

Abstract
Trabecular bone formation is the last step in endochondral ossification. This remodeling process of cartilage into bone involves blood vessel invasion and removal of hypertrophic chondrocytes (HTCs) by chondroclasts and osteoclasts. Periosteal- and chondrocyte-derived osteoprogenitors utilize the leftover mineralized HTC matrix as a scaffold for primary spongiosa formation. Here, we show genetically that β-catenin (encoded by Ctnnb1), a key component of the canonical Wnt pathway, orchestrates this remodeling process at multiple levels. Conditional inactivation or stabilization of β-catenin in HTCs by a Col10a1-Cre line locally modulated osteoclastogenesis by altering the Rankl:Opg ratio in HTCs. Lack of β-catenin resulted in a severe decrease of trabecular bone in the embryonic long bones. Gain of β-catenin activity interfered with removal of late HTCs and bone marrow formation, leading to a continuous mineralized hypertrophic core in the embryo and resulting in an osteopetrotic-like phenotype in adult mice. Furthermore, β-catenin activity in late HTCs is required for chondrocyte-derived osteoblastogenesis at the chondro-osseous junction. The latter contributes to the severe trabecular bone phenotype in mutants lacking β-catenin activity in HTCs.
KEY WORDS: Rankl, Tnfsf11, Tnfrsf11b, Beta-catenin, Chondrocyte-derived osteoblasts, Trabecular bone, Transdifferentiation, Mouse
Summary: The conditional modulation of β-catenin activity in late hypertrophic chondrocytes locally regulates osteoclast differentiation and the transdifferentiation of chondrocytes into osteoblasts.
INTRODUCTION
Severe loss of trabecular bone leads to osteopenia/osteoporosis, while a gain in trabecular bone mass is termed osteopetrosis. Trabecular bone formation is the final step in endochondral bone formation during fetal development, occurring at the chondro-osseous front in conjunction with the formation of a bone marrow cavity. For this, terminally differentiated hypertrophic chondrocytes (HTCs) need to be turned over. They undergo apoptosis and are actively removed by osteoclasts and chondroclasts (Bianco et al., 1998; Shapiro et al., 2005) or, alternatively, transdifferentiate into osteoblasts (Park et al., 2015; Yang et al., 2014a,b; Zhou et al., 2014).
HTC differentiation is accompanied by extracellular matrix changes, such as type X collagen (Col10a1) production, matrix mineralization, as well as matrix remodeling due to the upregulation of matrix metalloproteinase 13 (MMP13) (Gack et al., 1995; Mattot et al., 1995). Mature HTCs also produce vascular endothelial growth factor (VEGF). VEGF induces blood vessel invasion into the transition zone between the two hypertrophic domains, facilitating bone marrow cavity formation and attracting blood vessels to the chondro-osseous front (Gerber and Ferrara, 2000; Gerber et al., 1999; Maes et al., 2002; Zelzer et al., 2004, 2002). Along with the blood vessels, precursors of osteoclasts and perichondrial-derived osteoblasts invade. The latter utilize, together with the chondrocyte-derived osteoblast precursors, the mineralized matrix remnants of late HTCs as a template for trabecular bone formation. The HTC matrix is degraded by matrix metalloproteases such as MMP13 produced by late HTCs and MT1-MMP (also known as MMP14) and MMP9 produced by osteoclasts and chondroclasts (Zhou et al., 2000; Vu et al., 1998; Inada et al., 2004; Stickens et al., 2004; Holmbeck et al., 2003). Besides VEGF, which stimulates osteoclast recruitment, HTCs produce factors such as osteopontin (Opn; also known as Spp1), receptor activator of nuclear factor kappa-B ligand (Rankl; also known as Tnfsf11) and its antagonist osteoprotegerin (Opg; also known as Tnfrsf11b) that regulate osteoclastogenesis at the chondro-osseous front (Kishimoto et al., 2006; Nakashima et al., 2011; Silvestrini et al., 2005; Usui et al., 2008; Xiong et al., 2011). Osteoclast activity, and in particular MMP9, has been proposed to facilitate the release of matrix-bound bioactive VEGF (Bergers et al., 2000; Colnot et al., 2003; Engsig et al., 2000; Ortega et al., 2010).
The Wnt/β-catenin pathway regulates skeletogenesis at many levels, including by influencing cell lineage decisions (Day et al., 2005; Hill et al., 2005; Spater et al., 2006) and cell differentiation within the perichondrium-derived osteoblast lineage (Rodda and McMahon, 2006) or the chondrocyte lineage (Tamamura et al., 2005). The cytosolic levels of β-catenin determine pathway activity. Activation by a Wnt ligand results in increased cytosolic β-catenin levels and its subsequent translocation into the nucleus, where it acts as a transcriptional coactivator (MacDonald et al., 2009). Continuous Wnt/β-catenin signaling in immature chondrocytes leads to their dedifferentiation and blocks hypertrophy (Guo et al., 2004; Miclea et al., 2009; Tamamura et al., 2005). Inducible β-catenin activation in chondrocytes in vivo and β-catenin activation or Wnt9a overexpression in more mature chondrocytes in vitro stimulates hypertrophy and expression of the late hypertrophic markers Mmp13 and Vegfa, while slightly decreasing Col10a1 levels (Dao et al., 2012; Tamamura et al., 2005; Chen et al., 2008). Moreover, in perichondrium-derived osteoblasts the pathway postnatally influences bone homeostasis (Glass et al., 2005; Holmen et al., 2005; Kramer et al., 2010; Sato et al., 2009; Spencer et al., 2006). A recent study associated β-catenin signaling in Col2a1-expressing chondrocytes with the regulation of postnatal bone development (Wang et al., 2014).
Previous experimental settings might have obscured the requirement for β-catenin in differentiated HTCs. Hence, we specifically modulated β-catenin function in HTCs using a Col10a1-Cre line (Gebhard et al., 2008) and either conditionally deleted the Ctnnb1 gene or stabilized β-catenin at the protein level. Recently, Ctnnb1 deletion from HTCs has been shown to result postnatally in decreased trabecular bone density due to a local increase in osteoclast number through upregulation of Rankl (Golovchenko et al., 2013). Here, we show that this phenotype is already present in the embryo and that the augmented osteoclastogenesis as a result of increased Rankl does not fully account for the phenotype, as decreasing Rankl expression specifically in HTCs only partially reverts the phenotype. Our analysis uncovered an additional need for β-catenin in the differentiation of HTC-derived osteoblasts. Stabilization of β-catenin in HTCs, by contrast, interfered with late HTC turnover, affecting embryonic and postnatal bone marrow formation. The embryonic phenotype was associated with a reduction in osteoclast number due to reduced Rankl expression and could be reverted in part by additional removal of Opg. In the long bones of adult mice, HTC-derived osteoblast differentiation was promoted at the more active growth plates.
RESULTS
Loss of β-catenin activity from HTCs results in reduced trabecular bone formation
Consistent with a previous report, conditional Ctnnb1 inactivation from HTCs using Col10a1-Cre resulted in viable mice (Ctnnb1lacZ/fl;Col10a1-Cre+, hereafter Ctnnb1LOFHTC) with an osteopenic phenotype (Golovchenko et al., 2013). The phenotype was analyzed from embryonic day (E) 16.5 onwards, focusing primarily on the humerus. At this stage, the primary spongiosa begins to form in the wild type. Alcian Blue and von Kossa staining revealed a severe reduction of calcified structures in the bone marrow cavity of Ctnnb1LOFHTC mutants (Fig. 1A). Based on histology and Col10a1 in situ staining, the hypertrophic domains were not altered in size by the loss of Ctnnb1 (Fig. 1A,B). In situ hybridization for the osteoclast markers cathepsin K (Ctsk) and tartrate-resistant acid phosphatase (Trap; also known as Acp5) revealed an accumulation of cells positive for Ctsk and Trap at the chondro-osseous front (Fig. 1C,D). Staining for Mmp13 and Opn, which are both expressed in terminal mature HTCs and osteoblasts, appeared slightly increased at the chondro-osseous front in Ctnnb1LOFHTC embryos (Fig. 1E, Fig. S1H). By contrast, staining for the osteoblastic marker Col1a1 was restricted to the central region of the maturation zone, aside from its strong periosteal expression (Fig. 1F).
Fig. 1.

Phenotypic analysis of Ctnnb1LOFHTC mouse mutants. (A-F) Representative images of alternating sections through humeri of E16.5 control and Ctnnb1LOFHTC mutant littermates. (A) Alcian Blue/von Kossa staining. (B) Hypertrophic zones visualized by Col10a1 in situ hybridization (ISH). (C) Ctsk ISH. (D) Trap-positive osteoclasts visualized by ISH. (E) Mmp13 ISH. (F) Col1a1 ISH. (G-J) Representative alternating sections through humeri of P0 control and Ctnnb1LOFHTC mutant littermates. (G) Alcian Blue/von Kossa. (H) Col1a1 ISH. (I) Ctsk ISH. (J) Visualization of the vascular network by CD31 (Pecam1) immunostaining. Arrowheads point to the chondro-osseous border. The dashed line indicates the beginning of the diaphysis (DP). (K) qPCR analysis of P0 Ctnnb1LOFHTC relative to control long bone material (n=3). *P<0.05, **P<0.01. Genotype of control in A-K is Ctnnb1fl/+;Col10a1-Cre−. (L) qPCR analysis using E16.5 YFP sorted material from Ctnnb1LOFHTC;RosaYFP/+ long bones relative to material isolated from E16.5 Col10a1-Cre+;RosaYFP/+ limbs (n=2). Error bars indicate s.d.
Histology and marker expression in E18.5 and postnatal day (P) 0 humeri was similar to that at E16.5, showing reduced trabeculation, reduction in Col1a1-positive structures and an increase in cells positive for osteoclast markers at the chondro-osseous front (Fig. 1G-I, Fig. S1A-F). Mmp9-positive osteoclasts/chondroclasts were also increased in number at the chondro-osseous front (Fig. S1G). Staining for the prehypertrophic marker Indian hedgehog (Ihh) appeared to be slightly more intense (Fig. S1D). CD31 (also known as Pecam1) staining for endothelial cells revealed no apparent differences in vascularization close to the chondro-osseous front, but hypervascularity was apparent in the diaphysis (Fig. 1J). Vegf (Vegfa) expression in mature HTCs appeared to be similar to the control at E18.5 (Fig. S1C). Quantitative PCR (qPCR) analysis at P0 (Fig. 1K) and E16.5 (Fig. 1L) revealed that Rankl transcript levels were increased in Ctnnb1LOFHTC mutants compared with controls at both time points, as were Mmp13 and Opn transcript levels (Fig. 1K,L). Surprisingly, Vegf and Opg levels were both increased at E16.5, but not significantly altered at P0 (Fig. 1K,L). Ihh expression showed a slight, but not statistically significant, increase at P0 (Fig. 1K).
In summary, inactivation of β-catenin in HTCs results in an increase in osteoclast number, loss of mineralized structures and increased expression of Mmp13, Opn and Rankl – factors that promote osteoclastogenesis.
Stabilization of β-catenin in HTCs results in an expansion of the mineralized hypertrophic zone
Conditional stabilization of β-catenin in HTCs using Col10a1-Cre (Ctnnb1ex3fl/+;Col10a1-Cre+, hereafter Ctnnb1GOFHTC) revealed a massive expansion of the two mineralized HTC zones in E16.5 humeri, which remained connected (Fig. 2A). In the mutant, high levels of β-catenin were detected in all HTCs throughout the expanded zone (Fig. 2B). At the molecular level, the two Col10a1 expression domains were expanded in the mutant, but did not connect (Fig. 2C). In the centralmost region, where the cells still appeared to be chondrogenic in nature based on their rounded morphology and Alcian Blue-positive matrix, only a few cells expressed Col10a1 (Fig. 2C). These rounded, chondrocyte-like cells expressed Col1a1, which is not normally expressed by HTCs but rather by osteoblast precursors and mature osteoblasts, as seen in the control (Fig. 2D). The expression of Col10a1 and Col1a1 in the expanded HTC zone in the mutants was nearly mutually exclusive (Fig. S2A,C). These Col1a1-expressing chondrocyte-like cells also expressed the osteoblast marker parathyroid hormone 1 receptor (Ppr; also known as Pth1r) (Fig. S2B), but not the more mature osteoblast marker osteocalcin (also known as Bglap) (data not shown). Sox9 protein persisted in the expanded hypertrophic zone of the mutants (Fig. 2E). In the Ctnnb1GOFHTC mutants, osteoclasts positive for Ctsk and Mmp9 were restricted to the bone collar region and reduced in number (Fig. 2F; data not shown). Accordingly, blood vessel invasion visualized by CD31 staining was compromised in Ctnnb1GOFHTC mutants (Fig. 2G). Cells in the central region stained intensely for Opg (Fig. 2H). Analysis of histology and molecular markers in E18.5 mutant humeri revealed very similar results to E16.5 (Fig. 2I-P; data not shown). The Col10a1 and Ihh domains were expanded (Fig. 2J,K). Interestingly, late HTC markers such as Opn and Mmp13 were not expressed in the expanded hypertrophic zone (Fig. 2L,M). Staining for the aggrecan neoepitope DIPEN (Fosang et al., 1996), which was absent in the expanded region, together with a concurrent expansion of the collagen type II-positive region (Fig. 2N,O), confirmed the lack of Mmp13 in the central core. DIPEN-positive cells were only present at the edges of the expanded HTC zone and the two surrounding rows of cells, which by morphology appeared chondrocyte-like (Fig. 2N). Blood vessel invasion had proceeded but still appeared different to the control (Fig. 2P).
Fig. 2.

Phenotypic analysis of E16.5 and E18.5 Ctnnb1GOFHTC mutants. (A-H) Representative images of alternating sections through humeri from E16.5 Ctnnb1GOFHTC and control (Ctnnb1ex3fl/+;Col10a1-Cre−) littermates. (A) Alcian Blue/von Kossa staining. (B) β-catenin immunohistochemical staining. (C) Col10a1 ISH. (D) Col1a1 ISH showing the presence of chondrocyte-like, Col1a1-positive cells (red asterisk). (E) Sox9 immunohistochemical staining. (F) Ctsk ISH for osteoclasts. (G) CD31 immunohistochemical staining for blood vessels. (H) Opg immunohistochemical staining. (I-P) Representative images of E18.5 humerus sections of Ctnnb1GOFHTC and control (Ctnnb1ex3fl/+;Col10a1-Cre−) littermates. (I) Histological changes visualized by Alcian Blue/von Kossa staining. (J-M) ISH for Col10a1 (J), Ihh (K), Opn (L) and Mmp13 (M). (N) Immunohistochemical staining for the aggrecan neoepitope DIPEN, counterstained with Methyl Green. (O) Immunohistochemical staining for type II collagen (Col II). (P) Immunohistochemical staining for CD31. (Q) qPCR analysis of P0 Ctnnb1GOFHTC relative to control long bone material (n=3) ±s.d. *P<0.05, **P<0.01. (R) qPCR analysis using E16.5 YFP sorted material from Ctnnb1GOFHTC;RosaYFP/+ long bones relative to material isolated from E16.5 Col10a1-Cre+;RosaYFP/+ limbs (n=2).
qPCR using mRNA from P0 skeletal elements (Fig. 2Q) and E16.5 sorted chondrocytes (Fig. 2R) revealed that, upon stabilization of β-catenin, Mmp13, Opn and Rankl levels were decreased, whereas Ihh and Sox9 levels were slightly increased, as were Opg levels but, to our surprise, not all cells that produced stabilized β-catenin produced Opg (Fig. 2H). Although the overall phenotype of Ctnnb1GOFHTC mutants resembled that of transgenic animals expressing Sox9 under the control of the Col10a1 promoter (Hattori et al., 2010), we did not observe downregulation of Vegf; instead, the levels of anti-angiogenic factors such as chondromodulin 1 (ChM-I; also known as Lect1) and tissue inhibitor of metalloproteinase 1-3 (Timp1-3), were increased (Fig. 2Q). In addition to their function as metalloproteinase inhibitors, Timp2 and Timp3 can directly inhibit VEGF signaling (Qi et al., 2003; Seo et al., 2003). The upregulation of these anti-angiogenic factors might contribute to the failure of blood vessels to invade the hypertrophic region.
We next asked whether the mineralized HTCs would be removed after birth. In the long bones of P3 mutant pups the mineralized hypertrophic zones of the proximal and distal growth plates were still expanded and connected (Fig. 3A). The mutant long bones were slightly shorter. This shortening was more pronounced in P28 mice, which overall were smaller than their littermate controls (Ctnnb1ex3fl/+; Col10a1-Cre−) (Fig. S3A,B). MicroCT-analysis and histological examination of 4-week-old humeri and femora revealed a locally restricted osteopetrotic phenotype in Ctnnb1GOFHTC compared with littermate controls (Fig. 3B,C, Fig. S3C). Closer examination uncovered differences between distal and proximal regions of the skeletal elements: in the humerus, bone marrow space was present proximally but almost completely replaced by abnormally mineralized tissue distally (Fig. 3B,C). In the femur this pattern was reversed (Fig. S3C). Interestingly, bone remodeling and the formation of bone marrow space was always associated with the side of the growth plate that has been described in humans to be the more active, e.g. the proximal humeral and the distal femoral growth plate (Fig. 3B,C, Fig. S3C; data not shown) (Pritchett, 1991, 1992). The mineralized HTC zone was increased in the proximal growth plates of humeri and femora and the distal femoral growth plate (Fig. 3C, Fig. S3C). Furthermore, in the proximal humeri and distal femora, trabecular bone volume with respect to total volume (BV/TV) and trabecular number were increased and, accordingly, trabecular spacing was decreased, while trabecular thickness was unaltered (Fig. 3D, Fig. S3D).
Fig. 3.
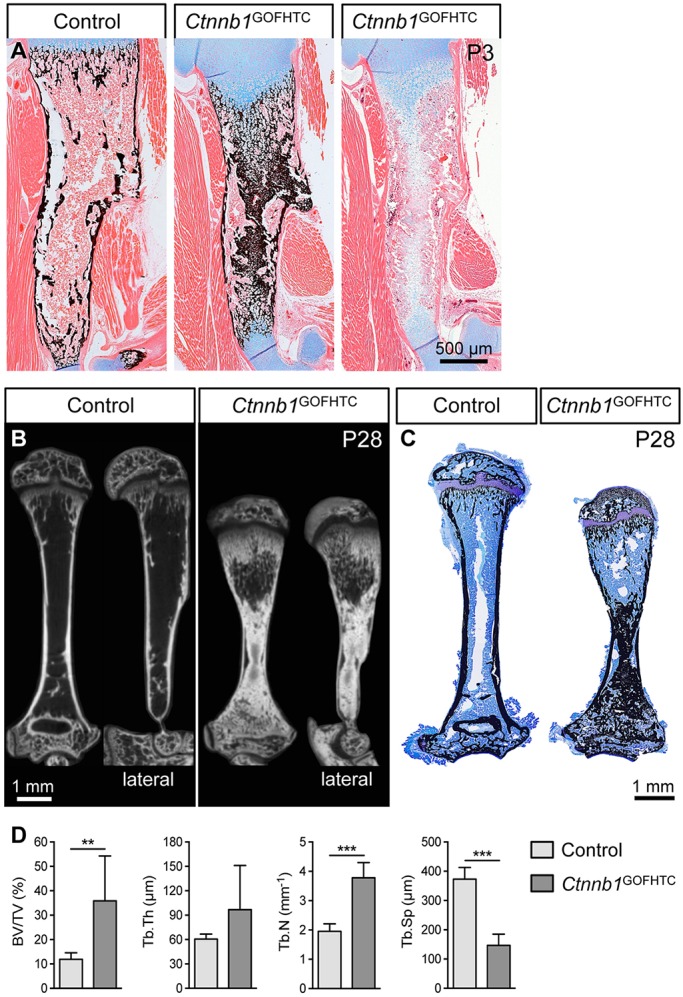
Postnatal phenotypic analysis of Ctnnb1GOFHTC mutants. (A) Representative images of humeri from P3 control and Ctnnb1GOFHTC littermates stained with Alcian Blue/von Kossa or Alcian Blue. (B) MicroCT images of control and Ctnnb1GOFHTC humeri (P28, frontal and lateral view). The lateral view reveals anterior-posterior differences in the trabecular density underneath the proximal growth plate in the mutant humerus. (C) Toluidine Blue/von Kossa-stained sections of control and Ctnnb1GOFHTC P28 humeri. Orientation is proximal up and distal down. (D) Histomorphometric quantification of the bone volume (BV) to total volume (TV) ratio, trabecular thickness (Tb.Th), trabecular number (Tb.N) and trabecular spacing (Tb.Sp) of P28 proximal humeri comparing Ctnnb1GOFHTC (n=8) and littermate controls (n=9). **P<0.01, ***P<0.001. Error bars indicate s.d. Genotype of control is Ctnnb1ex3fl/+;Col10a1-Cre−.
In summary, with the exception of the active growth plates in juveniles, stabilization of β-catenin in HTCs interferes with the removal of mineralized HTCs and blood vessel invasion. This is associated with the downregulation of pro-osteoclastic factors, a reduced number of osteoclasts and the upregulation of anti-angiogenic factors.
Genetic alteration of osteoclastogenesis partially rescues phenotypic changes caused by loss or stabilization of β-catenin in late HTCs
Conditional loss or stabilization of β-catenin in late HTCs led to an alteration in expression of osteoclastogenesis modulating factors, such as Opg and Rankl, and as a result altered the location and/or number of osteoclasts. Using a genetic approach we examined whether local downregulation or increased activation of osteoclastogenesis can rescue the phenotypic changes that occur upon conditional loss or stabilization of β-catenin. Examination of E19.5 Ctnnb1GOFHTC;Opg−/− double-mutant humeri (Ctnnb1ex3fl/+;Col10a1-Cre+;Opg−/−) revealed a partially rescued phenotype in all specimens examined (5/5) (Fig. 4). Bone marrow space was restored but trabecular bone formation was still abnormal and the mineralized HTC zones were still enlarged (Fig. 4A,B). Col1a1 and Mmp13 staining revealed the presence of osteoblasts and Ctsk staining the presence of osteoclasts in the bone marrow of Ctnnb1GOFHTC;Opg−/− humeri (Fig. 4C-E). Unlike in the Ctnnb1LOFHTC mutants, no pronounced reduction of trabeculae was observed in Opg mutants at P0 (Fig. S4A). According to the Col10a1 and Ctsk staining, the hypertrophic zone appeared normal and osteoclast numbers appeared not to be substantially increased (Fig. S4A).
Fig. 4.
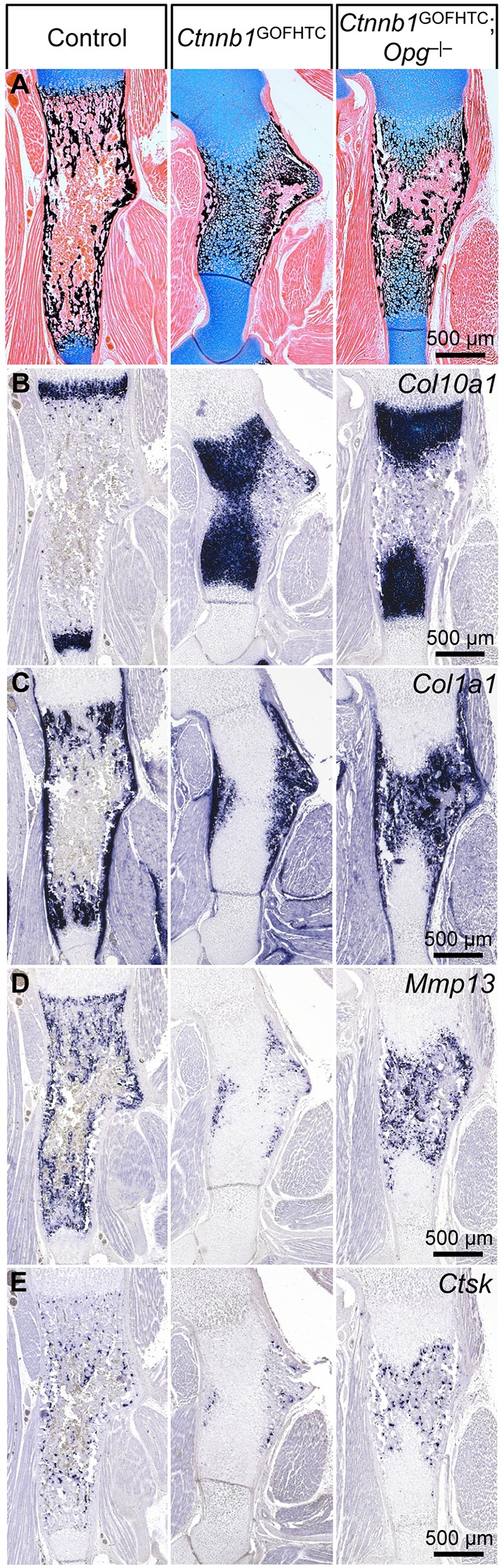
Partial rescue of the Ctnnb1GOFHTC phenotype by the loss of Opg. (A-E) Representative images of sections through E19.5 humeri of control (Ctnnb1ex3fl/+;Col10a1-Cre−), Ctnnb1GOFHTC and Ctnnb1GOFHTC;Opg−/− specimens. (A) Histological changes visualized by Alcian Blue/von Kossa staining showing remodeling of the extended zone of mineralized HTCs in the Ctnnb1GOFHTC upon loss of Opg. (B-E) ISH for Col10a1 (B), Col1a1 (C), Mmp13 (D) and Ctsk (E).
Given our hypothesis that β-catenin activity represses Rankl expression in HTCs, we decided to downregulate Rankl specifically in HTCs in the Ctnnb1 mutant background. Control, Ctnnb1LOFHTC and conditional Ctnnb1 and Rankl double mutants (Ctnnb1lacZ/fl;Ranklfl/fl;Col10a1-Cre+, hereafter Ctnnb1LOFHTC;RanklΔHTC) were examined by microCT (see Fig. S5A). BV/TV measurements revealed that trabecular bone formation was restored to a variable degree in the Ctnnb1LOFHTC;RanklΔHTC specimens and that this restoration was statistically significant (Fig. 5A). Based on the microCT data, the eight Ctnnb1LOFHTC;RanklΔHTC specimens fell into two groups: four with an almost ‘complete’ phenotypic rescue and four with a very ‘local rescue effect’ restricted to the chondro-osseous front (Fig. 5A, Fig. S5A). The average BV/TV ratio for the four ‘complete’ rescued specimens increased to 39.56±2.40% as compared with ∼18% in the Ctnnb1LOFHTC mutants, but was still at the low end of the range of controls with a BV/TV ratio of 45.90±4.51% (Fig. S5B). Histological and osteoblast marker analysis confirmed the variability of the rescue effect (Fig. 5B-F; data not shown). In ‘complete’ Ctnnb1LOFHTC;RanklΔHTC mutants increased trabeculae numbers and an increase in Col1a1-positive cells and trabeculae-associated cells positive for osterix (Osx; also known as Sp7) were observed (Fig. 5B-D). Local modulation of Rankl levels did not alter the vascular pattern (Fig. 5E). However, local loss of Rankl in the Ctnnb1LOFHTC;RanklΔHTC specimens decreased the overall osteoclast number compared with Ctnnb1LOFHTC littermates (Fig. S5C). The BV/TV ratio in RanklΔHTC (Ranklfl/fl;Col10a1-Cre+) newborns was increased to 61.16±5.89% (Fig. 5A, Fig. S5A,B), but no obvious decrease in Ctsk-stained cells was observed (Fig. S5C). In contrast to previous reports in adult mice (Nakashima et al., 2011; Xiong et al., 2011), the width of the hypertrophic zone was not altered upon deletion of Rankl in HTCs (Fig. 5F, Fig. S5C). In mutants lacking the receptor Rank (also known as Tnfrsf11a), no osteoclasts are present (Fig. S4B; data not shown), while some Mmp9-positive cells can still be detected primarily at the chondro-osseous border (Fig. S4B). Here, the Col10a1 staining revealed a noticeable enlargement of the HTC zone (Fig. S4B). Nevertheless, in contrast to the Ctnnb1GOFHTC mutants, remodeling of the hypertrophic zone was only slightly affected and bone marrow cavity formation was normal (compare Fig. S4B with Fig. 2I,J).
Fig. 5.
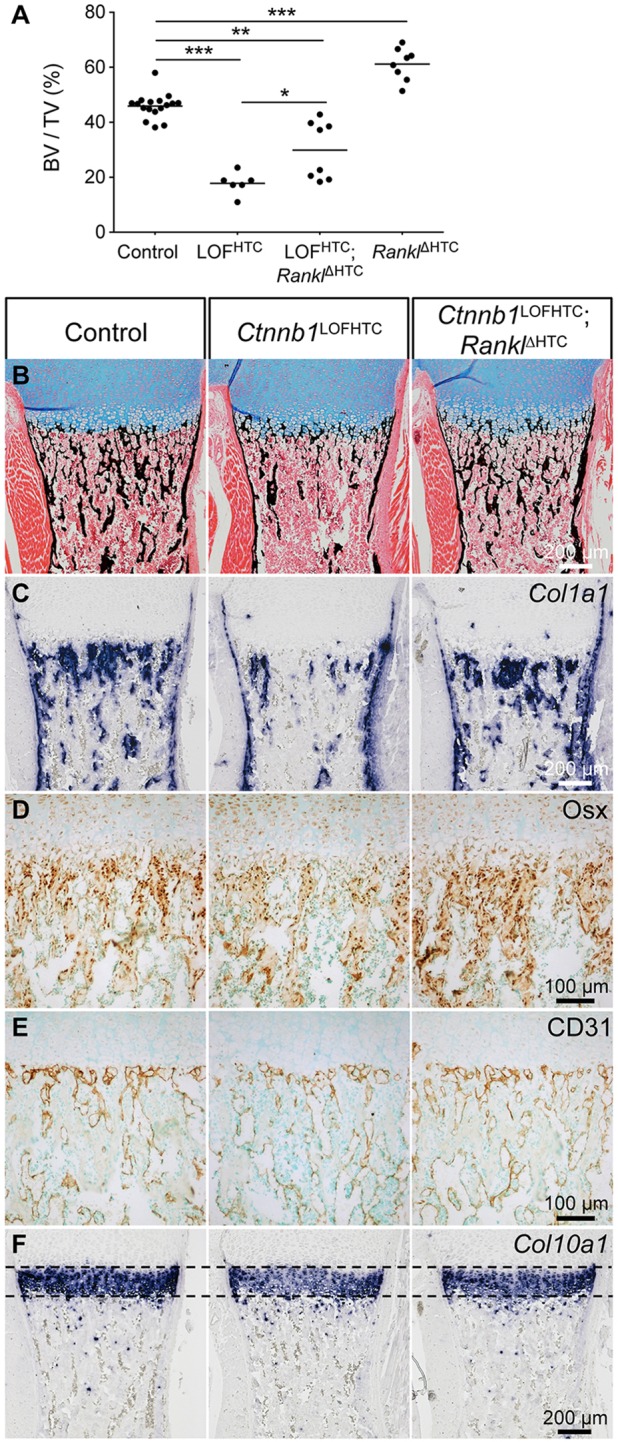
Partial reversal of the Ctnnb1LOFHTC phenotype by conditional deletion of Rankl in HTCs. (A) Scatter plot showing the distribution (and mean) of the bone volume (BV) to total volume (TV) (%) of the different controls (n=17), Ctnnb1LOFHTC (n=6), Ctnnb1LOFHTC;RanklΔHTC (Ctnnb1lacZ/fl;Ranklfl/fl;Col10a1-Cre+) (n=8) and RanklΔHTC (Ranklfl/fl;Col10a1-Cre+) (n=8) specimens. *P<0.05, **P<0.01, ***P<0.001. (B-F) Representative images of sections through control, Ctnnb1LOFHTC and ‘complete’ rescue Ctnnb1LOFHTC;RanklΔHTC humeri. (B) Alcian Blue/von Kossa staining. (C) Col1a1 ISH. (D) Osx immunohistochemical staining. (E) CD31 immunohistochemical staining. (F) Col10a1 ISH.
Taken together, genetic modulation of osteoclastogenesis is able to partially revert the β-catenin loss- and gain-of-function phenotypes, demonstrating that the alterations in osteoclast numbers are in part responsible for the phenotypic changes.
β-catenin activity in late HTCs is required for the differentiation of chondrocyte-derived osteoblasts
Given that the phenotypic rescue achieved by deleting Rankl in HTCs in the Ctnnb1LOFHTC mutant background was not 100%, and that β-catenin plays a role in periosteal osteoblast differentiation (Day et al., 2005; Hill et al., 2005), as well as in the light of recent publications demonstrating that HTC descendants can differentiate into osteoblasts (Yang et al., 2014a,b; Zhou et al., 2014), we addressed the question of whether chondrocyte-derived osteoblastogenesis is altered in Ctnnb1LOFHTC mutants. We examined Ctnnb1LOFHTC;RosaYFP/+ humeri and determined the percentage of all osteoblasts [Osx+(total)], all chondrocyte-derived cells [YFP+(total)], of chondrocyte-derived (Osx+ YFP+) and perichondrial-derived (Osx+ YFP−) osteoblasts, as well as the osteogenic (YFP+ Osx+) and non-osteogenic (YFP+ Osx−) subpopulations of YFP+ cells in the subchondral growth plate region compared with littermate controls (Fig. 6).
Fig. 6.

Chondrocyte-derived osteoblastogenesis is affected by the loss of Ctnnb1 from HTCs. (A) Representative images of control and Ctnnb1LOFHTC P0 specimens taken from the region below the chondro-osseous front. Immunofluorescent staining for Osx (red) and YFP (green), and DAPI staining for nuclei (blue). The dashed line indicates the position of the chondro-osseous border. The increase in DAPI-stained cells in the Ctnnb1LOFHTC mutant reflects an increase in cellularity. Higher magnification images of the boxed regions with only the Osx (red) and YFP (green) signals are provided on the right, with double-positive cells marked by arrows. (B) Mean percentage of Osx+(total) and YFP+(total) cells relative to control. (C) The distribution of Osx+ YFP− (perichondrial-derived) and Osx+ YFP+ (chondrocyte-derived) cells normalized with respect to the Osx+(total) population (100%) of the control. (D) The distribution of YFP+ Osx+ (osteoblastic) and YFP+ Osx− (non-osteoblastic) cells normalized with respect to the YFP+(total) population (100%) of the control. (E) The mean percentage of Osx+(total) and Runx2+(total) cells relative to control. (F) The distribution of Runx2+ YFP− (perichondrial-derived) and Runx2+ YFP+ (chondrocyte-derived) cells normalized with respect to the Runx2+(total) population (100%) of the control. (G) The distribution of YFP+ Runx2+ (osteoblastic) and YFP+ Runx2− (non-osteoblastic) cells normalized with respect to the YFP+(total) population (100%) of the control. Cells for B-D were counted within the region indicated by the thin white line in A, and for E-G in a square of the same size on three or four sections per specimen and genotype. Control is Ctnnb1fl/+;Col10a1-Cre+;RosaYFP/+; Ctnnb1LOFHTC is Ctnnb1lacZ/fl;Col10a1-Cre +;RosaYFP/+. (B,E) **P<0.01, ***P<0.001. The number of specimens analyzed (n) is indicated within the bars. Error bars indicate s.d.
The overall cellularity (DAPI+ cells) was increased in the Ctnnb1LOFHTC mutant bone marrow (Fig. 6A). The Osx+(total) and YFP+(total) populations were both reduced by ∼50% compared with controls (Fig. 6B). The perichondrial-derived Osx+ YFP− population decreased by 18%, and the chondrocyte-derived Osx+ YFP+ population dropped from 34% in the controls to 2% in Ctnnb1LOFHTC mutants (Fig. 6C). By contrast, the non-osteoblastic YFP+ Osx− population increased by 18%, whereas only 4% of all YFP+ cells differentiated towards the osteoblastic lineage (YFP+ Osx+) in the mutant (Fig. 6D).
These findings were corroborated by the analysis of Runx2 (also known as Cbfa1), a second osteoblastic marker, which acts upstream of Osx in osteoblastogenesis (Nakashima et al., 2002). Compared with the control, the Runx2+(total) population dropped by 44% (Fig. 6E), the chondrocyte-derived Runx2+ YFP+ population from 25% to 6%, and the perichondrial-derived Runx2+ YFP− population by 24% in the Ctnnb1LOFHTC mutants (Fig. 6F). Interestingly, unlike the non-osteogenic YFP+ Osx− population, the YFP+ Runx2− population did not increase and the osteogenic YFP+ Runx2+ subpopulation did not decrease to the same extent as the YFP+ Osx+ population (Fig. 6D,G). The fact that osteoblastic chondrocyte-derived bone marrow cell numbers decreased dramatically in the long bones lacking β-catenin activity in late HTCs suggests that β-catenin is required for the differentiation of chondrocyte-derived osteoblasts and that the remaining osteoblasts are essentially all periosteal derived.
Next, we asked whether this decrease in chondrocyte-derived osteoblast number is due to the fact that the precursor cells upregulated Sox9, similar to what is observed in perichondrial-derived osteoblasts that lack β-catenin (Hill et al., 2005). Sox9 protein expression and levels were similar to the control (Fig. S6). Furthermore, no increase in TUNEL-positive or cleaved caspase 3-positive cell numbers was observed in the growth plate or at the chondro-osseous front in Ctnnb1LOFHTC mutants (Fig. S6; data not shown). According to Ki67 staining, proliferation in the growth plate or of YFP+ cells was not altered (Fig. S6).
We then investigated whether the increased number of non-osteoblastic YFP+ Osx− cells is due to a cell lineage shift into adipocytes. Adipocytes were increased in number in 4-month-old Ctnnb1LOFHTC mutant tibiae (Fig. S7A). Similarly, the overall number of fatty acid-binding protein 4 (FABP4)-positive cells was increased in newborn Ctnnb1LOFHTC limbs compared with controls (Fig. S7B). However, none of the FABP4+ cells, located in close proximity to blood vessels, stained positively for YFP (Fig. S7B-D). Thus, we conclude that the increase in adipocyte number is not caused by a lineage shift of chondrocyte-derived osteoblast precursors.
Increased β-catenin activity promotes transdifferentiation of chondrocyte-derived osteoblasts
Finally, we analyzed whether increased β-catenin activity in late HTCs affects the number of chondrocyte-derived osteoblasts. Counting the YFP+ as well as the YFP+ Osx+ cells in an area spanning 250 µm below the chondro-osseous border (Fig. 7A) at the proximal growth plates of P28 humeri revealed a significant increase in the total number of YFP+ cells and in the number of YFP+ Osx+ cells by ∼70% each compared with the controls (Fig. 7C). At the growth plates undergoing active remodeling in humeri and femora, TRAP+ osteoclasts were detected but, compared with the control, the number of osteoclasts lining the cartilage erosion zone was decreased by ∼35% (Fig. 7B,D). Thus, the combination of increased chondrocyte-derived osteoblast precursor formation and decreased osteoclastogenesis observed at the growth plate undergoing remodeling is probably responsible for the increased BV/TV in the P28 Ctnnb1GOFHTC limbs (see Fig. 3D, Fig. S3D).
Fig. 7.

Alteration in chondrocyte-derived osteoblast and osteoclast numbers in P28 Ctnnb1GOFHTC specimens. (A) Representative images of control and Ctnnb1GOFHTC specimens below the chondro-osseous front at the proximal humeral growth plate. Immunofluorescent staining for Osx (red) and YFP (green), and DAPI staining for nuclei (blue). The dashed line indicates the position of the chondro-osseous border. (B) TRAP staining on control and Ctnnb1GOFHTC littermates. Asterisks mark the osteoclasts at the chondro-osseous border. (C) The mean percentage of YFP+(total) (which are progeny of Col10a1-Cre-expressing HTCs) and YFP+ Osx+ (the chondrocyte-derived osteogenic population) cells determined within the area 250 µm below the chondro-osseous front. (D) The mean number of osteoclasts lining the chondro-osseous front (marked by asterisks in B) in humerus and femur of control and Ctnnb1GOFHTC specimens. Control is Col10a1-Cre+;RosaYFP/+; Ctnnb1GOFHTC is Ctnnb1ex3fl/+;Col10a1-Cre+;RosaYFP/+. (C,D) **P<0.01, ***P<0.001. The number of samples analyzed (n) is indicated within the bars. Error bars indicate s.d.
DISCUSSION
During the late phase of fetal development, remarkable remodeling processes occur in long bones – resorption of hypertrophic calcified cartilage, bone marrow cavity and trabecular bone formation. Our data show that the severe reduction of trabecular bone in mutants lacking β-catenin activity in late HTCs is the result of a combinatorial mechanism: decreased chondrocyte-derived osteoblast differentiation and increased recruitment of osteoclasts to the chondro-osseous front. The latter is due to local dysregulation of the Rankl:Opg ratio, and probably further supported by Opn and Mmp13 upregulation, as molecules potentiating osteoclast function (Franzen et al., 2008; Pivetta et al., 2011; Ross et al., 1993). Our results concerning Rankl regulation are consistent with previous findings (Golovchenko et al., 2013; Wang et al., 2014) and resemble the situation in osteoblasts (Holmen et al., 2005; Kramer et al., 2010; Sato et al., 2009; Spencer et al., 2006). Yet, the molecular mechanisms may differ between the cell types, being direct in osteoblasts (Spencer et al., 2006) and potentially indirect in chondrocytes via interference with a glucocorticoid receptor (GR)-dependent mechanism (Wang et al., 2014). However, the latter is questioned by the fact that the cartilage-specific GR (Nr3c1) knockout has no apparent growth plate or bone phenotype (Tu et al., 2014). Wang and colleagues used Col2a1-CreER in their study to inactivate β-catenin. This Cre line is also active in mesenchymal precursors of perichondrium-derived osteoblasts (Ono et al., 2014), which also require β-catenin. Hence, some of the phenotypic changes might be due to altered β-catenin activity in perichondrial-derived osteoblasts (Wang et al., 2014). In contrast to Wang and colleagues, we did not observe a decrease in height upon Ctnnb1 loss or cellular disorganization upon β-catenin stabilization, suggesting that these changes correlate with β-catenin functions in Col2a1-expressing chondrocytes (Wang et al., 2014). Our hypothesis that increased Rankl expression in HTCs upon Ctnnb1 deletion contributes substantially to the Ctnnb1LOFHTC phenotype is supported by the reversal of the phenotype upon conditional removal of Rankl from HTCs in 50% of the specimens. The incomplete penetrance is likely to be explained by the fact that two Rankl alleles need to be recombined and Cre recombination efficiency at the cellular level varies between specimens (Nagy, 2000). Even in the four ‘complete’ Ctnnb1GOFHTC;RanklΔHTC specimens the average BV/TV was at the lower end of the control range. This indicates that Rankl dysregulation is not the sole cause of the phenotype.
The 17-fold decrease in the number of osteoblasts derived by chondrocyte transdifferentiation together with the 18% decrease in perichondrial-derived Osx+ YFP− precursor number in Ctnnb1LOFHTC mutants are likely to contribute to the slightly lower BV/TV ratio of the ‘complete’ Ctnnb1GOFHTC;RanklΔHTC specimens. Perichondrial-derived osteoblast precursors migrate along blood vessels into the bone marrow cavity (Maes et al., 2010). In the Ctnnb1LOFHTC mutants blood vessels reach the chondro-osseous border and there is even hypervascularity, which is noticeable primarily in the diaphysis. The latter might be due to the transient increase in Vegf observed at E16.5, or the liberation of matrix-bound VEGF due to increased osteoclastic activity at the chondro-osseous front (Bergers et al., 2000; Huang et al., 2002). Thus, perichondrial-derived osteoblast precursors should be able to migrate normally, but due either to the lack of mineralized matrix, the remnants of late HTCs, or to other environmental changes, not all of them may find the right environment to survive or to differentiate along the osteoblastic lineage. While both osteoblastic populations decreased, the number of non-osteoblastic, chondrocyte-derived bone marrow cells increased by ∼18% in the Ctnnb1LOFHTC mutants. The exact cellular nature of the YFP+ Osx− population observed here and in previous lineage-tracing transdifferentiation studies has not yet been clarified (Park et al., 2015; Yang et al., 2014a,b; Zhou et al., 2014). Some of the YFP+ Osx− cells in the Ctnnb1LOFHTC mutants apparently remained in a more undifferentiated state, being positive for Runx2. Given that Wnt/β-catenin signaling plays an important role in the osteoblastogenesis versus adipogenesis lineage decision process of bone marrow cells (Bennett et al., 2005; Gambardella et al., 2011; Krishnan et al., 2006; Song et al., 2012), that some YFP+ Osx− cells differentiated to adipocytes presented an intriguing possibility. However, the observed increase in adipocyte and adipocyte progenitor numbers in Ctnnb1LOFHTC mutants was not due to a lineage switch of chondrocyte-derived osteoprogenitors. Instead, our data suggest that the FABP4+ progenitors originate from perichondrium-derived osteoprogenitors, explaining in part their reduction. Osteoblast precursors in the perichondrium that lack β-catenin activity differentiate into chondrocytes and express Sox9 (Hill et al., 2005; Rodda and McMahon, 2006). Yet, the mechanism that blocks Ctnnb1-deficient cells from undergoing transdifferentiation appears to be distinct, as no alteration in Sox9 was observed. Neither altered apoptosis nor proliferation appears to cause the decrease in chondrocyte-derived osteoblastogenesis. Thus, the mechanism by which β-catenin controls this process is currently unclear.
In osteoblasts, Opg has been reported to be a direct, positively regulated β-catenin/Tcf target (Glass et al., 2005; Sato et al., 2009). However, this seems not to be the case in HTCs. In Ctnnb1GOFHTC specimens, Opg transcript levels were not increased and only the most centrally located cells, which also expressed osteoblast markers, stained positively for Opg. Hence, β-catenin might only be able to regulate Opg in osteoblastic cells. Nevertheless, genetic loss of Opg reverted the Ctnnb1GOFHTC phenotype to a certain extent, but only very late in embryonic development. Increased systemic Rankl levels in Opg mutants (Bennett et al., 2006) are likely to be responsible for the increase in osteoclast numbers. Yet, trabecular bone formation was abnormal and the mineralized HTC domains were still enlarged. The reason for the latter is probably multifactorial: despite Opg loss, Rankl expression by HTCs is probably still decreased and hence local osteoclastogenesis may not occur at the same rate as in wild type. This might contribute to the persistence of the enlarged mineralized hypertrophic zone in the Ctnnb1LOFHTC;Opg−/− animals, as the hypertrophic zone is also increased in mutants with compromised osteoclastogenesis. In addition, Opn and Mmp13 expression in late HTCs was still altered. As Mmp13 mutants also display a widening of the HTC zone, the lack of Mmp13 expression in late HTCs possibly contributes to the shortcomings of the phenotypic reversal (Inada et al., 2004).
Chondrocyte remodeling was absent in Ctnnb1GOFHTC mice and late hypertrophic markers were not expressed, whereas the domains of immature markers such as Ihh and Sox9 were expanded. These data indicate that stabilization of β-catenin in HTCs, among others, delays terminal HTC differentiation. However, a dramatic expansion of mineralized HTCs was observed. Here, the centralmost chondrocytes started to express osteoblastic markers such as Col1a1 and Ppr and produced Opg. We interpret this as an attempt by the cells to undergo osteoblastic transdifferentiation. Yet, these cells did not express osteocalcin, a marker of mature osteoblasts. Similar to the situation in periosteal osteoblast precursors, their attempt to mature might be stalled at an intermediate stage due to the high levels of stabilized β-catenin (Rodda and McMahon, 2006).
Although no true bone marrow cavity was formed in Ctnnb1GOFHTC skeletal elements up to age P3, partial bone marrow cavity formation had occurred in 4-week-old mice. Interestingly, its occurrence was always associated with the more active growth plate (Farnum, 1994; Pritchett, 1991, 1992). In all postnatal growth plates, the number of new chondrocytes produced was equal to the HTCs lost, but in the more active growth plates these numbers were 2- to 3-fold higher (Wilsman et al., 1996). The factors responsible for this phenomenon are not yet known. Nevertheless, given that more chondrocytes are produced and turned over in the same amount of time in active growth plates, this means that chondrocytes spend less time in the hypertrophic zone (Wilsman et al., 1996). Consequently, Col10a1-Cre efficacy may decrease as the activity window becomes narrower and, as such, fewer cells recombine the Ctnnb1 exon 3 floxed allele. As a result, Rankl would not be repressed in all HTCs and could support local osteoclastogenesis. Hence, bone marrow formation can reoccur and, due to increased chondrocyte transdifferentiation, trabecular bone formation is enhanced at these growth plates.
In conclusion, we have shown that in late HTCs β-catenin has dual functions in trabecular bone formation: first, it locally regulates osteoclastogenesis by repressing the expression of the pro-osteoclastic factor Rankl, and second it is involved in the newly discovered transdifferentiation process by which HTCs give rise to osteoblast precursors (Fig. 8). As such, β-catenin is a key player in early trabecular bone formation during embryonic skeletal development.
Fig. 8.

Model summarizing the roles of β-catenin in late HTCs. β-catenin locally regulates the Rankl:Opg ratio controlling osteoclastogenesis and negatively influences the expression of Mmp13 and Opn. Inactivation of Ctnnb1 from late HTCs leads to an increased Rankl:Opg ratio, increased expression of Mmp13 and Opn and increased osteoclast numbers, but simultaneously affects the differentiation of osteoblasts derived from HTCs. Thus, this leads to a severe defect in trabecular bone formation. Stabilization of β-catenin results on the one hand in a decrease in the Rankl:Opg ratio and a subsequent reduction in osteoclast number and, on the other hand, affects the terminal differentiation of HTCs as reflected in the absence of Mmp13 and Opn and their overall reduced expression, whereas anti-angiogenic factors are increased in expression. Thus, endochondral bone formation is severely impaired upon stabilization of β-catenin in HTCs owing to impaired cartilage resorption and bone marrow formation. GP, growth plate; WT, wild type.
MATERIALS AND METHODS
Mouse husbandry
The following strains were used: Ctnnb1 floxed (Huelsken et al., 2001), Ctnnb1 lacZ knock-in null (Huelsken et al., 2000), Ctnnb1 exon 3 floxed (Harada et al., 1999), BAC-Col10a1-Cre (Col10a1-Cre, #1465 and #1421) (Gebhard et al., 2008), Opg (Tnfrsf11b) knockout (Mizuno et al., 1998), Rankl floxed (Xiong et al., 2011), Rank (Tnfrsf11a)Δ/Δ (Hanada et al., 2009), and Rosa26-flox-Stop-flox-EYFP (RosaYFP) (Srinivas et al., 2001). For breeding details see the supplementary Materials and Methods. For timed pregnancies, the plug date was considered to be E0.5. Genotyping was performed using previously published PCR protocols. Animals were sacrificed by cervical dislocation, embryos and newborns by decapitation. All mouse experiments were performed in accordance with local, institutional and national regulations and licenses (84-02.05.20.12.261).
Microcomputed tomography (microCT) and histomorphometric analysis
Long bones were fixed for 24 h in 4% paraformaldehyde followed by two PBS washes and one wash in 70% ethanol for 24 h each. Prior to the scan, tissue was stored in 70% ethanol at 4°C and wrapped in Parafilm for the scan. Scans were performed using a SkyScan 1176 scanner (SkyScan, Kontich, Belgium). For scanning details see the supplementary Materials and Methods.
Processing of specimens
For a detailed description of specimen processing for histology, immunohistochemistry and immunofluorescence see the supplementary Materials and Methods.
Histology
Histological stainings, including Alcian Blue, Alcian Blue with von Kossa, Hematoxylin and Eosin (H&E) and TRAP were performed on 5 µm paraffin sections as described in the supplementary Materials and Methods.
Immunohistochemistry and immunofluorescence
Staining for CD31, Opg, Osx, Sox9, β-catenin, collagen type II, DIPEN, Runx2, YFP, Ki67, FABP4, endomucin, cleaved caspase 3 and TUNEL was performed on deparaffinized and rehydrated 5 µm sections as described in the supplementary Materials and Methods.
Alkaline phosphatase-based and double fluorescent in situ hybridization
In situ hybridization using digoxigenin (DIG)-labeled RNA probes was performed according to Murtaugh et al. (1999). Double-fluorescent in situ hybridization on paraffin sections was performed using antisense biotin-labeled Col1a1 probe and DIG-labeled Col10a1 or Ppr probes according to Taschner et al. (2008). Probes for Col10a1, Col1a1, Ihh, Osc (Hill et al., 2005), Mmp13 (Yamagiwa et al., 1999), Ppr (Spater et al., 2006b), Opn, Mmp9, Vegfa (Golovchenko et al., 2013), Catk (Bozec et al., 2008) and Trap (Lubberts et al., 2002) have been published previously and are available upon request.
RNA isolation from FACS-sorted primary HTCs and total skeletal elements
RNA was isolated from dissociated whole P0 skeletal elements or from YFP-positive cells isolated from E16.5 skeletal elements using the RNeasy Kit (Qiagen) according to the manufacturer's instructions, including a DNase I treatment. RNA concentration was determined by OD. For details, see the supplementary Materials and Methods.
cDNA synthesis
For first-strand cDNA synthesis, 0.5-1 µg total RNA was reverse transcribed using oligo(dT) primers. For qPCR analysis, cDNA was diluted 1:10. For details see the supplementary Materials and Methods.
Real-time PCR analysis
First-strand cDNA (2-3 µl of 1:10 diluted) was mixed with either Fast Start SBG Master Mix (Roche, #04673484001) or SYBR Premix Ex Taq II (TaKaRa, #RR820Q). Gene expression was monitored using a Bio-Rad CFX96 cycler. For details see the supplementary Materials and Methods and Table S1 for primer sequences.
Image acquisition
Histological images were acquired using either a Zeiss AxioPlan2 equipped with a Leica DFC320 3.45 µm pixel color camera or a Zeiss AxioImager.M2 equipped with a Zeiss AxioCam MRc 6.45 µm color camera. Immunofluorescent images were acquired using an AxioImager.M2 equipped with an ApoTome.2 and an AxioCam MRm 6.45 µm monochromatic camera using Zen software (all Zeiss).
Statistical analysis
Statistical analysis was performed by two-tailed, unpaired Student's t-test using GraphPad Prism 6.0. Data are displayed as mean ±s.d.
Acknowledgements
We thank Charles O'Brien for the Rankl floxed, Josef Penninger for the RankΔ/+ and Opg+/−, Mark Taketo for the Ctnnb1 exon 3 floxed, Walter Birchmeier for the Ctnnb1 lacZ and exon 3-6 floxed mice. We thank Petra Köckemann and Christine Fabritius for technical assistance, Iska Leifert for helpful advice regarding immunohistochemistry, and Joanna Sherwood for checking the English in the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.H.; Formal analysis and investigations: A.H., D.K.-P., M.W., J.G., S.T.; Writing – original draft preparation: C.H.; Writing – reviewing and editing: C.H., A.H., K.v.d.M.; Funding acquisition: C.H.; Resources: K.v.d.M.; Supervision: C.H.
Funding
This work was supported by Boehringer Ingelheim funding the Institute of Molecular Pathology in Vienna and by a grant from the Deutsche Forschungsgemeinschaft [HA 4767/2-1]. S.T. is supported by a grant from the Interdisziplinäres Zentrum für Klinische Forschung, Westfälische Wilhelms-Universität Münster [Har2/002/14]. The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Deposited in PMC for immediate release.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.137489.supplemental
References
- Bennett C. N., Longo K. A., Wright W. S., Suva L. J., Lane T. F., Hankenson K. D. and MacDougald O. A. (2005). Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 102, 3324-3329. 10.1073/pnas.0408742102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. J., Scatena M., Kirk E. A., Rattazzi M., Varon R. M., Averill M., Schwartz S. M., Giachelli C. M. and Rosenfeld M. E. (2006). Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler. Thromb. Vasc. Biol. 26, 2117-2124. 10.1161/01.ATV.0000236428.91125.e6 [DOI] [PubMed] [Google Scholar]
- Bergers G., Brekken R., McMahon G., Vu T. H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z. et al. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737-744. 10.1038/35036374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P., Cancedda F. D., Riminucci M. and Cancedda R. (1998). Bone formation via cartilage models: the “borderline” chondrocyte. Matrix Biol. 17, 185-192. 10.1016/S0945-053X(98)90057-9 [DOI] [PubMed] [Google Scholar]
- Bozec A., Bakiri L., Hoebertz A., Eferl R., Schilling A. F., Komnenovic V., Scheuch H., Priemel M. Stewart C. L., Amling M. and Wagner E. F. (2008). Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature 454, 221-225. 10.1038/nature07019 [DOI] [PubMed] [Google Scholar]
- Chen M., Zhu M., Awad H., Li T. F., Sheu T. J., Boyce B. F., Chen D. and O'Keefe R. J. (2008). Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci. 121, 1455-1465. 10.1242/jcs.020362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C., Thompson Z., Miclau T., Werb Z. and Helms J. A. (2003). Altered fracture repair in the absence of MMP9. Development 130, 4123-4133. 10.1242/dev.00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao D. Y., Jonason J. H., Zhang Y., Hsu W., Chen D., Hilton M. J. and O'Keefe R. J. (2012). Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J. Bone Miner. Res. 27, 1680-1694. 10.1002/jbmr.1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day T. F., Guo X., Garrett-Beal L. and Yang Y. (2005). Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739-750. 10.1016/j.devcel.2005.03.016 [DOI] [PubMed] [Google Scholar]
- Engsig M. T., Chen Q.-J., Vu T. H., Pedersen A.-C., Therkidsen B., Lund L. R., Henriksen K., Lenhard T., Foged N. T., Werb Z. et al. (2000). Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 151, 879-889. 10.1083/jcb.151.4.879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnum C. E. (1994). Differential growth rates of long bones. In Bone: Mechanisms of Bone Development and Growth (ed. Hall B. K.), pp. 193-222. Boca Raton: CRC Press. [Google Scholar]
- Fosang A. J., Last K., Knäuper V., Murphy G. and Neame P. J. (1996). Degradation of cartilage aggrecan by collagenase-3 (MMP-13). FEBS Lett. 380, 17-20. 10.1016/0014-5793(95)01539-6 [DOI] [PubMed] [Google Scholar]
- Franzen A., Hultenby K., Reinholt F. P., Onnerfjord P. and Heinegard D. (2008). Altered osteoclast development and function in osteopontin deficient mice. J. Orthop. Res. 26, 721-728. 10.1002/jor.20544 [DOI] [PubMed] [Google Scholar]
- Gack S., Vallon R., Schmidt J., Grigoriadis A., Tuckermann J., Schenkel J., Weiher H., Wagner E. F. and Angel P. (1995). Expression of interstitial collagenase during skeletal development of the mouse is restricted to osteoblast-like cells and hypertrophic chondrocytes. Cell Growth Differ. 6, 759-767. [PubMed] [Google Scholar]
- Gambardella A., Nagaraju C. K., O'Shea P. J., Mohanty S. T., Kottam L., Pilling J., Sullivan M., Djerbi M., Koopmann W., Croucher P. I. et al. (2011). Glycogen synthase kinase-3alpha/beta inhibition promotes in vivo amplification of endogenous mesenchymal progenitors with osteogenic and adipogenic potential and their differentiation to the osteogenic lineage. J. Bone Miner. Res. 26, 811-821. 10.1002/jbmr.266 [DOI] [PubMed] [Google Scholar]
- Gebhard S., Hattori T., Bauer E., Schlund B., Bosl M. R., de Crombrugghe B. and von der Mark K. (2008). Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol. 27, 693-699. 10.1016/j.matbio.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H.-P. and Ferrara N. (2000). Angiogenesis and bone growth. Trends Cardiovasc. Med. 10, 223-228. 10.1016/S1050-1738(00)00074-8 [DOI] [PubMed] [Google Scholar]
- Gerber H.-P., Vu T. H., Ryan A. M., Kowalski J., Werb Z. and Ferrara N. (1999). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 5, 623-628. 10.1038/9467 [DOI] [PubMed] [Google Scholar]
- Glass D. A. II, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A. et al. (2005). Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751-764. 10.1016/j.devcel.2005.02.017 [DOI] [PubMed] [Google Scholar]
- Golovchenko S., Hattori T., Hartmann C., Gebhardt M., Gebhard S., Hess A., Pausch F., Schlund B. and von der Mark K. (2013). Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone 55, 102-112. 10.1016/j.bone.2013.03.019 [DOI] [PubMed] [Google Scholar]
- Guo X., Day T. F., Jiang X., Garrett-Beal L., Topol L. and Yang Y. (2004). Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 18, 2404-2417. 10.1101/gad.1230704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada R., Leibbrandt A., Hanada T., Kitaoka S., Furuyashiki T., Fujihara H., Trichereau J., Paolino M., Qadri F., Plehm R. et al. (2009). Central control of fever and female body temperature by RANKL/RANK. Nature 462, 505-509. 10.1038/nature08596 [DOI] [PubMed] [Google Scholar]
- Harada N., Tamai Y., Ishikawa T.-O., Sauer B., Takaku K., Oshima M. and Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931-5942. 10.1093/emboj/18.21.5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T., Muller C., Gebhard S., Bauer E., Pausch F., Schlund B., Bosl M. R., Hess A., Surmann-Schmitt C., von der Mark H. et al. (2010). SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137, 901-911. 10.1242/dev.045203 [DOI] [PubMed] [Google Scholar]
- Hill T. P., Spater D., Taketo M. M., Birchmeier W. and Hartmann C. (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727-738. 10.1016/j.devcel.2005.02.013 [DOI] [PubMed] [Google Scholar]
- Holmbeck K., Bianco P., Chrysovergis K., Yamada S. and Birkedal-Hansen H. (2003). MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J. Cell Biol. 163, 661-671. 10.1083/jcb.200307061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmen S. L., Zylstra C. R., Mukherjee A., Sigler R. E., Faugere M.-C., Bouxsein M. L., Deng L., Clemens T. L. and Williams B. O. (2005). Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 280, 21162-21168. 10.1074/jbc.M501900200 [DOI] [PubMed] [Google Scholar]
- Huang S., Van Arsdall M., Tedjarati S., McCarty M., Wu W., Langley R. and Fidler I. J. (2002). Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J. Natl. Cancer Inst. 94, 1134-1142. 10.1093/jnci/94.15.1134 [DOI] [PubMed] [Google Scholar]
- Huelsken J., Vogel R., Brinkmann V., Erdmann B., Birchmeier C. and Birchmeier W. (2000). Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 148, 567-578. 10.1083/jcb.148.3.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J., Vogel R., Erdmann B., Cotsarelis G. and Birchmeier W. (2001). beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105, 533-545. 10.1016/S0092-8674(01)00336-1 [DOI] [PubMed] [Google Scholar]
- Inada M., Wang Y., Byrne M. H., Rahman M. U., Miyaura C., Lopez-Otin C. and Krane S. M. (2004). Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. USA 101, 17192-17197. 10.1073/pnas.0407788101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto K., Kitazawa R., Kurosaka M., Maeda S. and Kitazawa S. (2006). Expression profile of genes related to osteoclastogenesis in mouse growth plate and articular cartilage. Histochem. Cell Biol. 125, 593-602. 10.1007/s00418-005-0103-z [DOI] [PubMed] [Google Scholar]
- Kramer I., Halleux C., Keller H., Pegurri M., Gooi J. H., Weber P. B., Feng J. Q., Bonewald L. F. and Kneissel M. (2010). Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 30, 3071-3085. 10.1128/MCB.01428-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V., Bryant H. U. and MacDougald O. A. (2006). Regulation of bone mass by Wnt signaling. J. Clin. Invest. 116, 1202-1209. 10.1172/JCI28551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E., Oppers-Walgreen B., Pettit A. R., Van Den Bersselaar L., Joosten L. A., Goldring S. R., Gravallese E. M., Van Den Berg W. B. (2002). Increase in expression of receptor activator of nuclear factor kappaB at sites of bone erosion correlates with progression of inflammation in evolving collagen-induced arthritis. Arthritis Rheum. 46, 3055-3064. 10.1002/art.10607 [DOI] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K. and He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9-26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C., Carmeliet P., Moermans K., Stockmans I., Smets N., Collen D., Bouillon R. and Carmeliet G. (2002). Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech. Dev. 111, 61-73. 10.1016/S0925-4773(01)00601-3 [DOI] [PubMed] [Google Scholar]
- Maes C., Kobayashi T., Selig M. K., Torrekens S., Roth S. I., Mackem S., Carmeliet G. and Kronenberg H. M. (2010). Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329-344. 10.1016/j.devcel.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattot V., Raes M. B., Henriet P., Eeckhout Y., Stehelin D., Vandenbunder B. and Desbiens X. (1995). Expression of interstitial collagenase is restricted to skeletal tissue during mouse embryogenesis. J. Cell Sci. 108, 529-535. [DOI] [PubMed] [Google Scholar]
- Miclea R. L., Karperien M., Bosch C. A. J., van der Horst G., van der Valk M. A., Kobayashi T., Kronenberg H. M., Rawadi G., Akcakaya P., Lowik C. W. G. M. et al. (2009). Adenomatous polyposis coli-mediated control of beta-catenin is essential for both chondrogenic and osteogenic differentiation of skeletal precursors. BMC Dev. Biol. 9, 26 10.1186/1471-213X-9-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno A., Amizuka N., Irie K., Murakami A., Fujise N., Kanno T., Sato Y., Nakagawa N., Yasuda H., Mochizuki S.-I. et al. (1998). Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Bioche Biophys. Res. Commun. 247, 610-615. 10.1006/bbrc.1998.8697 [DOI] [PubMed] [Google Scholar]
- Murtaugh L. C., Chyung J. H. and Lassar A. B. (1999). Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev. 13, 225-237. 10.1101/gad.13.2.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. (2000). Cre recombinase: the universal reagent for genome tailoring. Genesis 26, 99-109. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R. and de Crombrugghe B. (2002). The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17-29. 10.1016/S0092-8674(01)00622-5 [DOI] [PubMed] [Google Scholar]
- Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F. et al. (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231-1234. 10.1038/nm.2452 [DOI] [PubMed] [Google Scholar]
- Ono N., Ono W., Nagasawa T. and Kronenberg H. M. (2014). A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 16, 1157-1167. 10.1038/ncb3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega N., Wang K., Ferrara N., Werb Z. and Vu T. H. (2010). Complementary interplay between matrix metalloproteinase-9, vascular endothelial growth factor and osteoclast function drives endochondral bone formation. Dis. Model. Mech. 3, 224-235. 10.1242/dmm.004226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Gebhardt M., Golovchenko S., Perez-Branguli F., Hattori T., Hartmann C., Zhou X., de Crombrugghe B., Stock M., Schneider H. et al. (2015). Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 4, 608-621. 10.1242/bio.201411031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivetta E., Scapolan M., Pecolo M., Wassermann B., Abu-Rumeileh I., Balestreri L., Borsatti E., Tripodo C., Colombatti A. and Spessotto P. (2011). MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 13, R105 10.1186/bcr3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchett J. W. (1991). Growth plate activity in the upper extremity. Clin. Orthop. Relat. Res. 268, 235-242. [PubMed] [Google Scholar]
- Pritchett J. W. (1992). Longitudinal growth and growth-plate activity in the lower extremity. Clin. Orthop. Relat. Res. 275, 274-279. 10.1097/00003086-199202000-00041 [DOI] [PubMed] [Google Scholar]
- Qi J. H., Ebrahem Q., Moore N., Murphy G., Claesson-Welsh L., Bond M., Baker A. and Anand-Apte B. (2003). A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 9, 407-415. 10.1038/nm846 [DOI] [PubMed] [Google Scholar]
- Rodda S. J. and McMahon A. P. (2006). Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231-3244. 10.1242/dev.02480 [DOI] [PubMed] [Google Scholar]
- Ross F. P., Chappel J., Alvarez J. I., Sander D., Butler W. T., Farach-Carson M. C., Mintz K. A., Robey P. G., Teitelbaum S. L. and Cheresh D. A. (1993). Interactions between the bone matrix proteins osteopontin and bone sialoprotein and the osteoclast integrin alpha v beta 3 potentiate bone resorption. J. Biol. Chem. 268, 9901-9907. [PubMed] [Google Scholar]
- Sato M. M., Nakashima A., Nashimoto M., Yawaka Y. and Tamura M. (2009). Bone morphogenetic protein-2 enhances Wnt/beta-catenin signaling-induced osteoprotegerin expression. Genes Cells 14, 141-153. 10.1111/j.1365-2443.2008.01258.x [DOI] [PubMed] [Google Scholar]
- Seo D.-W., Li H., Guedez L., Wingfield P. T., Diaz T., Salloum R., Wei B.-Y. and Stetler-Stevenson W. G. (2003). TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114, 171-180. 10.1016/S0092-8674(03)00551-8 [DOI] [PubMed] [Google Scholar]
- Shapiro I. M., Adams C. S., Freeman T. and Srinivas V. (2005). Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res. C Embryo Today Rev. 75, 330-339. 10.1002/bdrc.20057 [DOI] [PubMed] [Google Scholar]
- Silvestrini G., Ballanti P., Patacchioli F., Leopizzi M., Gualtieri N., Monnazzi P., Tremante E., Sardella D. and Bonucci E. (2005). Detection of osteoprotegerin (OPG) and its ligand (RANKL) mRNA and protein in femur and tibia of the rat. J. Mol. Histol. 36, 59-67. 10.1007/s10735-004-3839-1 [DOI] [PubMed] [Google Scholar]
- Song L., Liu M., Ono N., Bringhurst F. R., Kronenberg H. M. and Guo J. (2012). Loss of wnt/beta-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 27, 2344-2358. 10.1002/jbmr.1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spater D., Hill T. P., Gruber M. and Hartmann C. (2006a). Role of canonical Wnt-signalling in joint formation. Eur. Cells Materials 12, 71-80. [DOI] [PubMed] [Google Scholar]
- Spater D., Hill T. P., O'Sullivan R. J., Gruber M., Conner D. A. and Hartmann C. (2006b). Wnt9a signaling is required for joint integrety and regulation of Ihh during chondrogenesis. Development 133, 3039-3049. 10.1242/dev.02471 [DOI] [PubMed] [Google Scholar]
- Spencer G. J., Utting J. C., Etheridge S. L., Arnett T. R. and Genever P. G. (2006). Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J. Cell Sci. 119, 1283-1296. 10.1242/jcs.02883 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.-S., William C. M., Tanabe Y., Jessell T. M. and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickens D., Behonick D. J., Ortega N., Heyer B., Hartenstein B., Yu Y., Fosang A. J., Schorpp-Kistner M., Angel P. and Werb Z. (2004). Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131, 5883-5895. 10.1242/dev.01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamura Y., Otani T., Kanatani N., Koyama E., Kitagaki J., Komori T., Yamada Y., Costantini F., Wakisaka S., Pacifici M. et al. (2005). Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. 280, 19185-19195. 10.1074/jbc.M414275200 [DOI] [PubMed] [Google Scholar]
- Taschner M. J., Rafigh M., Lampert F., Schnaiter S. and Hartmann C. (2008). Ca2+/Calmodulin-dependent kinase II signaling causes skeletal overgrowth and premature chondrocyte maturation. Dev. Biol. 317, 132-146. 10.1016/j.ydbio.2008.02.007 [DOI] [PubMed] [Google Scholar]
- Tu J., Henneicke H., Zhang Y., Stoner S., Cheng T. L., Schindeler A., Chen D., Tuckermann J., Cooper M. S., Seibel M. J. et al. (2014). Disruption of glucocorticoid signaling in chondrocytes delays metaphyseal fracture healing but does not affect normal cartilage and bone development. Bone 69, 12-22. 10.1016/j.bone.2014.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui M., Xing L., Drissi H., Zuscik M., O'Keefe R., Chen D. and Boyce B. F. (2008). Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J. Bone Miner. Res. 23, 314-325. 10.1359/jbmr.071025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu T. H., Shipley J. M., Bergers G., Berger J. E., Helms J. A., Hanahan D., Shapiro S. D., Senior R. M. and Werb Z. (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93, 411-422. 10.1016/S0092-8674(00)81169-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Jin H., Zhu M., Li J., Zhao L., Zhang Y., Tang D., Xiao G., Xing L., Boyce B. F. et al. (2014). Chondrocyte beta-catenin signaling regulates postnatal bone remodeling through modulation of osteoclast formation in a murine model. Arthritis Rheumatol. 66, 107-120. 10.1002/art.38195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsman N. J., Leiferman E. M., Fry M., Farnum C. E. and Barreto C. (1996). Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J. Orthop. Res. 14, 927-936. 10.1002/jor.1100140613 [DOI] [PubMed] [Google Scholar]
- Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C. and O'Brien C. A. (2011). Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235-1241. 10.1038/nm.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagiwa H., Tokunaga K., Hayami T., Hatano H., Uchida M., Endo N. and Takahashi H. E. (1999). Expression of metalloproteinase-13 (Collagenase-3) is induced during fracture healing in mice. Bone 25, 197-203. 10.1016/S8756-3282(99)00157-X [DOI] [PubMed] [Google Scholar]
- Yang G., Zhu L., Hou N., Lan Y., Wu X.-M., Zhou B., Teng Y. and Yang X. (2014a). Osteogenic fate of hypertrophic chondrocytes. Cell Res. 24, 1266-1269. 10.1038/cr.2014.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Tsang K. Y., Tang H. C., Chan D. and Cheah K. S. E. (2014b). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 111, 12097-12102. 10.1073/pnas.1302703111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer E., McLean W., Ng Y. S., Fukai N., Reginato A. M., Lovejoy S., D'Amore P. A. and Olsen B. R. (2002). Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development 129, 1893-1904. [DOI] [PubMed] [Google Scholar]
- Zelzer E., Mamluk R., Ferrara N., Johnson R. S., Schipani E. and Olsen B. R. (2004). VEGFA is necessary for chondrocyte survival during bone development. Development 131, 2161-2171. 10.1242/dev.01053 [DOI] [PubMed] [Google Scholar]
- Zhou Z., Apte S. S., Soininen R., Cao R., Baaklini G. Y., Rauser R. W., Wang J., Cao Y. and Tryggvason K. (2000). Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA 97, 4052-4057. 10.1073/pnas.060037197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., von der Mark K., Henry S., Norton W., Adams H. and de Crombrugghe B. (2014). Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10, e1004820 10.1371/journal.pgen.1004820 [DOI] [PMC free article] [PubMed] [Google Scholar]