

ABSTRACT
The association of desmin with the α-crystallin Β-chain (αΒ-crystallin; encoded by CRYAB), and the fact that mutations in either one of them leads to heart failure in humans and mice, suggests a potential compensatory interplay between the two in cardioprotection. To address this hypothesis, we investigated the consequences of αΒ-crystallin overexpression in the desmin-deficient (Des−/−) mouse model, which possesses a combination of the pathologies found in most cardiomyopathies, with mitochondrial defects as a hallmark. We demonstrated that cardiac-specific αΒ-crystallin overexpression ameliorates all these defects and improves cardiac function to almost wild-type levels. Protection by αΒ-crystallin overexpression is linked to maintenance of proper mitochondrial protein levels, inhibition of abnormal mitochondrial permeability transition pore activation and maintenance of mitochondrial membrane potential (Δψm). Furthermore, we found that both desmin and αΒ-crystallin are localized at sarcoplasmic reticulum (SR)–mitochondria-associated membranes (MAMs), where they interact with VDAC, Mic60 – the core component of mitochondrial contact site and cristae organizing system (MICOS) complex – and ATP synthase, suggesting that these associations could be crucial in mitoprotection at different levels.
KEY WORDS: Intermediate filaments, Small heat-shock protein, Cytoskeleton, Heart failure, Mitochondria, Cristae, MAMs
Highlighted Article: Both desmin and its partner chaperone αB-crystallin associate with mitochondria–sarcoplasmic-reticulum contact sites (MAMs), stabilizing MICOS super-complexes, and thus contributing to proper mitochondrial cristae structure–function.
INTRODUCTION
Desmin, the muscle-specific intermediate filament protein, forms a three-dimensional scaffold that interconnects the contractile apparatus to the nucleus, cellular organelles and the sarcolemma (Capetanaki et al., 2007). The desmin scaffold is a primary target for cardiomyopathy and heart failure both in mice and humans (Goldfarb et al., 1998; Capetanaki et al., 2007, 2015). Mice deficient for desmin develop both skeletal and myocardial defects that are strongly linked to impaired mitochondrial morphology and function (Milner et al., 1996, 1999, 2000; Li et al., 1996; Papathanasiou et al., 2015). These mitochondrial abnormalities lead to cardiomyocyte death and myocardial degeneration, accompanied by inflammation and fibrosis, resulting in dilated cardiomyopathy (DCM) and heart failure (Milner et al., 2000; Mavroidis and Capetanaki 2002; Capetanaki et al., 2007; Psarras et al., 2012; Mavroidis et al., 2015). In humans, over 70 desmin mutations have been linked to desmin-related cardiomyopathies, of which DCM is the most frequent (Capetanaki et al., 2015; van Spaendonck-Zwarts et al., 2011). In addition, desmin aggregate formation due to caspase cleavage is an important mediator of TNF-α-induced heart failure (Panagopoulou et al., 2008). Importantly, regardless of the specific mutations, human hearts undergoing DCM-linked failure display mitochondrial and other structural abnormalities, similar to those in desmin-deficient mice and mice undergoing TNF-α-induced failure. Furthermore, the R120G mutation in the α-crystallin Β-chain (αB-crystallin; encoded by CRYAB), an abundant small heat-shock protein, also causes the same desmin-aggregate-related DCM and heart failure with similar mitochondrial defects (Wang et al., 2001; Vicart et al., 1998). Mitochondrial abnormalities seem to be a common feature of diseases in which desmin and/or αB-crystallin deficiencies are implicated. Previous observations suggest a physical association of desmin with mitochondria, as well as an involvement of desmin in mitochondrial homeostasis (Stromer and Bendayan, 1990; Reipert et al., 1999; Hnia et al., 2011), but the underlying mechanisms remain elusive. αΒ-crystallin associates with all three cytoskeletal networks – microfilaments (Bennardini et al., 1992; Singh et al., 2007), microtubules (Arai and Atomi, 1997; Houck and Clark, 2010) and intermediate filaments (Nicholl and Quinlan, 1994; Iwaki et al., 1989; Wisniewski and Goldman, 1998) (reviewed in Wettstein et al., 2012) – and specifically desmin (Bennardini et al., 1992; Nicholl and Quinlan, 1994; Perng et al., 1999). There is also evidence that a fraction of αΒ-crystallin is located in mitochondria (Fountoulakis et al., 2005; Martindale et al., 2005; Maloyan et al., 2005; Mitra et al., 2013); however, its role there is not completely understood.
Mitochondria are double-membrane-bounded organelles that are involved in important cellular processes. Proper assembly, maintenance and function of the various mitochondrial complexes, including oligomers of optic atrophy 1 (OPA1) (Frezza et al., 2006), F1F0-ATP synthase (Strauss et al., 2008) and the recently identified mitochondrial contact site and cristae organizing system (MICOS) (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011) are vital to inner membrane organization, and to proper and efficient mitochondrial function. Mitofilin (Mic60; also known as IMMT) (Gieffers et al., 1997; Odgren et al., 1996), the core component of MICOS, interacts with several inner and outer mitochondrial membrane (IMM and OMM, respectively) proteins (von der Malsburg et al., 2011; Darshi et al., 2011; Xie et al., 2007; Harner et al., 2011; Hoppins et al., 2011; van der Laan et al., 2012), thus maintaining IMM morphology and contact sites between IMM and OMM, and affecting mitochondrial function, mitochondrial (mt)DNA integrity and protein and metabolite trafficking.
To address the role of desmin and αB-crystallin, and their potential interplay in mitochondrial homeostasis and cardioprotection, we investigated the consequences of αΒ-crystallin overexpression in the desmin-deficient cardiomyopathy model. We demonstrated that cardiac-specific αB-crystallin overexpression rescues desmin-deficient heart failure and that this takes place through maintenance of proper mitochondrial composition, structure and function, through inhibition of abnormal mitochondrial permeability transition pore (mPTP) activation and through cardiomyocyte death prevention. Moreover, we found that among other sites, both desmin and αΒ-crystallin localize at sarcoplasmic reticulum (SR)–mitochondria-associated membranes (MAMs), where they interact with VDAC. Furthermore, we have unraveled a potential link of desmin and αB-crystallin with mitochondrial morphology and bioenergetics through their interaction with Mic60 and ATP synthase.
RESULTS
Generation of Des−/− mice overexpressing αΒ-crystallin in the heart
In order to examine the effect of αΒ-crystallin overexpression in the Des−/− myocardium, we generated transgenic mice overexpressing αΒ-crystallin specifically in the heart under the control of the α-myosin heavy chain (αMHC) promoter. Among different lines generated, two were chosen for further investigation, namely αΒCry4 and αΒCry6, with high and low αB-crystallin expression levels respectively (Fig. 1A; Fig. S1A). These mice were crossed with Des−/− mice to generate αΒ-crystallin overexpressing Des−/− mice (Des−/−αΒCry). Quantification of total αB-crystallin protein levels from hearts of wild-type, Des−/− and transgenic lines revealed 1.3-fold higher αB-crystallin levels in Des−/− mice, a 5.4-fold increase in line αΒCry4 and a 2.6-fold increase in line αΒCry6 in comparison to wild type.
Fig. 1.

Increased αΒ-crystallin levels overcome its desmin-dependent stress-induced relocation. (A) Expression levels of αB-crystallin protein in the heart from 2-month-old wild-type (WT), Des−/− and αΒ-crystallin-overexpressing (des−/−αBCry4 and des−/−αBCry6) transgenic mice. GAPDH, loading control. (B) Western blot analysis of total heart lysates (a) and subcellular fractions (b) from mice in control conditions and after a swimming exercise reveals increased levels of αΒ-crystallin (αΒCry) in the mitochondria of Des−/−αBCry4 hearts; GAPDH, cytosolic marker; cytochrome c oxidase IV (COXIV), mitochondrial marker. The arrowhead in Bb indicates nonspecific bands. (C) Cardiac protein levels of (a) desmin and (b) αB-crystallin under normal and stressed (swimming) conditions, and (c) the protein levels of αB-crystallin located in the cytosol (cyt) and mitochondria (mit). Error bars show mean±s.e.m. (a) ***P<0.001 vs wild-type control, *P≤0.05 vs wtαBCry control; (b) **P<0.001 vs wild-type control, ***P<0.0001 vs Des−/− control and swimming, *P<0.05 vs Des−/−αBCry control (Student's unpaired t-test). (D) Double immunofluorescence staining for αΒ-crystallin and α-actinin in frozen myocardial tissue sections from 6-month-old mice (des−/−αBCry4), fixed right after swimming (blue staining of nuclei, DAPI; arrows, intercalated discs). Scale bars: 10 μm.
Overexpression of αΒ-crystallin rescues its compromised Z-disc localization and allows for more efficient mitochondrial localization in Des−/− hearts
It has been shown that, upon stress, αΒ-crystallin translocates from a cytosolic pool to Z-discs (Golenhofen et al., 1998), where it colocalizes with desmin and α-actinin. Immunofluorescence microscopy was performed to determine whether desmin deficiency affects the Z-disc localization of αΒ-crystallin. We found that, in the unstressed wild-type myocardium, αB-crystallin colocalized only partially with desmin and α-actinin at the Z-disc, and was also located at the intercalated discs (Fig. S1B). This localization pattern was enhanced under stress as a result of the mice swimming (Fig. 1D). In the absence of desmin, the Z-disc localization of αB-crystallin was mostly lost, but its localization at intercalated discs was retained (Fig. 1D), possibly owing to other potential binding partners at the intercalated discs. Overexpression of αB-crystallin in the Des−/−αBCry4 myocardium rescued its Z-disc localization (Fig. 1D), and more importantly, a large amount of αB-crystallin was located in mitochondria (Fig. 1Bb). Mitochondrial localization of αB-crystallin also occurred in wild-type cardiomyocytes but could not be easily detected. Under the stress of swimming, the total protein levels of both desmin and αB-crystallin were increased (Fig. 1Ba,Ca,Cb), and this allowed for easier detection of αB-crystallin in the mitochondrial fraction in both wild-type and Des−/− myocardium (Fig. 1Cc). This was more obvious in the corresponding αB-crystallin-overexpressing myocardium [wild-type (wt)αBCry4 and Des−/−αBCry4]; however, the ratio of mitochondrial to cytosolic levels remained the same.
Complete rescue of adverse remodeling in desmin-deficient hearts overexpressing αΒ-crystallin
Inflammation, fibrosis and calcification are hallmarks of Des−/− mouse myocardial degeneration and necrosis (Milner et al., 1996; Mavroidis and Capetanaki, 2002; Psarras et al., 2012). Dissection of Des−/−αΒCry4 mice revealed a complete absence of extensive calcification in the myocardium. Further histological analysis revealed no inflammatory infiltration and a minimal amount of fibrosis in the ventricular walls (Fig. 2A–D). Quantification of histological section images confirmed the significant reduction of fibrosis in the Des−/−αΒCry myocardium (Fig. 2C). Comparison of the two transgenic lines Des−/−αΒCry4 and Des−/−αΒCry6 revealed that a 5.4-fold overexpression of αB-crystallin was more beneficial when compared to a 2.6-fold overexpression. Therefore, the subsequent studies were done using line Des−/−αΒCry4, designated from now on as Des−/−αΒCry.
Fig. 2.

Complete amelioration of adverse remodeling in Des−/− hearts overexpressing αΒ-crystallin. (A) Histological Masson's trichrome staining of heart sections from 3.5-month-old wild-type (wt; n=7), Des−/− (n=7), Des−/−αBCry4 (n=10) and Des−/−αBCry6 (n=7) mice. Fibrosis was detected by blue staining of collagen deposition. (B) Hematoxylin and eosin staining of 23-day-old wild-type (n=6), Des−/− (n=12), Des−/−αΒCry (n=15) and wtαΒCry (n=6) mice, showing the inflammatory infiltration in the Des−/− myocardium compared to that in the other genotypes. Scale bars: 20 µm. (C) Semi-quantitative determination of myocardial fibrosis using ImageJ software analysis. **P<0.01 vs wild-type and Des−/−αBCry6; ***P<0.0001 vs Des−/−; #P<0.001 vs wild type and Des−/−αBCry4. (D) Assessment of cardiac inflammation using 23-day-old hearts and a conventional grading system (see Psarras et al., 2012). ***P<0.0001 vs wild type, Des−/−αΒCry and wtαΒCry. (E) Evaluation of cardiomyocyte membrane permeability by Evan's-Blue (red) and α-actinin (green) staining of frozen heart sections from 23-day-old mice; blue staining of nuclei, DAPI. Scale bars: 100 µm. (F) A quantification of the EBD-positive area by using ImageJ software analysis. ***P<0.0001. In the box-and-whisker plots in D and F, the boxes indicate the interquartile range of the samples, the lines represent the median values and the whiskers represent the outliers. All error bars show mean±s.e.m. (unpaired Student's t-test).
Evans-Blue dye (EBD) analysis was performed to detect cellular membrane permeability. Wild-type cardiomyocytes were impermeable to EBD because their membranes were intact (Fig. 2E,F). In contrast, Des−/− hearts appeared to have considerably large areas of EBD-positive cells, especially in regions with inflammatory infiltration. In Des−/−αBCry myocardium there were no EBD-positive cardiomyocytes, leading to the conclusion that αB-crystallin overexpression protects Des−/− myocardium from necrotic cell death.
αΒ-crystallin overexpression considerably improves cardiac function and results in 100% survival during obligatory swimming exercise
Heart failure development with aging is characteristic of Des−/− hearts. To explore the effect of αΒ-crystallin overexpression on cardiac function, we performed standard morphometric analysis and two-dimensional directed M-mode echocardiography (Fig. 3; Table S1). Des−/−αBCry mice showed a significant improvement in all parameters tested in comparison to Des−/− mice. The improvement in left ventricular function of Des−/−αBCry mice was very extensive, reaching almost wild-type levels (96% of the wild-type fractional shortening; Fig. 3Bc), consistent with the improvement in left-ventricular-end systolic diameter, which was also comparable to wild-type levels (Fig. 3Bb). Posterior wall thickness was also increased by 35% in the Des−/−αBCry compared to the Des−/− group, consistent with the absence of degeneration in these hearts (Fig. 3Bd). Finally, the ratio of left ventricular radius to posterior wall thickness, an indicator of left ventricular wall stress, was significantly lower in Des−/−αBCry mice compared to that in Des−/− mice (Fig. 3Be).
Fig. 3.

αΒ-crystallin overexpression considerably improves cardiac function, survival rate during obligatory exercise and Des−/− cardiomyocyte ultrastructural defects. Mice at 7 months of age were subjected to echocardiography. Des−/−αBCry mice showed a significant improvement in left ventricular (LV) function reaching a fractional shortening (FS) value that was 96% of that of wild type (wt). (A) Representative M-mode echocardiograms from each mouse genotype. (B) Group data (mean±s.e.m) for left ventricular end diastolic diameter (a, LVEDD), end systolic diameter (b, ESD), FS (c), posterior wall dimension (d, PWd), ratio of left ventricular radius to PWd (e, r/h), ejection fraction (f, EF); *P<0.01, **P<0.001 and ***P<0.0001 vs Des−/− (ANOVA with Bonferroni–Dunn post-hoc test). (C) Survival curve of 3-month-old wild-type (n=12), Des−/− (n=12) and Des−/−αBCry (n=13) mice during a swimming protocol. The error bars correspond to mean±s.e.m.; P<0.0001 Des−/− vs wild type and Des−/−αBCry at all time points (unpaired Student's t-test). (D) Myocardial tissue ultra-structure from 3-month-old mice that were examined by using transmission electron microscopy. Scale bar: 2 µm. (E) Higher magnification images of the myocardial mitochondria. Desmin deficiency affects the ultrastructure of the IMM, leading to excessive disorganization of cristae. The mitochondria from Des−/−αΒCry transgenic myocardium appear normal. m, mitochondria; N, nucleus; c, cristae. Scale bars: 0.2 µm.
In order to investigate whether this cardioprotection also occurs under stress conditions, wild-type, Des−/− and Des−/−αΒCry mice were subjected to an obligatory swimming exercise protocol for 24 days. The Des−/− mice had a considerably reduced ability to handle stress because 50% of them died during swimming (in comparison to 0% of wild type) (Fig. 3C). The survival rate of Des−/−αΒCry mice was 100%, strongly demonstrating the powerful cardioprotective action of αΒ-crystallin overexpression against stress.
Rescue of Des−/− cardiomyocyte ultrastructural defects by αΒ-crystallin overexpression
In an effort to understand the mechanism by which the overexpression of αΒ-crystallin protects Des−/− myocardium against degeneration and improves cardiac function, we initially examined its effects on cardiomyocyte ultrastructure. Des−/− cardiomyocytes are characterized by various early-appearing mitochondrial defects (Milner et al., 2000), followed by other structural abnormalities and loss of muscle integrity (Fig. 3D). Mitochondrial abnormalities include disturbed shape and positioning, fragmentation, aggregation, swelling of the mitochondrial matrix, disruption and fragmentation of the cristae and degeneration (Fig. 3E). In contrast, none of these ultrastructural defects are present in Des−/−αΒCry mice. Myofibrils were more properly aligned and mitochondria, located parallel to myofibrils, appeared normal, without signs of fragmentation, cristae abnormalities or destruction. These data suggest that mitochondria are an important target of αΒ-crystallin cytoprotection.
αΒ-crystallin overexpression provides anti-oxidant protection to Des−/− cardiomyocytes
To further investigate the mechanism by which αΒ-crystallin protects against mitochondrial defects, we analyzed its effects on adult cardiomyocyte reactive oxygen species (ROS) levels using the molecular probe CM-H2DCFDA (Fig. 4A,B). Des−/− cardiomyocytes showed a significantly higher degree of ROS over those in the wild-type, whereas Des−/−αΒCry cardiomyocytes were characterized by a lower degree of ROS. Under oxidative stress conditions, simulated by incubation with H2O2, there was a significant decrease of ROS levels in both Des−/−αΒCry and wtαBCry compared to Des−/− and wild-type cells respectively, indicative of the protection provided by αΒ-crystallin overexpression. To further investigate the mechanism of anti-oxidant protection by αΒ-crystallin, we measured the levels of glutathione (GSH). In Des−/− hearts, glutathione levels are decreased by 33% and the GSH:glutathione-disulfide (GSSG) ratio by almost 50% in comparison to wild type (Fig. 4C). Overexpression of αΒ-crystallin increased glutathione and GSH:GSSG levels to 87% and 95%, respectively, of those in wild type, suggesting that, at least partially, the mechanism of anti-oxidant protection by αΒ-crystallin occurs through an increase of reduced glutathione levels.
Fig. 4.

αΒ-crystallin overexpression provides anti-oxidant protection to Des−/− cardiomyocytes, inhibits abnormal activation of the mPTP and the dissipation of Δψm. (A) Staining of live adult cardiomyocytes in culture with CM-H2DCFDA to detect ROS, in control conditions and after challenging with H2O2. Color graduation from light blue to brown corresponds to the intensity of CM-H2DCFDA fluorescence and consequently the level of ROS. Scale bar: 250 μm. (Ba) Semi-quantitative determination of CM-H2DCFDA intensity. Values are normalized to those of wild type (wt) (mean±s.e.m.), n=9; **P<0.01 vs wild type, *P<0.05 vs Des−/− (unpaired Student's t-test). (b) Same as panel Ba after incubation with H2O2; ***P<0.001 and #P≤0.05 vs wild type, **P<0.01 and *P<0.05 vs Des−/−; n=9. (C) Total glutathione levels (a) and GSH:GSSG ratio (b) in heart tissue homogenates from 6-month-old mice. Des−/− heart displays only 67%±4.7 (mean±s.e.m.) of the total GSH and 51.43%±11.10 of the reduced GSH compared to wild-type heart, wheteas αΒCry overexpression increases these levels up to 87%±9.3 and 95.39%±12.38, respectively. (a)***P≤0.001 vs the other genotypes, #P<0.001 vs wild type; (b) ***P≤0.001 vs wild type, *P≤0.05 vs Des−/−; n=4 (unpaired Student's t-test). (D) Determination of mitochondrial Δψm by labeling adult cardiomyocytes with the potentiometric probe TMRM. (E) Semi-quantitative determination of the initial TMRM fluorescence; *P<0.05 vs the other genotypes, #P≤0.05 vs untreated (unpaired Student's t-test). (F) Loss of mitochondrial Δψm analysis using 20-min time-lapse confocal imaging of cardiomyocytes loaded with TMRM probe (see also Fig. S2). The mPTP inhibitor cyclosporin A (CsA) was used to confirm the mPTP activation. The error bars show mean±s.e.m.; P<0.01 vs Des−/− and CsA; n=9.
αΒ-crystallin overexpression inhibits abnormal activation of the mitochondrial permeability transition pore and the dissipation of mitochondrial membrane potential
Mitochondrial swelling and increased ROS levels have been linked to abnormal activation of the mPTP. Therefore, we investigated whether the above demonstrated mitoprotection and cardioprotection by αΒ-crystallin overexpression takes place through inhibition of mPTP activation. First, we determined the sensitivity of isolated mitochondria to increased Ca2+ levels in the presence or absence of the mPTP inhibitor cyclosporin A (CsA). As expected, Des−/− mitochondria swelled more than wild-type mitochondria at the same Ca2+ concentration, as measured by the decrease in absorbance at 540 nm (Table S2). By contrast, Des−/−αBCry mitochondria showed a significant decrease in swelling, indicating that overexpression of αB-crystallin improves the diminished capacity of Des−/− mitochondria to resist exposure to Ca2+. Addition of CsA before the Ca2+ challenge significantly abrogated mitochondrial swelling. This finding demonstrates that first, the reduced ability of Des−/− mitochondria to resist increased Ca2+ levels (Weisleder et al., 2004) is mPTP dependent and second, the mechanism of mitoprotection and inhibition of necrotic cell death of Des−/− myocardium by αB-crystallin overexpression involves suppression of mPTP activation.
In the Des−/− heart, mitochondrial abnormalities, including swelling, can lead to dissipation of mitochondrial membrane potential (Δψm), a critical event in the process of cell death. To address this possibility, we incubated adult cardiomyocytes with tetramethylrhodamine methyl ester (TMRM), a potentiometric indicator that is targeted to mitochondria. The TMRM intensity was 21% lower in Des−/− cardiomyocytes compared to that in wild type, indicating reduced Δψm (Fig. 4D,E). Time-lapse confocal scanning showed a more rapid loss of TMRM in Des−/− mitochondria relative to that in wild-type mitochondria, consistent with mitochondrial membrane depolarization (Fig. 4F; Fig. S2). αB-crystallin overexpression in Des−/− myocardium significantly increased both the intensity and retention time of TMRM. To examine whether the accelerated TMRM loss in Des−/− cardiomyocytes was due to mPTP activation, we treated cardiomyocytes with CsA, significantly attenuating TMRM loss.
αΒ-crystallin overexpression restores changes in the levels of important mitochondrial proteins caused by desmin deficiency
In an effort to understand the underlying mechanisms responsible for both the observed mitochondrial defects in the absence of desmin and, most importantly, the unprecedented extensive rescue of these defects by αΒ-crystallin overexpression, we conducted proteomic analysis to identify any crucial alterations in the mitochondrial proteome of the different genotypes. The results revealed diminished levels of enzymes involved in the citric acid cycle, lipid metabolism and fatty acid β-oxidation in the Des−/− compared to wild-type mitochondria (Table 1; Table S3, Fig. S3). Moreover, in Des−/− mitochondria, the levels of many subunits and components of the electron transport chain were decreased, along with components of complexes linking different metabolic pathways. The most extensive change was found with the cristae junction protein Mic60, the levels of which were reduced by 78% compared to those in wild type (Table 1, Fig. 5A,B). On the contrary, in the mitochondria of Des−/−αΒCry myocardium, most of the proteins that showed decreased levels in Des−/− mice, including Mic60 protein, were apparently restored (Table 1, Fig 5; Table S4). In agreement with the western blotting data (Fig. 1B), proteomic analysis revealed that much higher levels of αΒ-crystallin localize to the mitochondria of the Des−/−αΒCry hearts in comparison to those of Des−/− hearts. The levels of some of the differentially expressed proteins were also evaluated by western blotting (Fig. 5A,B). In addition, we investigated the mitochondrial levels of adenine nucleotide translocator (ANT), an IMM protein with known mitochondrial and cardiomyocyte function (Narula et al., 2011; Vogelpohl et al., 2011) that cannot be easily detected by proteomic analysis. This analysis revealed that, in contrast to total ANT levels, mitochondrial ANT levels were reduced in Des−/− myocardium, a defect ameliorated by αB-crystallin overexpression (Fig. 5A,B).
Table 1.
αB-Crystallin overexpression restores several mitochondrial enzymes and proteins that are diminished in Des−/− hearts
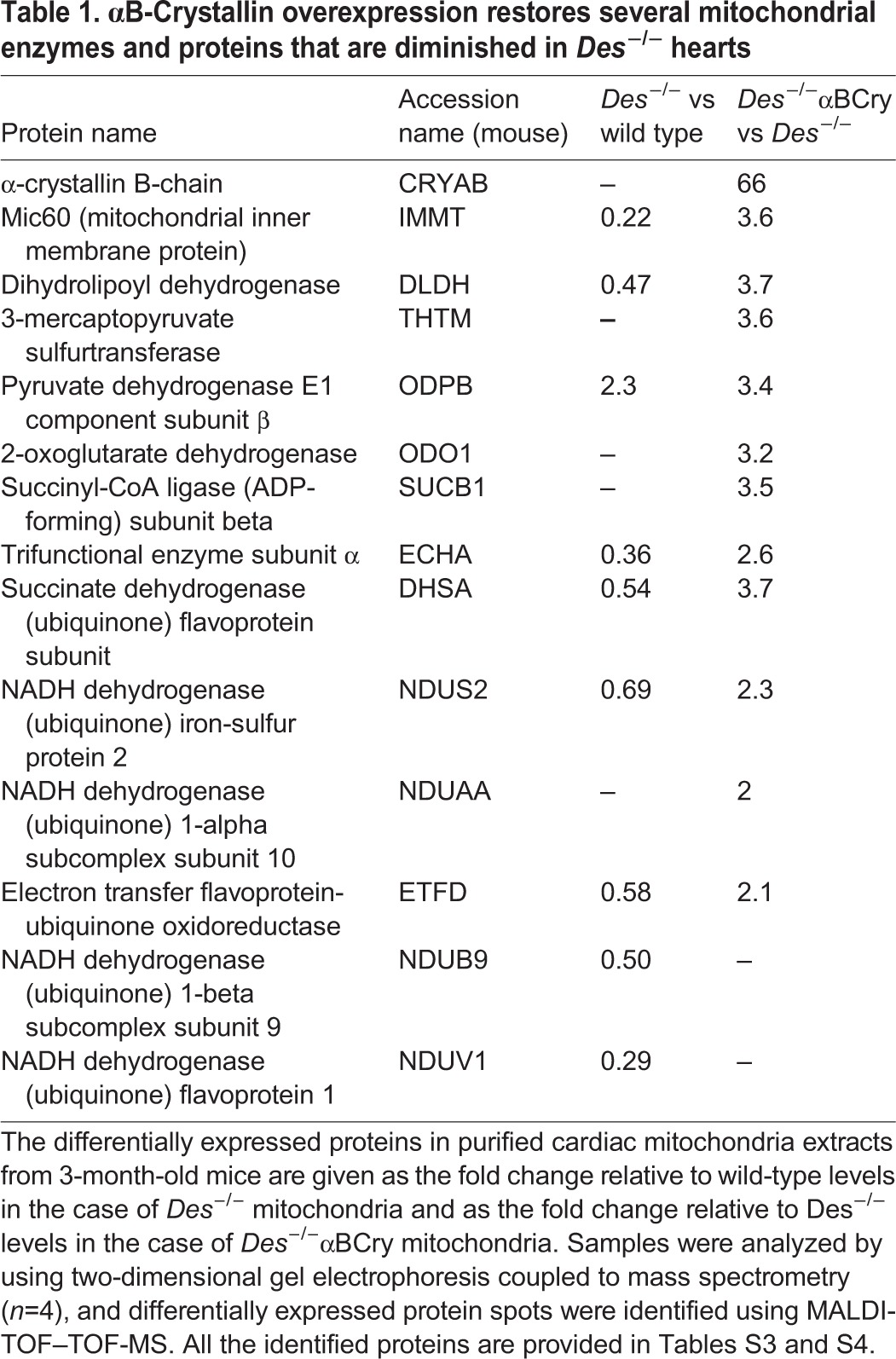
Fig. 5.

Desmin and αB-crystallin bind to Mic60 and ATP synthase β. (A) Western blot analysis of total and mitochondrial fractions isolated from hearts of the indicated genotypes. Mic60 is expressed as two isoforms of 88 and 90 kDa. Wt, wild type. (B) Densitometry analysis of mitochondrial (mit) protein levels. COXIV was used as loading control. Plots represent the mean±s.e.m. of more than five experiments; *P≤0.05 vs Des−/−, ***P≤0.001 vs Des−/−, #P<0.05 vs wild type, ^P<0.001 vs wild type (unpaired Student's t-test). (C) GST pulldown assay using wild-type mouse heart mitochondrial extract. Arrowhead, nonspecific binding of 55-KDa IgGs. (D) Co-immunoprecipitation (CO-IP) analysis using cardiac mitochondrial extracts. Arrowheads, nonspecific binding of 55- and 25-kDa IgGs. (E) ATP content of adult cardiomyocytes normalized to wild type (mean±s.e.m, n=8); ***P≤0.0001 vs wild type, *P<0.05 vs Des−/− (unpaired Student's t-test).
Desmin and αΒ-crystallin interact with proteins that are important for cristae morphology
As demonstrated above, one of the most interesting proteins downregulated in Des−/− mitochondria is Mic60 (Table 1, Fig. 5A), consistent with the shown cristae morphology defects (Fig. 3D,E). Given that both these defects are rescued by αΒ-crystallin overexpression, we aimed to further unravel the underlying mechanism. We used GST-tagged desmin and co-immunoprecipitation to examine whether desmin interacts with Mic60, and therefore, whether loss of this interaction in Des−/− cardiomyocytes affects its proper mitochondrial targeting and/or stabilization. Indeed, we found that desmin interacts with Mic60, and as expected, with αΒ-crystallin (Fig. 5C,D). More importantly, we demonstrated that αΒ-crystallin also associates with Mic60 (Fig. 5D), both in wild-type and Des−/− myocardial extracts, strongly suggesting that Mic60 is one of the αΒ-crystallin targets in the Des−/− mitochondria. Using similar immunoprecipitation studies, (and immunoprecipitation coupled to proteomic analysis in the case of desmin), we found that the β subunit of the F1 catalytic domain of ATP synthase, another protein important for determining cristae structure (Paumard et al., 2002) and mitochondrial function (Zick et al., 2009), interacts with desmin as well as with αΒ-crystallin (Fig. 5C,D). Loss of this interaction in the absence of desmin, combined with the disrupted cristae and depolarized Δψm, is expected to impact the ATP content, which we indeed confirmed to be 40% lower in the Des−/− cardiomyocytes compared to that in wild type (Fig. 5E). αΒ-crystallin overexpression increased this ATP content to 90% of the wild-type levels.
These findings support the view that the loss of desmin association with these proteins could be a key mediator of pathologies resulting from loss of desmin. Given that αΒ-crystallin also associates with these proteins, its higher levels in the Des−/−αΒCry cardiomyocytes, in comparison to its basal levels in Des−/− cells, might be able to compensate for the impact of desmin loss on Mic60 and ATP synthase function.
Desmin and αB-crystallin are located at the SR and MAMs, where they associate with VDAC
The question raised by the amelioration of Des−/− mitochondrial pathology by αB-crystallin overexpression is how can desmin and αB-crystallin, two mainly cytoplasmic proteins, interact with IMM proteins and impact mitochondrial homeostasis. To address this question, we first performed protease accessibility assays to examine whether a fraction of desmin and αB-crystallin is located inside the mitochondria. Crude mitochondria from wild-type hearts were isolated and digested with proteinase K. As shown in Fig. 6A, both desmin and αB-crystallin were degraded by proteinase K, whereas the IMM protein ANT and the OMM protein VDAC remain intact. The digestion pattern of desmin and αB-crystallin resembled that of mitofusin 2, an OMM protein, as detected by an antibody that recognizes a part of mitofusin 2 that is exposed to the cytosol. In contrast, overexpressed αB-crystallin appears to localize inside mitochondria (Fig. 6Ab).
Fig. 6.

Desmin and αB-crystallin are located at the SR and MAMs. (A) Cardiac mitochondria (mit) from wild-type (a) and Des−/−αBCry (b) mice were digested with proteinase K (prot K) under the indicated conditions. VDAC and mitofusin 2 (Mfn2) were used as OMM markers, whereas ANT was used as an IMM marker. Osmotic, 20 mM KCl. (B) Subcellular fractions from wild-type hearts were analyzed by western blotting for known markers of cytosol (α-actinin), mitochondria (MnSOD), MAMs and light membranes (LM; mostly containing SR, distinguished by VDAC, Mfn2, RYR). wt, wild type. (C) αB-crystallin subcellular localization in wild type (1), Des−/− (2) and Des−/−αBCry (3) hearts. (D) Electron micrographs of double immunogold labeling of desmin (10-nm particle) and VDAC (25-nm particle). Desmin is found at Z-discs (z, black arrows), as expected (a), and in intimate association with mitochondria (m, black arrowheads) (a–c′) and membrane structures attaching mitochondria, most possibly SR and MAMs (white arrows) (c, c′), where it colocalizes with VDAC (white arrowheads) (a, a′, b, b′). a′–c′ are higher magnification images of the boxed areas. Scale bars: 0.1 μm.
Next, we performed subcellular fractionation of rat and mouse hearts to further determine the localization of desmin. Interestingly, although we found desmin in the SR fraction, it was mainly enriched in the SR–MAMs (Fig. 6B). In contrast, αB-crystallin was found enriched more in the SR fraction and less so in the MAMs and pure mitochondrial fraction. Furthermore, overexpressed αB-crystallin was also enriched in the SR fraction but even more so in the MAMs and in pure mitochondria (Fig. 6C). The localization of desmin was confirmed by using immunogold labeling studies (Fig. 6D; Fig. S4). In addition to the expected places, such as intercalated discs (Fig. S4) and Z-lines, desmin was found to associate with mitochondria and membranous structures, most likely SR, in close proximity to mitochondria.
A previous yeast two-hybrid screening of a heart cDNA library using the entire desmin molecule as bait, conducted in our laboratory, had shown binding of desmin to VDAC (our unpublished results). In addition to its mitochondrial localization, VDAC is also enriched in MAMs, and furthermore it has been found to interact with Mic60 (Hoppins et al., 2011). Therefore, we reconfirmed the binding of VDAC to desmin using both co-immunoprecipitation studies as well as GST pulldown assays (Fig. 7A,B). To determine the corresponding binding regions of VDAC and desmin, we generated several deletion constructs and used them in reciprocal GST pulldown experiments. As demonstrated in Fig. 7D, the N-terminal domain of VDAC is necessary for its binding to desmin, whereas the C-terminal tail domain of desmin and, to a lesser extent, the rod domain, are sufficient for association of desmin with VDAC. The finding of desmin–VDAC binding was further strengthened by double immunogold labeling studies, which confirmed the close proximity of these two proteins in vivo (Fig. 6D; Fig. S4). αB-crystallin was also co-immunoprecipitated with VDAC, consistent with earlier reports (Mitra et al., 2013). The demonstrated associations of desmin and αB-crystallin with VDAC, Mic60 and ATP synthase could somehow facilitate the formation and/or stabilization of a scaffold-like super complex extending from SR–mitochondria contact sites to MICOS and ATP complexes.
Fig. 7.

Desmin and αB-crystallin are found in the same super-complex with Mic60 and VDAC. GST pulldown assay (A) and VDAC immunoprecipitation analysis (B) using cardiac mitochondrial (mit) lysates. Arrowheads, nonspecific binding of 25-kDa IgGs. BSA, bovine serum albumin; wt, wild type. (C) Schematic representation of desmin (a) and VDAC (b), and their deletion mutants. Des, full-length desmin; H, head; HR, head-rod; R, rod; RT, rod-tail; and T, tail domains. VDAC, full-length VDAC; ΔNterm, deletion of the α-helix N-terminus of VDAC; Δβ1β4, deletion of the first four β-strands. (D) GST pulldown analysis of desmin and VDAC domain interactions using mitochondrial lysates from wild-type and, in the case of VDAC, Des−/− hearts. (E) Heart mitochondria, solubilized with digitonin, were analyzed by blue native (BN) PAGE in the first dimension. The Coomassie-Blue dye contained in the loading buffer allows the visualization of the proteins, which serves as a loading control. (F) Analysis of solubilized mitochondria from animals in control conditions and after swimming exercise in the second dimension by using SDS-PAGE analysis. Red box, macromolecular complexes of Mic60 diminished in the absence of desmin and were restored by αB-crystallin overexpression; blue box, shift of Mic60 and VDAC macromolecular complexes to a lower molecular mass after swimming stress.
αB-crystallin restores the formation of Mic60-containing macromolecular complexes that are affected by desmin deficiency
We speculated that desmin and αB-crystallin association with Mic60 could affect the formation or stabilization of Mic60-containing complexes. We analyzed crude mitochondria from mouse heart that had been solubilized with digitonin by using blue native PAGE in the first dimension and SDS-PAGE in the second dimension, followed by detection of Mic60 by western blotting (Fig. 7E,F). In the absence of desmin, the number of the protein complexes containing Mic60 was much lower compared to those in wild type, consistent with the diminished Mic60 protein levels found in Des−/− mitochondria (Table 1, Fig. 5A). Importantly, as determined by using much lower protein levels, the higher-molecular-mass complexes of Mic60 were proportionally less than the corresponding lower-molecular-mass complexes, indicating a possible facilitating role of desmin in the formation or stabilization of these complexes (Fig. 7F; Fig. S4). All these data are consistent with the cristae defects of Des−/− mitochondria (Fig. 3). Overexpression of αB-crystallin in the absence of desmin restored the abundance of all the different-sized Mic60 complexes to their normal levels. Importantly, desmin and αB-crystallin appeared to be in the same complexes as Mic60 and VDAC, supporting the idea of a functional role in the interface of SR with OMMs and IMMs. Under swimming stress conditions, we obtained similar results with only a slight shift in the molecular mass of VDAC complexes, as observed in the Des−/− mitochondria (Fig. 7F). This potentially reflects a partial dissociation of Mic60 and VDAC complexes.
DISCUSSION
Mitoprotection as an important mechanism of αΒ-crystallin-mediated cardioprotection in desmin-deficient heart failure
We have demonstrated that αB-crystallin overexpression in the Des−/− myocardium completely halts the development of heart failure. The present results show that, although the mechanisms of cardioprotection might be occurring at various levels, αB-crystallin overexpression definitively provides extensive mitoprotection to Des−/− cardiomyocytes. The earliest features of the Des−/− pathology are mitochondrial structural perturbations, evident as early as the second week after birth, before any other cardiac dysfunction arises (Milner et al., 2000). These findings are further supported by additional studies showing that overexpression of Bcl-2, which has a known protective action on mitochondria, provides significant improvement of Des−/− cardiomyopathy (Weisleder et al., 2004). Our studies demonstrated an increase in oxidative stress, an increased activation of mPTP and a decreased Δψm in the absence of desmin. All these mitochondrial defects are corrected in the Des−/−αBCry myocardium, in which mitochondria have an apparently wild-type phenotype. It has been shown that the αB-crystallin mutation R120G, known to cause desminopathy characterized by desmin and αB-crystallin aggregation, also leads to redox imbalance (Rajasekaran et al., 2007). Defining the extent to which these aggregates contribute to the development and progression of the disease is of great importance. Clearing away the aggregates improves this model of proteotoxicity (Li et al., 2011; Su et al., 2015; Cabet et al., 2015; Pattison et al., 2011; Gupta et al., 2014), but it does not compensate for the loss of function of the aggregated proteins. Previous and present data with the Des−/− model – which shows no desmin and αB-crystallin aggregates but displays similar cellular and functional defects with those of desmin- and αB-crystallin-related cardiomyopathy (Milner et al., 1996, 1999, 2000; Li et al., 1996; Pattison et al., 2011; Wang et al., 2003; Wang and Robbins, 2006; McLendon and Robbins, 2011) – suggest alternative causes for this pathology, in addition to the gain of toxic function (Bhuiyan et al., 2013).
αB-crystallin and desmin – old partners in a new game
The association of desmin with αΒ-crystallin and the fact that mutations in either one of them lead to heart failure with common characteristics suggest a potential compensatory interplay between these two proteins in mitoprotection and consequently cardioprotection. Indeed, we found that both proteins are protecting mitochondria by, among other possible mechanisms, maintaining proper composition of mitochondrial proteins. This could take place either though facilitation of efficient protein targeting and transport of the nucleus-encoded mitochondrial proteins into the mitochondria, or through the support of the formation and/or stabilization of mitochondrial protein complexes. Up to now, the former possibility was considered more probable for a desmin-based scaffold, which, although it does associate with mitochondria through plectin (Winter et al., 2008), is mainly cytosolic. The present study identifies a new way by which desmin can regulate mitochondrial homeostasis. The contact sites where SR links to the OMMs and IMMs are established sites for multiple cellular processes, including Ca2+ and metabolite transfer, lipid metabolism, mitochondrial shape regulation, and autophagosome and inflammasome formation (Naon and Scorrano, 2014). The desmin scaffold, located at these contact sites, could contribute to all of the above processes, and particularly to the maintenance of SR–mitochondria proximity. This proximity allows the desmin scaffold, together with αΒ-crystallin, to facilitate directed protein and metabolite targeting to mitochondria. As shown here, and as previously suggested by others (Fountoulakis et al., 2005; Martindale et al., 2005; Maloyan et al., 2005), αB-crystallin does localize in and outside of mitochondria, and thus, could support multiple processes. The potential compensatory interplay between desmin and αΒ-crystallin is further supported by the fact that both of them associate with important mitochondrial proteins. Our data indicate that desmin plays a possible role in the formation and/or stabilization of Mic60 macromolecular complexes. In the absence of desmin, this role is restored by overexpressed αB-crystallin. As discussed above, Mic60 is crucial for the formation and function of the extended cristae membrane structures, the main sites of ATP synthesis by oxidative phosphorylation (Gilkerson et al., 2003; Vogel et al., 2006; Wurm and Jakobs, 2006). In addition, it is located at the contact sites (Harner et al., 2011) and has recently been implicated in mitochondrial protein import (van der Laan et al., 2012). It is thus conceivable that the decrease of Mic60 in the absence of desmin could compromise protein import into mitochondria. Our proteomic analysis revealed that the levels of many enzymes involved in aerobic respiration are diminished in the Des−/− cardiomyocytes (Table 1; Tables S3, S4), whereas αΒ-crystallin overexpression restores them, thus conferring to the cell high-energy electrons in the form of NADH and FADH2, which can be used for ATP generation by oxidative phosphorylation.
In addition to Mic60, F1F0-ATP synthase affects cristae membrane structure by imposing curvature through dimerization and oligomerization (Strauss et al., 2008; Dudkina et al., 2006; Minauro-Sanmiguel et al., 2005), thus providing the required conditions to enhance the catalytic capacity of the oxidative phosphorylation system (Strauss et al., 2008). Recently, mitochondrial cristae shape has been found to determine respiratory chain super-complex assembly and respiratory efficiency (Cogliati et al., 2013). Such efficiency minimizes the downstream ROS production discussed above, consistent with the proposed upstream action of αΒ-crystallin. All these data are also consistent with the significant decrease of ATP levels in Des−/− hearts and their restoration by αΒ-crystallin overexpression. Moreover, recent work indicates that the mPTP can be formed from ATP synthase dimers (Giorgio et al., 2013). The interaction of desmin and αΒ-crystallin with both ATP synthase and VDAC suggests a possible role of these proteins in the regulation of mitochondrial bioenergetics and mPTP opening.
The present data suggest that in normal heart the desmin cytoskeletal network could facilitate the mitoprotective and cytoprotective roles of the endogenous αΒ-crystallin. In the absence of desmin, αB-crystallin loses its Z-disc localization and thus the ability to reach the required high concentrations at a Z-disc-proximal SR– or endoplasmic reticulum (ER)–mitochondrial sites for its proper function. Indeed, much higher levels of αB-crystallin are required to facilitate the same level of protection in the absence of desmin. This is consistent with the presently proposed involvement of both proteins in the ER–mitochondria organizing network, which might constitute only part of the function of these partners in mitochondrial behavior.
MATERIALS AND METHODS
Transgenic animal generation
Des−/− mice had been previously generated in the 129SV inbred background (Milner et al., 1996). Transgenic mice overexpressing αΒ-crystallin were generated in the C57BL/6J background, using the αMHC promoter (Genbank ID U71441) (Subramaniam et al., 1991), driving expression of full-length murine αΒ-crystallin cDNA, followed by the bovine growth hormone poly-A sequence and backcrossed onto a pure 129SV mouse strain for several generations. The animals were used independent of sex. The approved procedures for animal care and treatment were followed, according to institutional guidelines following those of the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the recommendations of Federation of European Laboratory Animal Science Association (FELASA).
Southern and western blotting
For the identification of transgenes by southern blotting, a fragment of αMHC was isolated and used to generate 32P-labeled probes by random priming.
For western blotting analysis, proteins were transferred to polyvinylidene fluoride (PVDF) membrane (Whatman) and probed with the following antibodies: anti-αB-Crystallin (Stressgen, SPA223, 1:2000; Santa Cruz, sc-22744, 1:1000), anti-desmin (Abcam, ab8592, 1:1000), anti-GAPDH (Ambion, AM4300, 1:4000), anti-ANT (Santa Cruz, sc-9299, 1:200), anti-ATP5B (Mitosciences, ab5432, 1:500), anti-mitofilin (Novus Biologicals, NB100-1919, 1:1000; Abcam, ab110329, 1:1000), anti-NDUFS2 (Thermo Scientific, PA5-19342, 1:500), anti-SDHA (ab14715, 1:1000), anti-COXIV (ab33985, 1:1000), anti-VDAC1 (ab14734, 1:1000; ab15895, 1:1000; both Abcam), anti-mitofusin-2 (Sigma, M6444, 1:1000) and anti-MnSOD (BD Biosciences, M9920, 1:500) antibodies. Secondary horseradish peroxidase (HRP)-conjugated antibodies were purchased from Sigma-Aldrich. Proteins were visualized using chemiluminescence (ECL kit, Amersham). Films were scanned using a GS800 densitometer (Bio-Rad) and quantified using Quantity one software (Bio-Rad).
Histology
For the evaluation of fibrosis, Masson's-trichrome-stained paraffin sections were photographed and subjected to ImageJ software analysis. The results were expressed as the percentage of fibrosis in a given tissue section. For the evaluation of inflammation, hearts were observed under a Stemi 2000-C stereomicroscope (Zeiss) and were graded 0–4, according to the presence of inflammatory infiltrate, as previously described (Psarras et al., 2012) – 0=no inflammatory infiltrate; 1=one inflammatory infiltrate; 2=more than one inflammatory infiltrates appearing in limited areas of the myocardial wall; 3=multiple inflammatory infiltrates extended in multiple areas; 4=multiple extended inflammatory infiltrates eventually covering both the anterior and posterior wall.
Evans Blue staining
The mice were injected intraperitoneally with EBD (250 μg/g of body weight in 1×PBS). After 24 h, the heart was embedded in optimum-cutting temperature (OCT) compound (VWR) and snap frozen in liquid nitrogen. EBD staining was evaluated by using the ImageJ software, and the results were expressed as the percentage of EBD-stained area in the image.
Electron microscopy
Mice were treated with heparin for 30 min and euthanized, and the heart was perfused with 2.5% glutaraldehyde in 0.1 M phosphate puffer, pH 7.4, fixed overnight and post-fixed with 1% osmium tetroxide for 1 h at 4°C, dehydrated, embedded in epon–araldite resin mixture and allowed to polymerize at 60°C for 24 h. Ultrathin sections (65–70 nm) (Leica EM-UC7 ultramicrotome, Leica) were examined with a Philips 201C transmission electron microscope and photographed on Agfa Copex HDP13 microfilm.
For immunogold labeling, mice were perfused with cold 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Hearts were post fixed in the same fixative for 2 h and rinsed in 0.1 M phosphate buffer. The dehydration and embedding of tissue samples was performed using the progressive lowering of temperature (PLT) method using a Leica EM AFS (Leica), as described previously (Robertson et al., 1992).
Double immunogold labeling
Mounted ultrathin sections were subjected to the post-embedding immunogold procedure, as previously described (Havaki et al., 2003; Diokmetzidou et al., 2016). All incubations were performed using the automated immunogold labeling Leica EM IGL instrument. Primary antibodies (VDAC, Abcam, ab147234, 1:10; desmin, Abcam, ab8592, 1:20) were incubated overnight at 4°C, and samples were incubated for 1 h at room temperature with 10-nm-gold-conjugated secondary antibodies against polyclonal IgG particles (1:40) or 25-nm-gold-conjugated secondary antibodies against monoclonal IgG (1:40). Sections were examined with a Philips 420 transmission electron microscope at 60 kV and photographed with a Megaview G2 CCD camera (Olympus SIS).
Immunofluorescence
Frozen mouse cardiac sections were used for immunolabeling, as previously described (Diokmetzidou et al., 2016). Primary antibodies against αB-crystallin (Stressgen, SPA223, 1:100; Santa Cruz, sc-22744, 1:50) and α-actinin (Sigma-Aldrich, Α7811, 1:1000) were incubated overnight at 4°C, and with secondary antibodies (conjugated to Alexa-Fluor-594 or Alexa-Fluor-488, Invitrogen) for 1 h at room temperature. The sections were examined using a Leica TCS SP5 confocal microscope equipped with a HC-PL-APO-CS 63× oil objective and LAS-AF acquisition software (Leica).
Swimming trial
Mice were exercised twice a day using a swimming protocol, as previously described (Milner et al., 1999). The swimming program began at 10 min/day and was increased by 10 min daily to a maximum of 1 h. The twice-a-day 1 h exercise was continued for 9 days, and subsequently was increased to 90 min per session. The twice-daily 90 min exercise was continued for 9 days. In the case of αB-crystallin localization studies, a three-day protocol, during which the mice were exercised twice for 30 min the first day and twice daily for 1 h the next two days.
Echocardiography
Mice were anesthetized through intraperitoneal injection of ketamine (100 mg/kg of bodyweight). Echocardiographic studies were performed using a Vivid 7 GE ultrasound system with a 13-MHz linear transducer. Two-dimensional targeted M-mode imaging was obtained from the short-axis view at the level of greatest left ventricular dimension. Images were analyzed using the Echopac PC SW 3.1.3 software (GE Healthcare). Data were expressed as mean±s.d. for continuous variables and analyzed by using Statview 5.0 (Abacus Concepts). Statistical comparisons were performed using ANOVA with Bonferroni or Dunn post-hoc test, or the unpaired Student's t-test where appropriate.
Adult cardiomyocyte isolation
Isolation was performed using a modified protocol (Diokmetzidou et al., 2016) based on standard procedures (O'Connell et al., 2007).
Measurement of mitochondrial membrane potential, mPTP opening and ROS
Adult cardiomyocytes were incubated with 20 nM TMRM (Invitrogen) for 15 min at 37°C, washed and examined by using time-lapse confocal scanning for 20 min with a 1-min interval (TCS SP5, Leica) by excitation at 568 nm and emission at 580–640 nm. 5 μM cyclosporin A (Invitrogen) was added with TMRM to confirm mPTP opening. For the ROS measurement, the cardiomyocytes were incubated with 20 μΜ CM-H2DCFDA (Invitrogen) for 30 min at 37°C, washed and examined using a 488-nm argon-ion laser excitation and emission at 503–555 nm. A HCX-PL-APO-CS-20 ×0.70 dry UV objective and LAS-AF acquisition software (Leica) were used in both cases. The acquired images were analyzed using the Volocity 5 software (Perkin Elmer).
Ca2+-induced swelling of mitochondria was performed as previously described (Weisleder et al., 2004). 5 μΜ CsA was added to mitochondria 5 min before CaCl2.
Tissue fractionation, mitochondrial purification and isolation of MAMs
Fractionation of mouse or rat hearts and the isolation of MAMs and pure mitochondria were performed by differential centrifugation at 4°C, as previously described (Wieckowski et al., 2009; Diokmetzidou et al., 2016; Frezza et al., 2007).
GSH measurements
The GSH concentration was measured in hearts of 6-month-old mice, as previously described (Rahman et al., 2006). The hearts were weighed, homogenized in 5% sulfosalicilic acid and centrifuged at 10,000 g for 10 min at 4°C. The supernatant was removed and used for the GSH assay based on that of Griffith (1980).
Determination of ATP levels
ATP content was measured using the Aposensor ATP assay kit (Biovision) in a luminometer (Berthold) according to the manufacturer's instructions.
Two-dimensional native blue and SDS PAGE
Mouse cardiac mitochondria were solubilized in native solubilization buffer pH 7 (20 mM Bis-Tris, 500 mM aminocaproic acid, 20 mM NaCl, 2 mM EDTA pH 8, 10% glycerol, 2 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail) with 4 g digitonin per g of protein for 30 min on ice and analyzed in a 3–12% native gel (Serva). Individual lanes were excised, incubated in equilibration buffer (125 mM Tris-HCl, pH 6.8, and 1% SDS) for 20 min and analyzed in the second dimension by 10% gel SDS-PAGE. Proteins were transferred to PVDF membrane (Whatman) and probed with the desired antibodies.
Two-dimensional gel electrophoresis
Two-dimensional gel electrophoresis was performed as previously described (Makridakis et al., 2010) with the following specifications. Mitochondrial fractions were solubilized in the isoelectric focusing (IEF) sample buffer [7 M urea, 2 M thiourea, 4% CHAPS, 1% dithioerythreitol, 2% ampholytes (pH range: 3–10)] and resolved (100 μg per sample) on 7-cm strips, pH range 3–10 non-linear (Bio-Rad), using the in gel rehydration method. IEF was conducted for about 12,500 volt-hours. Strips were then incubated in equilibration buffer (6 M urea, 50 mM Tris-HCl pH 8.8, 30% glycerol, 2% SDS) containing 0.5% dithioerythreitol for 45 min with gentle agitation, followed by a second incubation with equilibration buffer containing 4.32% iodoacetamide. Second dimensional analysis was performed on a 12% SDS-PAGE gel, and the gels were stained with Coomassie Colloidal Blue stain (Candiano et al., 2004).
Quantification of spots
Gels were scanned on a GS-800 imaging densitometer (Bio-Rad) in transmission mode, and image analysis was performed with the PD Quest 8 software package (Bio-Rad). Individual protein spot quantification was normalized based on the total intensity of the spots in the gel and expressed in ppm. Statistical analysis was conducted using the Mann–Whitney and Student's t-test.
Protein identification with MALDI-TOF MS
Protein spots were excised manually or automatically with the use of the ProteineerSp Protein picker (BrukerDaltonics), destained (30% acetonitrile, 50 mM NH4HCO3), washed, dried and trypsinized overnight (trypsin, proteomics-grade, Roche). Peptides were extracted (50% acetonitrile, 0.1% trifluoroacetic acid) and applied onto the matrix-assisted laser desorption ionization (MALDI) target using the dry droplet method. Peptide masses were determined by using MALDI time-of-flight mass spectrometry (MALDI-TOF)–TOF-MS (Ultraflex TOF/TOF instrument, BrukerDaltonics). The peak list was created with Flexanalysis v3.3 software (Bruker); smoothing was applied with the Savitzky–Golay algorithm (width 0.2 m/z, cycle number 1), and a signal:noise threshold ratio of 2.5 was allowed. For peptide matching (Mascot Server 2; Matrix Science), the following settings were used: monoisotopic mass, one miscleavage site, carbamidomethylation of cysteine as fixed and oxidation of methionine as variable modifications. Stringent criteria were used for protein identification with a maximum allowed mass error of 25 ppm and a minimum of four matching peptides. False identity probability was usually lower than 10−5. Data analyzed with the same settings as described above, using a sequence scrambled version of Swiss-Prot data base generated by the decoy-generating script that is available at Matrix Science, provided no identifications.
Expression and purification of GST-tagged proteins
Desmin mutants – head, amino acids 1–108 (1–324 bp); head-rod, amino acids 1–412 (1–1236 bp); rod, amino acids 109–412 (324–1236 bp); rod-tail, amino acids 109–470 (324–1410 bp); tail, amino acids 413–470 (1237–1410 bp). VDAC open reading frame and mutants – ΔNterm, amino acids 33–296 (99–888 bp) lacking the N-terminal α-helix; Δβ1–β4, lacks amino acids 34–81 (102–243 bp), thus lacking β-barrels 1–4. Sequences were isolated by using PCR with wild-type mouse heart cDNA as template and DNA Q5 high fidelity polymerase and sub-cloned into pGEX-4T-3. The recombinant polypeptides were expressed in BL21 bacteria, and the proteins were isolated as previously described (Kouloumenta et al., 2007; Diokmetzidou et al., 2016)
GST pull down and co-immunoprecipitation
Mouse cardiac mitochondria (5 mg) were solubilized in native buffer pH 7 (20 mM Bis-Tris, 500 mM aminocaproic acid, 20 mM NaCl, 2 mM EDTA, pH 8, 10% glycerol, 2 mM PMSF, protease inhibitor cocktail), containing 4 g digitonin/g of protein, for 30 min on ice and were pre-cleared for 3 h at 4°C and used as previously described (Diokmetzidou et al., 2016). The bound proteins were eluted by heating at 97°C for 10 min in 2× electrophoresis loading buffer or, in the case of GST pull down, with 20 mM reduced GSH in 50 mM Tris-HCl, pH 8, for 30 min at room temperature. The eluates were analyzed by western blotting.
Protease accessibility assay
One hundred μg (1 mg/ml) of crude mouse cardiac mitochondria were suspended in MSE buffer (225 mM mannitol, 75 mM sucrose, 1 mm EGTA, 1 mM Tris-HCl pH 7.4) or osmotic buffer (20 mM KCl) with or without 0.5% Triton X-100 and incubated with 2 μg/ml proteinase K for 1 h on ice. The reaction was stopped with 4 mM (final) PMSF. Samples were loaded with 2× SDS-PAGE loading buffer and analyzed by western blotting.
Statistical analysis
All data are expressed as mean±s.e.m. unless otherwise stated. Statistical comparisons were performed using ANOVA with Bonferroni or Dunn post-hoc test, or unpaired Student's t-test where appropriate. A P-value<0.05 was considered significant. Graphics and statistical analysis were performed using Sigma Plot 10.0 (Systat Software).
Acknowledgements
We thank very much S. Pagkakis and E. Rigana for their help with imaging. We are grateful to E. Mavroidis and I. Kostavasili for constant assistance throughout this work. We also thank Prof. William James Craigen and N. Flytzanis for valuable comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
A.D. designed and performed experiments, analyzed data and wrote the manuscript; E.S. and M.T. performed experiments; I.K. performed the electron microscopy experiments; M.M. and A. Vlahou performed and analyzed the proteomic experiments; S.G. helped with transgenic mice generation; A. Varela and C.H.D. performed and analyzed the echocardiography; Y.C. directed the research project, analyzed the data and wrote the manuscript. I.K. and M.T. made equal contributions to the work.
Funding
This work was supported by ESPA 2007-2013 grants from the Greek General Secretariat of Research and Development – Ministerium of Education [grant numbers PENED 01ED371, EPAN YB-22, PEP ATT-39 and ESPA 09SYN-21-965 to Y.C.], including a grant of Excellence II/ARISTEIA II 5342 (to Y.C.); a grant from the US National Institutes of Health [grant number RO1 AR39617 to Y.C.]; and in part by a European Commission grant [grant number FP7/REGPOT-2008-1 (Transmed)] to A.V. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.192203.supplemental
References
- Arai H. and Atomi Y. (1997). Chaperone activity of alpha B-crystallin suppresses tubulin aggregation through complex formation. Cell Struct. Funct. 22, 539-544. 10.1247/csf.22.539 [DOI] [PubMed] [Google Scholar]
- Bennardini F., Wrzosek A. and Chiesi M. (1992). Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ. Res. 71, 288-294. 10.1161/01.RES.71.2.288 [DOI] [PubMed] [Google Scholar]
- Bhuiyan M. S., Pattison J. S., Osinska H., James J., Gulick J., Mclendon P. M., Hill J. A., Sadoshima J. and Robbins J. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284-5297. 10.1172/JCI70877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabet E., Batonnet-Pichon S., Delort F., Gausserès B., Vicart P. and Lilienbaum A. (2015). Antioxidant treatment and induction of autophagy cooperate to reduce desmin aggregation in a cellular model of desminopathy. PLoS ONE 10, e0137009 10.1371/journal.pone.0137009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L. and Righetti P. G. (2004). Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327-1333. 10.1002/elps.200305844 [DOI] [PubMed] [Google Scholar]
- Capetanaki Y., Bloch R. J., Kouloumenta A., Mavroidis M. and Psarras S. (2007). Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 313, 2063-2076. 10.1016/j.yexcr.2007.03.033 [DOI] [PubMed] [Google Scholar]
- Capetanaki Y., Papathanasiou S., Diokmetzidou A., Vatsellas G. and Tsikitis M. (2015). Desmin related disease: a matter of cell survival failure. Curr. Opin. Cell Biol. 32, 113-120. 10.1016/j.ceb.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S., Frezza C., Soriano M. E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L. C. et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160-171. 10.1016/j.cell.2013.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshi M., Mendiola V. L., Mackey M. R., Murphy A. N., Koller A., Perkins G. A., Ellisman M. H. and Taylor S. S. (2011). ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 286, 2918-2932. 10.1074/jbc.M110.171975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diokmetzidou A., Tsikitis M., Nikouli S., Kloukina I., Tsoupri E., Papathanasiou S., Psarras S., Mavroidis M. and Capetanaki Y. (2016). Strategies to study Desmin in cardiac muscle and culture systems. Methods Enzymol. 568, 427-459. 10.1016/bs.mie.2015.09.026 [DOI] [PubMed] [Google Scholar]
- Dudkina N. V., Sunderhaus S., Braun H.-P. and Boekema E. J. (2006). Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett. 580, 3427-3432. 10.1016/j.febslet.2006.04.097 [DOI] [PubMed] [Google Scholar]
- Fountoulakis M., Soumaka E., Rapti K., Mavroidis M., Tsangaris G., Maris A., Weisleder N. and Capetanaki Y. (2005). Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J. Mol. Cell. Cardiol. 38, 461-474. 10.1016/j.yjmcc.2004.12.008 [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Martins De Brito O., Micaroni M., Beznoussenko G. V., Rudka T., Bartoli D., Polishuck R. S., Danial N. N., De Strooper B. et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177-189. 10.1016/j.cell.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S. and Scorrano L. (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2, 287-295. 10.1038/nprot.2006.478 [DOI] [PubMed] [Google Scholar]
- Gieffers C., Korioth F., Heimann P., Ungermann C. and Frey J. (1997). Mitofilin is a transmembrane protein of the inner mitochondrial membrane expressed as two isoforms. Exp. Cell Res. 232, 395-399. 10.1006/excr.1997.3539 [DOI] [PubMed] [Google Scholar]
- Gilkerson R. W., Selker J. M. L. and Capaldi R. A. (2003). The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 546, 355-358. 10.1016/S0014-5793(03)00633-1 [DOI] [PubMed] [Google Scholar]
- Giorgio V., Von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., Glick G. D., Petronilli V., Zoratti M., Szabo I. et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 110, 5887-5892. 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb L. G., Park K.-Y., Cervenaková L., Gorokhova S., Lee H.-S., Vasconcelos O., Nagle J. W., Semino-Mora C., Sivakumar K. and Dalakas M. C. (1998). Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 19, 402-403. 10.1038/1300 [DOI] [PubMed] [Google Scholar]
- Golenhofen N., Ness W., Koob R., Htun P., Schaper W. and Drenckhahn D. (1998). Ischemia-induced phosphorylation and translocation of stress protein alpha B-crystallin to Z lines of myocardium. Am. J. Physiol. 274, H1457-H1464. [DOI] [PubMed] [Google Scholar]
- Griffith O. W. (1980). Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 106, 207-212. 10.1016/0003-2697(80)90139-6 [DOI] [PubMed] [Google Scholar]
- Gupta M. K., Gulick J., Liu R., Wang X., Molkentin J. D. and Robbins J. (2014). Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ. Res. 115, 721-729. 10.1161/CIRCRESAHA.115.304760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F. and Neupert W. (2011). The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 30, 4356-4370. 10.1038/emboj.2011.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havaki S., Kittas C., Marinos E., Dafni U., Sotiropoulou C., Goutas N., Voloudakis-Baltatzis I., Vassilaros S. D., Athanasiou E. and Arvanitis D. L. (2003). Ultrastructural immunostaining of infiltrating ductal breast carcinomas with the monoclonal antibody H: a comparative study with cytokeratin 8. Ultrastruct. Pathol. 27, 393-407. 10.1080/01913120390209875 [DOI] [PubMed] [Google Scholar]
- Hnia K., Tronchère H., Tomczak K. K., Amoasii L., Schultz P., Beggs A. H., Payrastre B., Mandel J. L. and Laporte J. (2011). Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Invest. 121, 70-85. 10.1172/JCI44021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S., Collins S. R., Cassidy-Stone A., Hummel E., Devay R. M., Lackner L. L., Westermann B., Schuldiner M., Weissman J. S. and Nunnari J. (2011). A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 195, 323-340. 10.1083/jcb.201107053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck S. A. and Clark J. I. (2010). Dynamic subunit exchange and the regulation of microtubule assembly by the stress response protein human αB crystallin. PLoS ONE 5, e11795 10.1371/journal.pone.0011795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T., Kume-Iwaki A., Liem R. K. H. and Goldman J. E. (1989). αB-crystallin is expressed in non-lenticular tissues and accumulates in Alexander's disease brain. Cell 57, 71-78. 10.1016/0092-8674(89)90173-6 [DOI] [PubMed] [Google Scholar]
- Kouloumenta A., Mavroidis M. and Capetanaki Y. (2007). Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J. Biol. Chem. 282, 35211-35221. 10.1074/jbc.M704733200 [DOI] [PubMed] [Google Scholar]
- Li Z., Colucci-Guyon E., Pinçon-Raymond M., Mericskay M., Pournin S., Paulin D. and Babinet C. (1996). Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 175, 362-366. 10.1006/dbio.1996.0122 [DOI] [PubMed] [Google Scholar]
- Li J., Horak K. M., Su H., Sanbe A., Robbins J. and Wang X. (2011). Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J. Clin. Invest. 121, 3689-3700. 10.1172/JCI45709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makridakis M., Roubelakis M. G., Bitsika V., Dimuccio V., Samiotaki M., Kossida S., Panayotou G., Coleman J., Candiano G., Anagnou N. P. et al. (2010). Analysis of secreted proteins for the study of bladder cancer cell aggressiveness. J. Proteome Res. 9, 3243-3259. 10.1021/pr100189d [DOI] [PubMed] [Google Scholar]
- Maloyan A., Sanbe A., Osinska H., Westfall M., Robinson D., Imahashi K.-i., Murphy E. and Robbins J. (2005). Mitochondrial dysfunction and apoptosis underlie the pathogenic process in α-B-crystallin desmin-related cardiomyopathy. Circulation 112, 3451-3461. 10.1161/CIRCULATIONAHA.105.572552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale J. J., Wall J. A., Martinez-Longoria D. M., Aryal P., Rockman H. A., Guo Y., Bolli R. and Glembotski C. C. (2005). Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J. Biol. Chem. 280, 669-676. 10.1074/jbc.M406690200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavroidis M. and Capetanaki Y. (2002). Extensive induction of important mediators of fibrosis and dystrophic calcification in desmin-deficient cardiomyopathy. Am. J. Pathol. 160, 943-952. 10.1016/S0002-9440(10)64916-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavroidis M., Davos C. H., Psarras S., Varela A., Athanasiadis N. C., Katsimpoulas M., Kostavasili I., Maasch C., Vater A., Van Tintelen J. P. et al. (2015). Complement system modulation as a target for treatment of arrhythmogenic cardiomyopathy. Basic Res. Cardiol. 110, 27 10.1007/s00395-015-0485-6 [DOI] [PubMed] [Google Scholar]
- McLendon P. M. and Robbins J. (2011). Desmin-related cardiomyopathy: an unfolding story. Am. J. Physiol. Heart Circ. Physiol. 301, H1220-H1228. 10.1152/ajpheart.00601.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner D. J., Weitzer G., Tran D., Bradley A. and Capetanaki Y. (1996). Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 134, 1255-1270. 10.1083/jcb.134.5.1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner D. J., Taffet G. E., Wang X., Pham T., Tamura T., Hartley C., Gerdes A. M. and Capetanaki Y. (1999). The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J. Mol. Cell. Cardiol. 31, 2063-2076. 10.1006/jmcc.1999.1037 [DOI] [PubMed] [Google Scholar]
- Milner D. J., Mavroidis M., Weisleder N. and Capetanaki Y. (2000). Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 150, 1283-1298. 10.1083/jcb.150.6.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minauro-Sanmiguel F., Wilkens S. and Garcia J. J. (2005). Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. USA 102, 12356-12358. 10.1073/pnas.0503893102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A., Basak T., Datta K., Naskar S., Sengupta S. and Sarkar S. (2013). Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 4, e582 10.1038/cddis.2013.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naon D. and Scorrano L. (2014). At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 1843, 2184-2194. 10.1016/j.bbamcr.2014.05.011 [DOI] [PubMed] [Google Scholar]
- Narula N., Zaragoza M. V., Sengupta P. P., Li P., Haider N., Verjans J., Waymire K., Vannan M. and Wallace D. C. (2011). Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging 4, 1-10. 10.1016/j.jcmg.2010.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholl I. D. and Quinlan R. A. (1994). Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO J. 13, 945-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'connell T. D., Rodrigo M. C. and Simpson P. C. (2007). Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 357, 271-296. 10.1385/1-59745-214-9:271 [DOI] [PubMed] [Google Scholar]
- Odgren P. R., Toukatly G., Bangs P. L., Gilmore R. and Fey E. G. (1996). Molecular characterization of mitofilin (HMP), a mitochondria-associated protein with predicted coiled coil and intermembrane space targeting domains. J. Cell Sci. 109, 2253-2264. [DOI] [PubMed] [Google Scholar]
- Panagopoulou P., Davos C. H., Milner D. J., Varela E., Cameron J., Mann D. L. and Capetanaki Y. (2008). Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J. Cell Biol. 181, 761-775. 10.1083/jcb.200710049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanasiou S., Rickelt S., Soriano M. E., Schips T. G., Maier H. J., Davos C. H., Varela A., Kaklamanis L., Mann D. L. and Capetanaki Y. (2015). Tumor necrosis factor-α confers cardioprotection through ectopic expression of keratins K8 and K18. Nat. Med. 21, 1076-1084. 10.1038/nm.3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison J. S., Osinska H. and Robbins J. (2011). Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 109, 151-160. 10.1161/CIRCRESAHA.110.237339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumard P., Vaillier J., Coulary B., Schaeffer J., Soubannier V., Mueller D. M., Brèthes D., Di Rago J.-P. and Velours J. (2002). The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21, 221-230. 10.1093/emboj/21.3.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng M. D., Cairns L., Van Den I. P., Prescott A., Hutcheson A. M. and Quinlan R. A. (1999). Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J. Cell Sci. 112, 2099-2112. [DOI] [PubMed] [Google Scholar]
- Psarras S., Mavroidis M., Sanoudou D., Davos C. H., Xanthou G., Varela A. E., Panoutsakopoulou V. and Capetanaki Y. (2012). Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 33, 1954-1963. 10.1093/eurheartj/ehr119 [DOI] [PubMed] [Google Scholar]
- Rahman I., Kode A. and Biswas S. K. (2006). Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 1, 3159-3165. 10.1038/nprot.2006.378 [DOI] [PubMed] [Google Scholar]
- Rajasekaran N. S., Connell P., Christians E. S., Yan L. J., Taylor R. P., Orosz A., Zhang X. Q., Stevenson T. J., Peshock R. M., Leopold J. A. et al. (2007). Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130, 427-439. 10.1016/j.cell.2007.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reipert S., Steinböck F., Fischer I., Bittner R. E., Zeöld A. and Wiche G. (1999). Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp. Cell Res. 252, 479-491. 10.1006/excr.1999.4626 [DOI] [PubMed] [Google Scholar]
- Robertson D., Monaghan P., Clarke C. and Atherton A. J. (1992). An appraisal of low-temperature embedding by progressive lowering of temperature into Lowicryl HM20 for immunocytochemical studies. J. Microsc. 168, 85-100. 10.1111/j.1365-2818.1992.tb03253.x [DOI] [PubMed] [Google Scholar]
- Singh B. N., Rao K. S., Ramakrishna T., Rangaraj N. and Rao Ch M. (2007). Association of αB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J. Mol. Biol. 366, 756-767. 10.1016/j.jmb.2006.12.012 [DOI] [PubMed] [Google Scholar]
- Strauss M., Hofhaus G., Schröder R. R. and Kühlbrandt W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154-1160. 10.1038/emboj.2008.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromer M. H. and Bendayan M. (1990). Immunocytochemical identification of cytoskeletal linkages to smooth muscle cell nuclei and mitochondria. Cell Motil. Cytoskeleton 17, 11-18. 10.1002/cm.970170104 [DOI] [PubMed] [Google Scholar]
- Su H., Li J., Zhang H., Ma W., Wei N., Liu J. and Wang X. (2015). COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ. Res. 117, 956-966. 10.1161/CIRCRESAHA.115.306783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam A., Jones W. K., Gulick J., Wert S., Neumann J. and Robbins J. (1991). Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J. Biol. Chem. 266, 24613-24620. [PubMed] [Google Scholar]
- Van Der Laan M., Bohnert M., Wiedemann N. and Pfanner N. (2012). Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol. 22, 185-192. 10.1016/j.tcb.2012.01.004 [DOI] [PubMed] [Google Scholar]
- Van Spaendonck-Zwarts K. Y., Van Hessem L., Jongbloed J. D. H., De Walle H. E. K., Capetanaki Y., Van Der Kooi A. J., Van Langen I. M., Van Den Berg M. P. and Van Tintelen J. P. (2011). Desmin-related myopathy. Clin. Genet. 80, 354-366. 10.1111/j.1399-0004.2010.01512.x [DOI] [PubMed] [Google Scholar]
- Vicart P., Caron A., Guicheney P., Li Z., Prévost M.-C., Faure A., Chateau D., Chapon F., Tomé F., Dupret J.-M. et al. (1998). A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 20, 92-95. 10.1038/1765 [DOI] [PubMed] [Google Scholar]
- Vogel F., Bornhövd C., Neupert W. and Reichert A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175, 237-247. 10.1083/jcb.200605138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelpohl I., Vetter R., Heger J., Ebermann L., Euler G., Schultheiss H. P. and Dörner A. (2011). Transgenic overexpression of heart-specific adenine nucleotide translocase 1 positively affects contractile function in cardiomyocytes. Cell. Physiol. Biochem. 27, 121-128. [DOI] [PubMed] [Google Scholar]
- Von der Malsburg K., Müller J. M., Bohnert M., Oeljeklaus S., Kwiatkowska P., Becker T., Loniewska-Lwowska A., Wiese S., Rao S., Milenkovic D. et al. (2011). Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 21, 694-707. 10.1016/j.devcel.2011.08.026 [DOI] [PubMed] [Google Scholar]
- Wang X. and Robbins J. (2006). Heart failure and protein quality control. Circ. Res. 99, 1315-1328. 10.1161/01.RES.0000252342.61447.a2 [DOI] [PubMed] [Google Scholar]
- Wang X., Osinska H., Klevitsky R., Gerdes A. M., Nieman M., Lorenz J., Hewett T. and Robbins J. (2001). Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 89, 84-91. 10.1161/hh1301.092688 [DOI] [PubMed] [Google Scholar]
- Wang X., Klevitsky R., Huang W., Glasford J., Li F. and Robbins J. (2003). AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ. Res. 93, 998-1005. 10.1161/01.RES.0000102401.77712.ED [DOI] [PubMed] [Google Scholar]
- Weisleder N., Taffet G. E. and Capetanaki Y. (2004). Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc. Natl. Acad. Sci. USA 101, 769-774. 10.1073/pnas.0303202101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein G., Bellaye P. S., Micheau O. and Bonniaud P. (2012). Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity? Int. J. Biochem. Cell Biol. 44, 1680-1686. 10.1016/j.biocel.2012.05.024 [DOI] [PubMed] [Google Scholar]
- Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J. and Pinton P. (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582-1590. 10.1038/nprot.2009.151 [DOI] [PubMed] [Google Scholar]
- Winter L., Abrahamsberg C. and Wiche G. (2008). Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 181, 903-911. 10.1083/jcb.200710151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T. and Goldman J. E. (1998). Alpha B-crystallin is associated with intermediate filaments in astrocytoma cells. Neurochem. Res. 23, 385-392. 10.1023/A:1022465702518 [DOI] [PubMed] [Google Scholar]
- Wurm C. A. and Jakobs S. (2006). Differential protein distributions define two sub-compartments of the mitochondrial inner membrane in yeast. FEBS Lett. 580, 5628-5634. 10.1016/j.febslet.2006.09.012 [DOI] [PubMed] [Google Scholar]
- Xie J., Marusich M. F., Souda P., Whitelegge J. and Capaldi R. A. (2007). The mitochondrial inner membrane protein mitofilin exists as a complex with SAM50, metaxins 1 and 2, coiled-coil-helix coiled-coil-helix domain-containing protein 3 and 6 and DnaJC11. FEBS Lett. 581, 3545-3549. 10.1016/j.febslet.2007.06.052 [DOI] [PubMed] [Google Scholar]
- Zick M., Rabl R. and Reichert A. S. (2009). Cristae formation-linking ultrastructure and function of mitochondria. Biochim. Biophys. Acta 1793, 5-19. 10.1016/j.bbamcr.2008.06.013 [DOI] [PubMed] [Google Scholar]