebrafish. Proc. Natl. Acad. Sci. USA 103, 6554-6559. 10.1073/pnas.0506886103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covassin L. D., Siekmann A. F., Kacergis M. C., Laver E., Moore J. C., Villefranc J. A., Weinstein B. M. and Lawson N. D. (2009). A genetic screen for vascular mutants in zebrafish reveals dynamic roles for Vegf/Plcg1 signaling during artery development. Dev. Biol. 329, 212-226. 10.1016/j.ydbio.2009.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte A., Hirashima M., Benedito R., Trindade A., Diniz P., Bekman E., Costa L., Henrique D. and Rossant J. (2004). Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 18, 2474-2478. 10.1101/gad.1239004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H.-P., McMurtrey A., Kowalski J., Yan M., Keyt B. A., Dixit V. and Ferrara N. (1998). Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 273, 30336-30343. 10.1074/jbc.273.46.30336 [DOI] [PubMed] [Google Scholar]
- Goll M. G., Anderson R., Stainier D. Y. R., Spradling A. C. and Halpern M. E. (2009). Transcriptional silencing and reactivation in transgenic zebrafish. Genetics 182, 747-755. 10.1534/genetics.109.102079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M., Guillermet-Guibert J., Foukas L. C., Phng L.-K., Cain R. J., Salpekar A., Pearce W., Meek S., Millan J., Cutillas P. R. et al. (2008). Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453, 662-666. 10.1038/nature06892 [DOI] [PubMed] [Google Scholar]
- Hellstrom M., Phng L.-K., Hofmann J. J., Wallgard E., Coultas L., Lindblom P., Alva J., Nilsson A.-K., Karlsson L., Gaiano N. et al. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776-780. 10.1038/nature05571 [DOI] [PubMed] [Google Scholar]
- Hogan B. M., Herpers R., Witte M., Helotera H., Alitalo K., Duckers H. J. and Schulte-Merker S. (2009). Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development 136, 4001-4009. 10.1242/dev.039990 [DOI] [PubMed] [Google Scholar]
- Hong C. C., Peterson Q. P., Hong J.-Y. and Peterson R. T. (2006). Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and MAP kinase/ERK signaling. Curr. Biol. 16, 1366-1372. 10.1016/j.cub.2006.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S., Lawson N. D., Torrealday S., Horiguchi M. and Weinstein B. M. (2003). Angiogenic network formation in the developing vertebrate trunk. Development 130, 5281-5290. 10.1242/dev.00733 [DOI] [PubMed] [Google Scholar]
- Itoh M., Kim C.-H., Palardy G., Oda T., Jiang Y.-J., Maust D., Yeo S.-Y., Lorick K., Wright G. J., Ariza-McNaughton L. et al. (2003). Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67-82. 10.1016/S1534-5807(02)00409-4 [DOI] [PubMed] [Google Scholar]
- Koch S. and Claesson-Welsh L. (2012). Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2, a006502 10.1101/cshperspect.a006502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N. D. and Weinstein B. M. (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318. 10.1006/dbio.2002.0711 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Scheer N., Pham V. N., Kim C. H., Chitnis A. B., Campos-Ortega J. A. and Weinstein B. M. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675-3683. [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Vogel A. M. and Weinstein B. M. (2002). sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127-136. 10.1016/S1534-5807(02)00198-3 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Mugford J. W., Diamond B. A. and Weinstein B. M. (2003). phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 17, 1346-1351. 10.1101/gad.1072203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie J. D., Ariza-McNaughton L., Bermange A. L., McAdow R., Johnson S. L. and Lewis J. (2007). Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 134, 839-844. 10.1242/dev.003244 [DOI] [PubMed] [Google Scholar]
- Marchetti S., Gimond C., Chambard J.-C., Touboul T., Roux D., Pouyssegur J. and Pages G. (2005). Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 25, 854-864. 10.1128/MCB.25.2.854-864.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J. C., Sheppard S., Shestopalov I. A., Chen J. K. and Lawson N. D. (2013). Post-transcriptional mechanisms contribute to Etv2 repression during vascular development. Dev. Biol. 384, 128-140. 10.1016/j.ydbio.2013.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muda M., Theodosiou A., Rodrigues N., Boschert U., Camps M., Gillieron C., Davies K., Ashworth A. and Arkinstall S. (1996). The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J. Biol. Chem. 271, 27205-27208. 10.1074/jbc.271.44.27205 [DOI] [PubMed] [Google Scholar]
- Pajusola K., Aprelikova O., Pelicci G., Weich H., Claesson-Welsh L. and Alitalo K. (1994). Signalling properties of FLT4, a proteolytically processed receptor tyrosine kinase related to two VEGF receptors. Oncogene 9, 3545-3555. [PubMed] [Google Scholar]
- Potente M., Gerhardt H. and Carmeliet P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873-887. 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- Quillien A., Moore J. C., Shin M., Siekmann A. F., Smith T., Pan L., Moens C. B., Parsons M. J. and Lawson N. D. (2014). Distinct Notch signaling outputs pattern the developing arterial system. Development 141, 1544-1552. 10.1242/dev.099986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y., Ohgimoto K., Kataoka Y., Yoshida N. and Shibuya M. (2005). Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. USA 102, 1076-1081. 10.1073/pnas.0404984102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer N. and Campos-Ortega J. A. (1999). Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech. Dev. 80, 153-158. 10.1016/S0925-4773(98)00209-3 [DOI] [PubMed] [Google Scholar]
- Shalaby F., Ho J., Stanford W. L., Fischer K.-D., Schuh A. C., Schwartz L., Bernstein A. and Rossant J. (1997). A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell 89, 981-990. 10.1016/S0092-8674(00)80283-4 [DOI] [PubMed] [Google Scholar]
- Shin M., Male I., Beane T. J., Villefranc J. A., Kok F. O., Zhu L. J. and Lawson N. D. (2016). Vegfc acts through ERK to induce sprouting and differentiation of trunk lymphatic progenitors. Development 143, 3785-3795. 10.1242/dev.137901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekmann A. F. and Lawson N. D. (2007). Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445, 781-784. 10.1038/nature05577 [DOI] [PubMed] [Google Scholar]
- Srinivasan R., Zabuawala T., Huang H., Zhang J., Gulati P., Fernandez S., Karlo J. C., Landreth G. E., Leone G. and Ostrowski M. C. (2009). Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 4, e8283 10.1371/journal.pone.0008283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalmans I., Ng Y.-S., Rohan R., Fruttiger M., Bouche A., Yuce A., Fujisawa H., Hermans B., Shani M., Jansen S. et al. (2002). Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109, 327-336. 10.1172/JCI0214362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Yamaguchi S., Chida K. and Shibuya M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768-2778. 10.1093/emboj/20.11.2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villefranc J. A., Amigo J. and Lawson N. D. (2007). Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn. 236, 3077-3087. 10.1002/dvdy.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villefranc J. A., Nicoli S., Bentley K., Jeltsch M., Zarkada G., Moore J. C., Gerhardt H., Alitalo K. and Lawson N. D. (2013). A truncation allele in vascular endothelial growth factor c reveals distinct modes of signaling during lymphatic and vascular development. Development 140, 1497-1506. 10.1242/dev.084152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wythe J. D., Dang L. T. H., Devine W. P., Boudreau E., Artap S. T., He D., Schachterle W., Stainier D. Y. R., Oettgen P., Black B. L. et al. (2013). ETS factors regulate Vegf-dependent arterial specification. Dev. Cell 26, 45-58. 10.1016/j.devcel.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]