

Abstract
Endothelial barrier function is tightly regulated by plasma membrane receptors and is crucial for tissue fluid homeostasis; its dysfunction causes disease, including sepsis and inflammation. The ubiquitous activation of Ca2+ signaling upon phospholipase C-coupled receptor ligation leads quite naturally to the assumption that Ca2+ signaling is required for receptor-regulated endothelial barrier function. This widespread hypothesis draws analogy from smooth muscle and proposes the requirement of G protein-coupled receptor (GPCR)-generated Ca2+ signaling in activating the endothelial contractile apparatus and generating interendothelial gaps. Notwithstanding endothelia being non-excitable in nature, the hypothesis of Ca2+-induced endothelial contraction has been invoked to explain actions of GPCR agonists that either disrupt or stabilize endothelial barrier function. Here, we challenge this correlative hypothesis by showing a lack of causal link between GPCR-generated Ca2+ signaling and changes in human microvascular endothelial barrier function. We used three endogenous GPCR agonists: thrombin and histamine, which disrupt endothelial barrier function, and sphingosine-1-phosphate, which stabilizes barrier function. The qualitatively different effects of these three agonists on endothelial barrier function occur independently of Ca2+ entry through the ubiquitous store-operated Ca2+ entry channel Orai1, global Ca2+ entry across the plasma membrane, and Ca2+ release from internal stores. However, disruption of endothelial barrier function by thrombin and histamine requires the Ca2+ sensor stromal interacting molecule-1 (STIM1), whereas sphingosine-1-phosphate-mediated enhancement of endothelial barrier function occurs independently of STIM1. We conclude that although STIM1 is required for GPCR-mediated disruption of barrier function, a causal link between GPCR-induced cytoplasmic Ca2+ increases and acute changes in barrier function is missing. Thus, the cytosolic Ca2+-induced endothelial contraction is a cum hoc fallacy that should be abandoned.
Keywords: calcium release-activated calcium channel protein 1 (ORAI1), endothelial cell, endothelial dysfunction, G protein-coupled receptor (GPCR), stromal interaction molecule 1 (STIM1), calcium signaling
Introduction
The endothelial layer of blood vessels is a highly regulated barrier between the bloodstream and the interstitial tissue, controlling transvascular passage of fluids, solutes, and cells. A significant contribution to endothelial permeability resides in the paracellular diffusion pathway facilitated via intercellular gaps (1–3). Paracellular permeability is essentially mediated by cell-cell junctional proteins, which are in turn regulated by intracellular signaling pathways that impact on cytoskeletal architecture (2, 4). The balance between competing tethering and disassembling mechanisms determines the degree of endothelial barrier function and thus the extent of vascular leakage. Disruption of endothelial barrier function causes increased vascular permeability and is associated with reorganization of the actin cytoskeleton and disassembly of adherens junctions which are contributed by vascular endothelial cadherin (VE-cadherin)·catenin2 complexes (2, 4). These events are under the control of various signaling pathways that are activated by diverse paracrine and autocrine mediators in blood and interstitial tissue, many of which act on heterotrimeric G protein-coupled receptors (GPCR) or receptor tyrosine kinase and play crucial roles in the control of vascular permeability, tone, angiogenesis and inflammation (2–4).
Barrier disrupting GPCR agonists including thrombin and histamine activate downstream signaling that regulate actin reorganization and VE-cadherin localization triggered by activation of the small GTPase RhoA (1, 2). GPCR-mediated activation of the heterotrimeric G protein Gα12/13 family subtype causes the association and activation of p115Rho guanine nucleotide exchange factor (p115RhoAGEF), leading to RhoA activation (1, 2). RhoA then activates Rho kinase (ROCK), which phosphorylates the regulatory subunit of the myosin light chain phosphatase and inhibits its activity. This in turn enhances the phosphorylation of myosin light chain (MLC), causing actin-myosin interactions and disruption of endothelial barrier function (5). Furthermore, ROCK can directly phosphorylate MLC to cause barrier disruption (6). The signaling pathways activated downstream barrier-stabilizing GPCR agonists such as the sphingosine-1-phosphate (S1P) are less understood. S1P acts on heteromultimeric Gαi proteins leading to the activation of the small GTPase Rac and focal adhesion kinase, thus promoting formation of focal adhesions, cortical actin, and adherens junction assembly (3, 7).
Although it is obvious that endothelial cells develop force in response to barrier-disrupting agonists such as thrombin and histamine, the tension developed by endothelial cells in response to the most powerful agonist thrombin is at least 50-fold weaker than the bona fide tension generated by smooth muscle cells during contraction (8). Nevertheless, during the past three decades Ca2+-dependent endothelial contraction, a concept extrapolated from studies on muscle cells, has been invoked to explain changes in endothelial barrier function downstream GPCR agonists. Barrier disrupting GPCR agonists such as thrombin and histamine activate Gαq,11 protein and induce the production of inositol 1,4,5-trisphosphate (IP3) through the action of phospholipase C. This will result in Ca2+ release from the IP3-sensitive internal stores of the endoplasmic reticulum (ER) and activation of Ca2+ entry across the plasma membrane through the ubiquitous store-operated Ca2+ entry (SOCE) pathway activated by ER store depletion (9). It is now appreciated that ER store depletion causes the ER-resident Ca2+ sensor stromal-interacting molecule 1 (STIM1) to move toward ER-plasma membrane junctional spaces to trap and directly activate Orai1 Ca2+ entry channels (10–17). According to the Ca2+-dependent model, the sustained Ca2+ entry signal thus generated (but not Ca2+ release) activates a key Ca2+- and calmodulin-dependent kinase, the myosin light chain kinase (MLCK) leading to MLC phosphorylation, formation of actin stress fibers, and endothelial contraction resulting in formation of intercellular gaps (3, 18–21). For the barrier-stabilizing agonist S1P, Ca2+ release from internal stores, but not Ca2+ entry, was proposed to induce Rac activation, thus promoting assembly of adherens junctions and strengthening of endothelial barrier function (22).
Early studies from our group and others demonstrated that in endothelial cells from various vascular beds (human pulmonary artery, human dermal microvasculature, and human umbilical vein) thrombin, VEGF, and the store-depleting drug thapsigargin activate SOCE encoded by STIM1 and Orai1 (11, 23–25). In a recent study, we have challenged the hypothesis that SOCE is required for endothelial contraction in response to the powerful barrier-disrupting agonist thrombin (23). We demonstrated using molecular tools that thrombin-mediated endothelial barrier disruption required the ER-resident STIM1 protein but occur independently of SOCE, Orai1, and MLCK (23). We also showed that STIM1 is required for RhoA activation, MLC phosphorylation, actin reorganization, and disruption of intercellular adhesions (23).
In the current study, we set out to determine whether these findings are unique to thrombin or shared by other barrier-altering or barrier-enhancing GPCR agonists and whether Ca2+ release from the ER is required for agonist-mediated effects on endothelial barrier function. We thus used high throughput impedance measurements to determine the role of Ca2+ release and Ca2+ entry mechanisms in regulating endothelial barrier function downstream of three GPCR agonists, namely thrombin, histamine, and S1P. Thrombin and histamine are two typical inflammatory agonists that cause transient barrier disruption, whereas the platelet-derived agonist S1P enhances endothelial barrier function. These three agonists are of major relevance to vascular pathologies such as inflammation, allergy, and atherosclerosis. We compared side by side the effects of these three agonists on endothelial barrier function using electrical measurements and fluorescence microscopy. We also monitored the Ca2+ release from stores and Ca2+ entry across the plasma membrane induced by these agonists. We report that although Ca2+ signaling in response to these agonists coincides with changes in barrier function, neither Ca2+ entry nor Ca2+ release are necessary for GPCR-mediated disruption or enhancement of barrier function. We show that Orai1 plays no significant role in thrombin-, histamine-, and S1P-induced changes in endothelial barrier function. However, STIM1 is required for disruption of endothelial barrier function by thrombin and histamine but is not involved in S1P-mediated enhancement of endothelial barrier function.
Results
Thrombin, Histamine, and S1P Evoke Distinct Impedance Response Profiles in HDMECs
Impedance measurements were performed in monolayers of human dermal microvascular endothelial cells (HDMECs) before and after addition of GPCR agonists using ECIS protocols as described in methods and outlined in details recently (26). Upon stimulation of confluent HDMEC monolayers with GPCR agonists, thrombin, histamine, and S1P, each agonist showed a typical response as shown in Fig. 1. Although thrombin and histamine lead to a transient disruption of endothelial barrier function (Fig. 1, A and B), S1P enhanced (or stabilized) endothelial barrier function (Fig. 1C). Furthermore, the barrier-disrupting response of HDMEC monolayers to thrombin and histamine differed in severity and kinetics; thrombin effects were more profound, and the barrier took over an hour to recover, whereas the histamine effects were modest in comparison, and recovery of the barrier to pre-agonist levels occurred within 10 min (Fig. 1, A and B). Changes in electrical resistance on ECIS electrodes are caused by changes in cell morphology and changes at the intercellular junctions caused by rearrangements of actin and adherens junction proteins. The opening of cell-cell junctions increases paracellular current flow and thus causes decreases in resistance. Reciprocally, strengthening of cell-cell junctions reduces paths for current flow and leads to resistance increases. These results are consistent with cell morphology changes that were assessed microscopically: immunofluorescence staining for actin (green) and VE-cadherin (red) under control conditions and 2 min after addition of the respective agonists showed that although thrombin caused a disruption of adherens junctions, spanning the full length of cell-cell contact sites, histamine caused localized small oval-shaped openings that seem lacking VE-cadherin staining (Fig. 1D). S1P on the other hand led to increased VE-cadherin staining at the cell-cell junctions (Fig. 1D). To illustrate the changes in VE-cadherin and actin distribution during the time course of HDMEC response to thrombin and histamine, cell layers were stained at specific time points after agonist addition as indicated by red arrows in Fig. 1E for thrombin and Fig. 1F for histamine. HDMEC immunofluorescence images representing time points at 2, 5, 10, and 30 min after thrombin stimulation and 1, 5, and 20 min after histamine stimulation are shown in Fig. 1 (G and H), respectively. For comparison, typical control non-stimulated HDMEC monolayers are shown in Fig. 1I. According to the Ca2+-induced contractility model, tension produced through actin stress fiber formation upon agonist stimulation of endothelial cells is required for barrier disruption (3). The fluorescence micrographs show that within the first 2–5 min after thrombin stimulation, actin redistributes into the cytosol away from cell-cell contacts, which also leaves a staining pattern of interrupted VE-cadherin/actin co-localization (yellow) at the cell borders (Fig. 1G). Increased proportion of distinct actin bundles became visible after 10 min of thrombin stimulation, during the monolayer recovery phase (Fig. 1G). Additional quantification of fluorescence intensity from actin and VE-cadherin across cell cross-sections reveals actin accumulation in central regions of the cell at 2–10 min after thrombin stimulation, whereas in untreated and S1P-stimulated cell layers actin is predominantly co-localized with VE-cadherin at the cell borders, as indicated by overlap of intensity peaks (Fig. 1J). After 30 min of thrombin stimulation, at which time HDMEC monolayer resistance was typically restored by ∼80% of basal levels (Fig. 1E), accumulation of actin toward the cell borders was evident (Fig. 1, G and J). Similarly, in HDMEC monolayers stimulated with histamine, the appearance of stress fibers occurred 20 min after agonist stimulation (Fig. 1H), but was clearly absent in early stages, where decrease in resistance was maximal (Fig. 1F) and fluorescence intensity of actin and VE-cadherin staining decreased at the cell borders (Fig. 1J). These data suggested to us that the rapid decrease in barrier function in response to thrombin and histamine precedes stress fiber formation. Initial breakdown of endothelial barrier might be rather due to redistribution of actin away from the cell-cell junctional sites, thereby weakening junctional complexes. In turn, actual actin fiber formation might be more important during the barrier recovery phase when actin redistributes back toward the cell junctions for their stabilization. Thus, actin stress fiber tension might not be necessary for GPCR-mediated barrier breakdown.
FIGURE 1.


Typical responses of HDMEC to GPCR agonists thrombin, histamine, and S1P. A–C, electrical resistance of confluent HDMEC cell layers after stimulation with 5 nm thrombin (A), 10 μm histamine (B), and 1 μm S1P (C) where indicated by arrows. Electrical resistance was normalized to the last values before agonist addition. Absolute baseline resistances were 4202 Ω (A), 3841 Ω (B), and 2198 Ω (C). Note that for thrombin and histamine stimulation, mature HDMEC cell layers were used after 4 days in culture, whereas S1P experiments were intentionally conducted with cell layers after 2 days in culture, which explains the lower initial baseline resistance. S1P experiments were performed in less mature HDMEC monolayers to better resolve S1P-mediated enhancement of barrier function. D, confocal fluorescence images of confluent HDMEC cell layers 2 min after stimulation with thrombin, histamine, or S1P compared with the control. The cells were stained for VE-cadherin (red), actin (green), and Hoechst 33342 (blue). E–I, kinetics of changes in HDMEC cell layer morphology and junctions after 5 nm thrombin and 10 μm histamine stimulation were followed at different time points as indicated by red arrows in ECIS traces (E and F). Thrombin and histamine addition, respectively, is indicated by black arrows. Distribution of VE-cadherin and actin is shown for the selected time points after thrombin (G) or histamine (H) stimulation and compared with an untreated control (I). The white arrows point to areas with increased amount of actin bundles and fiber structures. Asterisks indicate cells used for quantification of actin and VE-cadherin distribution in J. J, quantification of actin (green traces) and VE-cadherin (red traces) fluorescence after thrombin, histamine, or S1P stimulation. Intensity histograms (in arbitrary units, a.u.) were generated for the green (actin) and red (VE-cadherin) fluorescence channel images as function of pixel position (a.u.) along cell cross-sections indicated by the white solid lines drawn into the corresponding fluorescence image. Within the intensity plots, cell borders are indicated by dashed lines.
Thrombin, Histamine, and S1P Activate SOCE in HDMECs
We tested whether Ca2+ signaling is activated in HDMECs by our three model agonists and whether Orai1 and STIM1 mediate SOCE in response to these agonists as was shown for virtually all cell types, including endothelial cells (10, 11, 13, 27, 28). We and others have shown that GPCR and receptor tyrosine kinase agonists activated Ca2+ entry into endothelial cells (from various vascular beds including HDMEC) through SOCE bearing typical pharmacological signature and mediated by Orai1 and STIM1 (11, 12, 23, 24). Like thrombin, the other two GPCR agonists considered under this study (histamine and S1P) also couple to phospholipase C activation with generation of IP3, activation of Ca2+ release from internal stores, and subsequent activation of Ca2+ entry across the plasma membrane presumably through the SOCE pathway. One of the most common means to activate SOCE without stimulating membrane receptors and without generating second messengers is to passively deplete the internal Ca2+ stores using the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (29). Treatment of HDMEC with thapsigargin in the absence of extracellular Ca2+ led to a typical transient response corresponding to Ca2+ release from internal stores (Fig. 2A, black trace). Restoration of 2 mm Ca2+ to the external buffer revealed a second response corresponding to Ca2+ entry across the plasma membrane (Fig. 2A, black trace). These typical Ca2+ release and Ca2+ entry profiles can be readily observed when HDMEC are stimulated with thrombin, histamine, or S1P under the same recording protocol (Fig. 2, B–D, black traces). Note that although 5 nm thrombin induces maximal changes in endothelial barrier function (Fig. 1A), concentrations of 100 nm thrombin were required to obtain significant Ca2+ responses to thrombin.
FIGURE 2.

SOCE inhibitors inhibit thapsigargin-, thrombin-, histamine-, and S1P-activated Ca2+ entry in HDMEC. Fura-2 recordings showing changes in intracellular free Ca2+ concentrations represented as ratio 340/380 (mean ± S.D.) after stimulation with 2 μm thapsigargin (A, E, and I), 100 nm thrombin (B, F, and J), 10 μm histamine (C, G, and K), and 1 μm S1P (D, H, and L). Effect of 2 min of preincubation with 5 μm Gd3+ (A–D, red traces) on changes in intracellular free Ca2+ mobilization compared with controls (black traces) after stimulation with 2 μm thapsigargin (A), 100 nm thrombin (B), 10 μm histamine (C), and 1 μm S1P (D). In the control black traces, 5 μm Gd3+ was added after Ca2+ entry reached a plateau. E–H, effects of 2 min of preincubation with 10 μm BTP2 (red traces) on changes in intracellular free Ca2+ mobilization compared with controls (black traces) after stimulation with 2 μm thapsigargin (E), 100 nm thrombin (F), 10 μm histamine (G), and 1 μm S1P (H). In the control black traces, 10 μm BTP2 was added after Ca2+ entry has reached a plateau. I–L, effects of 2 min of preincubation with 25 μm 2-APB on Ca2+ mobilization induced by thapsigargin (I), thrombin (J), histamine (K), and S1P (L) compared with controls (black traces). In the control black traces and after Ca2+ entry has reached a plateau, 5 μm 2-APB was added first to cells followed by 25 μm 2-APB to assess potentiation of Ca2+ entry with 5 μm 2-APB and subsequent inhibition with 25 μm 2-APB. In the blue traces, a 2-min preincubation with 5 μm 2-APB was performed to reveal potential enhancement of Ca2+ entry followed by addition of 25 μm 2-APB after Ca2+ entry has reached a plateau. The data are represented as means ± S.D., and the number of cells/condition is depicted in each figure panel and color-coded for each trace.
To show involvement of SOCE upon GPCR activation, as a first step, we systematically tested side by side the effect of different commonly used SOCE inhibitors on the HDMEC response to thapsigargin and the three GPCR agonists, thrombin, histamine, and S1P (Fig. 2). We used three different SOCE channel inhibitors; the lanthanide ion Gd3+ at relatively low concentrations of 5 μm is a specific inhibitor of SOCE (30, 31), BTP2 (32–35), and 2-APB (36, 37). Ca2+ entry (but not Ca2+ release) in response to thapsigargin and all three agonists was inhibited by 5 μm Gd3+ (Fig. 2, A–D, red traces), 10 μm BTP2 (Fig. 2, E–H, red traces), and 25 μm 2-APB (Fig. 2, I–L). As shown in Fig. 2, these commonly used SOCE inhibitors inhibited the Ca2+ entry phase when cells were either treated with drugs 2 min before the addition of agonist (red traces) or when drugs were added after Ca2+ entry has developed (control black traces; arrow indicates addition of drug). Specifically, Ca2+ entry in response to thapsigargin, thrombin, and histamine was potentiated by 2-APB at 5 μm and inhibited at 25 μm (Fig. 2, I–K, black traces), a defining peculiarity of Orai1-mediated SOCE (36, 38). Furthermore, when cells were preincubated with 5 μm 2-APB before the addition of thapsigargin, thrombin, or histamine (Fig. 2, I–K, blue traces), the Ca2+ entry phase was enhanced, but when 25 μm 2-APB was subsequently added, Ca2+ entry was inhibited. In conclusion, all three model GPCR agonists thrombin, histamine, and S1P can activate Orai1-mediated SOCE in HDMEC in a manner reminiscent of passive store depletion with thapsigargin.
Effect of SOCE Inhibitors on GPCR-mediated Endothelial Barrier Regulation
We previously showed that blockade of SOCE in HDMEC with 5 μm Gd3+ did not affect thrombin-mediated disruption of barrier function (23). This original finding led to the hypothesis that Ca2+ entry is not necessary for mediating typical acute barrier responses to GPCR agonists. Therefore, we decided here to systematically test the effects of all three SOCE inhibitors (Gd3+, 2-APB, and BTP2) on the barrier-modulating effects of the three GPCR agonists, thrombin (5 nm), histamine (10 μm), and S1P (1 μm) by using impedance measurements. HDMEC cell layers grown onto ECIS electrodes were subjected to the three SOCE inhibitors: Gd3+, BTP2, and 2-APB. Dose-response curves showed that when these inhibitors were added to HDMEC monolayers, without additional GPCR stimulation, they significantly altered the baseline resistance of the monolayers at higher concentrations, 25 μm for Gd3+, 5 μm for BTP2, and 25 μm for 2-APB (Fig. 3, A–C). BTP2 effects were transient with a recovery to baseline values within 2–3 h (Fig. 3B). Because of these intrinsic inhibitor effects, in all subsequent data related to effects of inhibitors on GPCR-mediated changes in barrier function, for the sake of transparency, we thought it more appropriate to represent raw traces that are not background subtracted (i.e. without subtracting from these data resistance values obtained with inhibitors alone).
FIGURE 3.

Concentration-dependent effect of SOCE inhibitors on HDMEC without GPCR stimulation. A–C, changes in basal electrical resistance over time after addition of different concentrations of Gd3+ (A), BTP2 (B), and 2-APB (C) in the absence of stimulation with GPCR agonists are represented. The data are represented as means ± S.D. of four independent experiments (for Gd3+, except 1 μm Gd3+ condition where n = 3) and three independent experiments (for all concentrations of BTP2 and 2-APB) per condition. The typical concentrations as used in Fura-2 recordings are highlighted in red.
The addition of 5 μm Gd3+ to HDMEC monolayers, which fully blocked SOCE without affecting the basal monolayer resistance, did not affect the typical changes in cell layer resistance upon stimulation with thrombin, histamine, or S1P (Fig. 4, A–C). All traces from Fig. 4 represent averages from 3–4 wells/condition from a single HDMEC isolation. Even higher concentrations of Gd3+ (10–50 μm) were without effect on changes in barrier function induced by all three GPCR agonists (Fig. 4, A–C). Statistical analysis on maximum change in normalized resistance in the absence or presence of Gd3+ at concentrations of 5, 10, or 50 μm from several independent experiments, 4–12, involving independent HDMEC isolations showed no statistical significance for all agonists (Fig. 4, D–F).
FIGURE 4.

Gd3+ effects on barrier function upon stimulation with thrombin, histamine, and S1P. A–C, changes in electrical resistance after stimulation with thrombin (A), histamine (B), and S1P (C) in the presence of 5 μm (red), 10 μm (magenta), or 50 μm (black) Gd3+ compared with vehicle controls (0 μm, blue). The data represent means ± S.D. from 3–4 independent wells (Gd3+; thrombin: n = 3, histamine: n = 4 and S1P: n = 4, for all Gd3+ concentrations). D–F, quantification of the maximum change in normalized resistance (Δ Normalized Resistance) upon addition of thrombin (D), histamine (E), and S1P (F) stimulation in the absence or presence of 5, 10, or 50 μm Gd3+ are shown. The data are represented as means ± S.D. from 4–12 independent experiments as marked in the bar graphs.
The decrease in monolayer resistance in response to thrombin and histamine was enhanced in the presence of BTP2 (Fig. 5, A and B). The transient decrease of resistance observed when BTP2 is added alone (Fig. 3B) is likely responsible for this enhanced decrease in resistance seen when HDMEC monolayers are stimulated with either thrombin or histamine in the presence of BTP2. Indeed, when this effect of BTP2 on basal monolayer resistance is taken into consideration, BTP2 even at higher concentrations that fully inhibit SOCE has virtually no effect on thrombin- and histamine-mediated disruption of HDMEC barrier function (Figs. 5, A and B, and 3B). The lowest concentration of 1 μm BTP2 used in experiments reduced SOCE, without causing full block (data not shown). This was consistent with reported BTP2 effect on Jurkat cells where 1 μm BTP2 resulted in ∼25% inhibition of Orai1-mediated Ca2+ release-activated Ca2+ current (39). With all concentrations of BTP2 used, the S1P response profile of HDMEC monolayers was not affected (Fig. 5C). The effect of BTP2 at different concentrations of 1, 5, and 10 μm in 6–12 experiments involving HDMEC from several independent isolations is summarized in Fig. 5 (D–F).
FIGURE 5.

BTP2 effects on barrier function in response to thrombin, histamine and S1P. A–C, changes in electrical resistance after stimulation with thrombin (A), histamine (B), and S1P (C) in the presence of 1 μm (magenta), 5 μm (black), or 10 μm BTP2 (red) compared with controls (blue). The data represent means ± S.D. from four independent wells. D–F, quantification of the maximum change in normalized resistance (Δ Normalized Resistance) upon thrombin (D), histamine (E), and S1P (F) stimulation in the absence or presence of 1, 5, or 10 μm BTP2. The data are represented as means ± S.D. from 6–12 independent experiments as marked in the bar graphs.
As discussed in Fig. 3C, 2-APB at 25 μm led to a slow decrease of cell layer resistance over the time course of the measurements and thus seemed to be a side effect of 2-APB, independently of the GPCR agonist used (Fig. 6). Even when taking into consideration this slow effect of 2-APB on basal monolayer resistance (Fig. 3C), treatment of HDMEC on ECIS electrodes with 25 μm 2-APB significantly inhibited the acute and rather fast decrease in resistance after thrombin and histamine stimulation (Fig. 6, A and B) but had no significant effect on the S1P-induced increase in monolayer resistance (Fig. 6C). The effect of 2-APB at different concentrations of 5, 10, and 25 μm in 6–11 experiments involving HDMEC from several independent isolations is summarized in Fig. 6 (D–F).
FIGURE 6.

2-APB effects on barrier function in response to thrombin, histamine and S1P. A–C, changes in electrical resistance after stimulation with thrombin (A), histamine (B), and S1P (C) in the presence of 5 μm (magenta), 10 μm (black), or 25 μm (red) 2-APB compared with controls (blue). The data represent means ± S.D. from 3–6 independent wells (2-APB; thrombin: n = 4, except for 0 μm 2-APB, where n = 3, histamine: n = 6, S1P: n = 6 for all 2-APB concentrations). D–F, quantification of the maximum change in normalized resistance (Δ Normalized Resistance) upon thrombin (D), histamine (E), and S1P (F) stimulation in the absence or presence of 5, 10, or 25 μm 2-APB are shown. The data are represented as means ± S.D. from 6–11 independent experiments as marked in the bar graphs.
In summary, of the three well known SOCE inhibitors, only 2-APB reliably inhibited the initial fast decrease in HDMEC monolayer resistance induced by thrombin and histamine with marginal effect on S1P-induced enhancement of barrier function. Therefore, the effects of 2-APB are clearly not related to inhibition of SOCE and could be due either to nonspecific effects of 2-APB on endothelial barrier function or to the ability of 2-APB to block STIM1 oligomerization and reorganization (40), consistent with the reported STIM1-mediated control of barrier dysfunction in response to thrombin that occur independently of Orai1 and Ca2+ entry (23). To confirm results with SOCE inhibitors, we decided to complement these pharmacological data with molecular knockdown in HDMEC monolayers of SOCE components Orai1 and STIM1, as discussed in the following.
Effect of Knockdown of Orai1 and STIM1 on GPCR-mediated Endothelial Ca2+ Signaling and Barrier Regulation
For molecular knockdown of Orai1 and STIM1, we used siRNA sequences previously characterized in our laboratory, which achieved significant knockdown in 80–90% of HDMEC monolayers without off-target effects, specifically on other STIM/Orai isoforms (11, 23, 41–43). Non-targeting siRNA (siNT) were used as control, and knockdown efficiency was quantified by Western blot and shown for both STIM1 and Orai1 (Fig. 7, A and C); densitometry results from three (for STIM1) and four (for Orai1) independent transfections were quantified statistically (Fig. 7, B and D). Functional evaluation of SOCE activated by passive store depletion using thapsigargin (Fig. 7, E–H) or agonist stimulation with histamine (Fig. 7, I–L) using Fura-2 imaging showed significant inhibition of Ca2+ entry after knockdown of either STIM1 or Orai1. The effects of knockdown of STIM1 and Orai1 were quantified from a large number of cells originating from 3–4 independent transfections with 6–8 independent recordings (Fig. 7, F, H, J, and L); the two numbers on the bar graphs separated by comma “x, y” represent the number of independent runs (x) and the total number of cells averaged (y). STIM1 and Orai1 knockdown was performed using a second set of sequences (siSTIM1 # 2 and siOrai1 # 2), and effective inhibition of thrombin and histamine-mediated Ca2+ entry was confirmed with these additional sequences (Fig. 7, M–T).
FIGURE 7.


Molecular and functional quantification of STIM1 and Orai1 knockdown efficiency. A–L, validation of knockdown efficiency with siSTIM1 # 1 and siOrai1 # 1. A and C, representative Western blots comparing STIM1 (A) and Orai1 (C) expression 4 days after transfection with siRNA against STIM1 (A) and Orai1 (C), compared side by side with control siNT. B and D, densitometry quantifications of Western blots from 3 (STIM1) and 4 (Orai1) independent transfections similar to those in A and C. E and G, representative Fura-2 ratiometric Ca2+ imaging traces in response to 2 μm thapsigargin after STIM1 (E) or Orai1 (G) knockdown (red or blue traces) compared with siNT controls (black traces). F and H, quantification of the maximal change in thapsigargin-induced Ca2+ entry after STIM1 (F) or Orai1 (H) knockdown from 7–9 independent experiments per condition. I and K, representative ratiometric Ca2+ imaging traces in response to 10 μm histamine after STIM1 (I) or Orai1 (K) knockdown (red traces) compared with siNT controls (black traces). J and L, quantification of the maximal change in histamine-induced Ca2+ entry after STIM1 (J) or Orai1 (L) knockdown from 5–7 independent experiments per condition is represented. M–T, validation of knockdown efficiency with siSTIM1 # 2 and siOrai1 # 2. M and N, Western blots comparing STIM1 (M) and Orai1 (N) expression after transfection with siSTIM1 # 2 (M) and siOrai1 # 2 (N) to control siNT. O–Q, representative Fura-2 ratiometric Ca2+ imaging traces in response to 2 μm thapsigargin (O), 100 nm thrombin (P), or 10 μm histamine (Q) after STIM1 (red traces) or Orai1 (blue traces) knockdown compared with siNT controls (black traces). R–T, quantification of the maximal change in thapsigargin-induced (R), thrombin-induced (S), or histamine-induced (T) Ca2+ entry after STIM1 (red) or Orai1 (blue) knockdown compared with non-targeting control (siNT, black) from 3 independent experiments/condition. The data represent means ± S.E.
Knockdown of either STIM1 or Orai1 had a slight effect on the basal HDMEC monolayer resistance, but this effect was somewhat significant only for Orai1 knockdown (Fig. 8, A–D), suggesting a role for Orai1 in proper maturation of HDMEC monolayers. An important observation is that initial baseline values vary significantly depending on the primary cell isolation. The importance of paying attention to initial baseline values and its effect on change in resistance after GPCR activation has been recently discussed in detail (26). A lower initial resistance leads to a smaller resistance change after agonist stimulation, when compared with a cell layer with high initial resistance when all other conditions are equal (26). We quantified the response intensities (ΔR) for each agonist and found a clear dependence of the signal intensity on initial resistance values of the HDMEC cell layers for thrombin and histamine, which was also evident for all types of siRNA used (siNT, siOrai1, and siSTIM1) (Pearson's correlation coefficients 0.58–0.87; data not shown). Because siRNA treatment itself had no impact on basal HDMEC barrier development after cell seeding compared with siRNA-untreated control HDMEC (Fig. 8, E and F), we ascribed differences in baseline unstimulated resistance to variation in cell isolations between human donors. We intentionally chose to conduct knockdown experiments with HDMEC monolayers from different isolations generating different basal resistance values to show the general applicability of our findings. Therefore, and as discussed in the next paragraph, ΔR values showed wide variation among different individual experiments.
FIGURE 8.
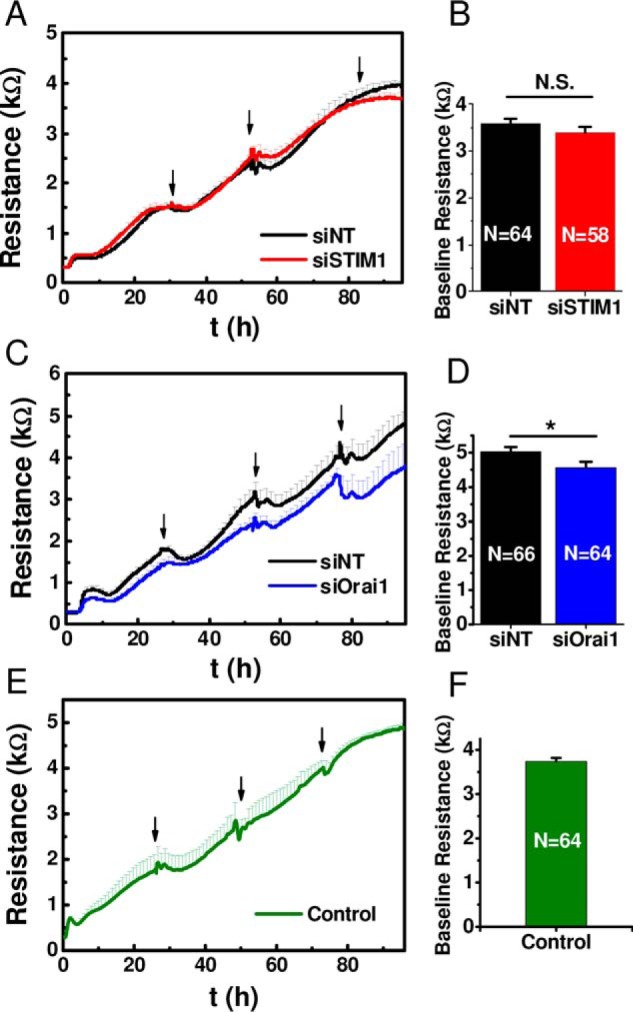
Dependence of GPCR agonist-mediated changes in HDMEC monolayer resistance on basal resistance values. A–D, effect of siRNA treatment on HDMEC monolayer resistance. E and F, siRNA-untreated control HDMEC cell layers. A, C, and E show the resistance increase of HDMEC cell layers over a 96-h period from initial seeding (100,000 cells/cm2) to full maturation. Typically, at full maturation cell monolayers were used in thrombin and histamine stimulation experiments as will be discussed in Fig. 10. Traces of basal resistance evolution in siSTIM1 #1-treated (A, red trace) or siOrai1 #1-treated (C, blue trace) cells are compared with control siNT (black traces) and shown as averages ± S.D. from an experiment with 8 wells/condition; arrows indicate medium changes. B and D, quantification of the maximal baseline resistance reached after 4 days post-transfection with siRNA and seeding into ECIS plates are shown as means ± S.E. for siSTIM1#1 (B) or siOrai1 #1(D) compared with control siNT from 58–66 wells/condition as indicated in the bar graphs. E, growth curve of siRNA-untreated control HDMEC cell layers (average ± S.D.) of four individual cell layers. F, quantification of the baseline resistance of control HDMEC cell layers reached after 4 days in culture shown as means ± S.E. of 64 individual cell layers. As stated earlier, S1P addition was performed on immature cell layers maintained only 2 days in culture to better resolve S1P enhancement of barrier function.
STIM1 knockdown inhibited the typical decrease in HDMEC monolayer resistance but only upon stimulation with thrombin and histamine with no effect on S1P-induced enhancement of HDMEC monolayer resistance (Fig. 9, A–C). However, Orai1 knockdown failed to affect the changes in endothelial barrier function in response to all three GPCR agonists (Fig. 9, D–F). Traces in Fig. 9 (A–F) represent averages from n = 4–8/condition from a single HDMEC isolation. To establish solid statistical coverage over different measurements from different cell isolations, statistical analyses were performed on 4–10 independent HDMEC isolations for each experimental condition (with 23–86 independent wells/condition). The collected data are shown as density plots (Fig. 9, G–I). High densities indicate frequent occurrence of respective response intensities (ΔR), and vertical lines represent the mean response intensity. As shown in Fig. 9 (G and H), STIM1 knockdown caused a left shift in response intensity upon thrombin and histamine stimulation toward lower ΔR values. The respective means of ΔR values from siSTIM1-treated cell layers were significantly different from those of siNT-treated cells, as revealed by ANOVA analysis. In contrast, Orai1 knockdown does not affect the response intensity to thrombin or histamine (Fig. 9, G and H). Neither STIM1 nor Orai1 knockdown significantly influenced ΔR values in response to S1P stimulation (Fig. 9I). Qualitatively the same results were obtained using a second set of siRNAs (siSTIM1 # 2 and siOrai1 # 2) (Fig. 9, J–O).
FIGURE 9.

Effect of STIM1 and Orai1 knockdown on changes in HDMEC cell layer resistance upon stimulation with thrombin, histamine, and S1P. A–C, changes in resistance of HDMEC monolayers transfected with either siSTIM1 #1 (red traces) or control siNT (black traces) before and after stimulation (indicated by arrow) with 5 nm thrombin (A), 10 μm histamine (B), and 1 μm S1P (C). D–F, changes in resistance of HDMEC monolayers transfected with either siOrai1 #1 (blue traces) or control siNT (black traces) before and after stimulation with 5 nm thrombin (D), 10 μm histamine (E), and 1 μm S1P (F). All traces shown are from representative experiments reflecting averages ± S.D. of n = 4–8 wells/condition as indicated in the respective plots. G–I, density plots with statistical analysis on a large set of experiments similar to those in A–F performed on HDMEC monolayers transfected with siSTIM1 #1, siOrai1 #1, siNT, and control non-transfected cells and stimulated with thrombin (G), histamine (H), and S1P (I). The data are from 4–10 independent HDMEC isolations and reflect 23–86 independent wells/experimental condition. Density plots represent the relative distribution of the maximum response intensities to agonist addition. Maximum response intensities were quantified as the difference in resistance between baseline and the maximal response obtained after GPCR stimulation, ΔR (Ω). For all agonists, responses are shown as positives. Density plots were generated by Kernel density smoothing for histograms. High densities indicate frequent occurrence of these values. In addition to HDMEC populations treated with siNT, data of siRNA-untreated cells (control) were also included. Vertical lines represent the mean response intensity (G: siNT: 1107 Ω, siOrai1: 951 Ω, siSTIM1: 652 Ω, control: 1040 Ω; H: siNT: 482 Ω, siOrai1: 457 Ω, siSTIM1: 228 Ω, control: 564 Ω; I: siNT: 317 Ω, siOrai1: 229 Ω, siSTIM1: 351 Ω, control: 282 Ω). The data were subjected to ANOVA analysis, and significant differences are indicated by asterisks as described under “Experimental Procedures.” J–O, changes in resistance of HDMEC monolayers transfected with siSTIM1 #2 (J–L, red traces) or siOrai1 #2 (M–O, blue traces) compared with control siNT (black traces) before and after stimulation (indicated by arrow) with 5 nm thrombin (J and M), 10 μm histamine (K and N), or 1 μm S1P (L and O). All traces shown are from representative experiments reflecting averages ± S.D. of n = 4–8 wells/condition.
In summary, our statistical analysis confirmed that significant effects on GPCR-mediated changes in endothelial barrier function were only detectable upon STIM1 knockdown and only for stimulation with thrombin and histamine. STIM1 obviously plays an important role in mediating the opening of endothelial barrier upon GPCR stimulation, but its effect is independent from Ca2+ entry. In the following, we set out to further support our finding that HDMEC barrier breakdown is independent from extracellular Ca2+ rise.
Role of Extracellular Ca2+ in GPCR-mediated Endothelial Barrier Regulation
We have previously refrained from performing long term experiments that omit Ca2+ from the external solution as means to test the involvement of Ca2+ entry in endothelial barrier function (23). Ca2+ ions are required co-factors for adhesion molecules, and long term removal of external Ca2+ will inevitably lead to gradual decrease in HDMEC monolayer resistance. Therefore, here we used a protocol involving short term removal of external Ca2+ (for ∼8 min) while monitoring changes in cytosolic Ca2+ in HDMEC, the monolayer resistance by ECIS, and visual inspection of monolayers by fluorescence microscopy. As seen in Fig. 10, the addition of thrombin in the absence of external Ca2+ caused an even bigger decrease in HDMEC monolayer resistance than obtained when thrombin was added in presence of external Ca2+ (Fig. 10A; 11 min zoom in is shown in Fig. 10B). Restoration of 2 mm Ca2+ to the external solution did not cause additional decrease in monolayer resistance (Fig. 10B). Fluorescence images showed that the disruption of cell-cell junctions after thrombin is added in the absence of external Ca2+ was much more substantial than that observed in the presence of 2 mm Ca2+ (Fig. 10D). This is likely due to monolayers being less stable in the absence of external Ca2+ as a result of weak cell-cell and cell-substrate adhesion. The Ca2+ response of HDMEC in the absence and presence of external Ca2+ showed the expected Ca2+ release and Ca2+ entry phases (Fig. 10C). Our conclusion from this experiment is that although extracellular Ca2+ is required for proper cell adhesion and monolayer stability and integrity, GPCR-activated Ca2+ entry across the plasma membrane is not required to open the endothelial barrier.
FIGURE 10.

Changes in HDMEC monolayer resistance in response to thrombin in Ca2+-free and Ca2+-containing HBSS. A, time course of changes in HDMEC monolayer electrical normalized resistance upon addition of 100 nm thrombin in Ca2+-free extracellular HBSS and subsequent restoration of 2 mm Ca2+ to the extracellular solution. The arrow indicates time point of change to Ca2+-free HBSS. B, close-up of A showing with three consecutive arrows the time of switch to Ca2+-free HBSS (−Ca2+), addition of thrombin at 100 nm in Ca2+-free HBSS and change to HBSS containing 2 mm Ca2+ (+Ca2+). For this specific experiment, specialized ECIS arrays were used (2W4 × 1E PC). These arrays had wells with the same dimensions as the chambers used for Ca2+ imaging, and ECIS measurements were performed at room temperature. C, corresponding Ca2+ imaging trace upon addition of 100 nm thrombin using the same −Ca2+/+Ca2+ protocol. D, corresponding immunofluorescence staining of VE-cadherin (red), actin (green) and nuclei stain Hoechst (blue) on cells stimulated for 2 min with 100 nm thrombin in the Ca2+-free (upper panels) and Ca2+-containing (lower panels) HBSS.
Role of Ca2+ Release from Internal Stores in GPCR-mediated Endothelial Barrier Regulation
The use of Ca2+-free extracellular solutions, SOCE inhibitors, and STIM1/Orai1 molecular knockdown used thus far only prevent Ca2+ entry across the plasma membrane with no effect on Ca2+ release from IP3-sensitive stores. Therefore, we set out next to determine whether Ca2+ release from internal stores is required for changes in endothelial barrier function in acute response to GPCR agonists. We therefore incubated HDMEC monolayers with the membrane permeant form of the fast pH-insensitive Ca2+ chelator BAPTA (BAPTA-AM). Incubation of HDMEC monolayers with 10 μm of BAPTA-AM for 10 min caused a substantial inhibition of both Ca2+ release and Ca2+ entry in response to thapsigargin as measured with Fura-2 cytosolic Ca2+ imaging (Fig. 11A, red trace); 1 μm of BAPTA-AM had partial effects on inhibiting these thapsigargin-induced Ca2+ signals in HDMEC (Fig. 11A, blue trace) and was therefore not used any further. The incubation of HDMEC with 10 μm of BAPTA-AM for 10 min also substantially inhibited both Ca2+ release and Ca2+ entry in response to thrombin (Fig. 11B), histamine (Fig. 11C), and S1P (Fig. 11D). However, 10 min of preincubation of monolayers with 10 μm BAPTA-AM failed to inhibit the changes in endothelial barrier function in response to thrombin (Fig. 11E), histamine (Fig. 11F), and S1P (Fig. 11G). Thus, buffering of Ca2+ release from internal stores does not prevent the acute agonist-mediated changes in endothelial barrier function.
FIGURE 11.

Effect of chelation of intracellular Ca2+ with BAPTA-AM on changes in barrier function in response to thrombin, histamine, and S1P. A–D, representative recordings of changes in intracellular free Ca2+ in response to stimulation with 2 μm thapsigargin (A), 100 nm thrombin (B), 10 μm histamine (C), and 1 μm S1P (D). Recordings were initiated after 10 min of incubation with vehicle (DMSO control, black traces) or BAPTA-AM at either 1 μm (blue trace; performed only for thapsigargin) or 10 μm (red traces). Because 1 μm BAPTA-AM did not fully inhibit Ca2+ mobilization in response to thapsigargin, 10 μm BAPTA-AM was used in all subsequent experiments. E–G, changes in electrical resistance of confluent HDMEC monolayers after 10 min of incubation with 10 μm BAPTA-AM (red traces; BAPTA-AM addition indicated by B at the first arrow) compared with vehicle control (DMSO, black traces) and stimulation with 5 nm thrombin (E, second arrow labeled Thr), 10 μm histamine (F, second arrow labeled His), and 1 μm S1P (G, second arrow labeled S1P).
Discussion
Previous work from our laboratory showed that STIM1 and Orai1 are the molecular components of SOCE in several human endothelial cell types, including endothelial cells from the umbilical vein, the pulmonary artery, and the dermal microvasculature (HDMECs) (11, 23). We also showed that thrombin and VEGF trigger Ca2+ entry into endothelial cells through SOCE and its biophysical correlate, the Ca2+ release-activated Ca2+ current (11, 23). We performed ECIS studies using both human endothelial cells from the umbilical vein and HDMEC monolayers and showed that STIM1 controls thrombin-mediated disruption of endothelial barrier function independently of Orai1 and Ca2+ entry, suggesting that SOCE and Ca2+ entry in general were not required for thrombin-mediated disruption of endothelial barrier function.
In the current study, we aimed to determine whether the requirement of STIM1 and the dispensable aspect of Orai1 and Ca2+ entry in thrombin-mediated barrier disruption is generally applicable to other GPCR agonists, including those that enhance barrier function and whether Ca2+ release from internal stores is required for GPCR-mediated changes in barrier function. Thus, in addition to thrombin, we chose to focus on two additional GPCR agonists, histamine which disrupts endothelial barrier function but does so with less intensity and quicker recovery time than thrombin, and S1P, which enhances endothelial barrier function. We found that all three GPCR agonists, thrombin, histamine, and S1P activate Ca2+ entry, which displays the typical pharmacological profiles of SOCE. Inhibition of SOCE and Ca2+ entry in general with lanthanides or BTP2 had no effects on changes in barrier function caused by all three GPCR agonists. However, the uses of 25 μm 2-APB (a concentration that inhibits Ca2+ entry with no effect on Ca2+ release) inhibited thrombin- and histamine-mediated disruption of barrier function with no effect on S1P-mediated enhancement of barrier function. In light of the results obtained with Gd3+ and BTP2, the effect of 2-APB is likely unrelated to its ability of inhibiting Ca2+ entry. 2-APB was shown to inhibit STIM1 function by preventing STIM1 aggregation and movements in response to store depletion (40), which is consistent with our STIM1 molecular knockdown inhibiting barrier disruption in response to thrombin and histamine. STIM1 knockdown had no effect on the barrier-stabilizing function of S1P, suggesting that STIM1 plays a role in processes involving disruption of barrier function but not in the molecular pathways that control stabilization of endothelial barrier function. Knockdown of Orai1 failed to affect the disruption of barrier function in response to thrombin and histamine and had no bearing on the enhancement of barrier function in response to S1P.
We also showed that the use of a fast cytosolic Ca2+ buffer, BAPTA-AM on endothelial monolayers led to a strong inhibition of the Ca2+ release and Ca2+ entry signals in response to thapsigargin and GPCR agonists. However, this Ca2+ buffering failed to affect the typical response of endothelial monolayers to thrombin, histamine, and S1P, strongly arguing that Ca2+ release and Ca2+ entry are not necessary for GPCR-mediated changes in endothelial barrier function. These results are consistent with previous studies showing that thrombin-mediated disruption of endothelial barrier function occurs independently of Gq-mediated Ca2+ signaling (44) and a recent study showing that histamine-mediated disruption of endothelial barrier function requires RhoA and ROCK, whereas phospholipase C pharmacological inhibition had a marginal effect (45). Our previous studies showed that knockdown of STIM1 caused inhibition of RhoA activity and MLC phosphorylation in response to thrombin. We also showed that STIM1 knockdown led to increased basal phosphorylation of focal adhesion kinase and paxillin, consistent with a role for STIM1 in mediating GPCR-induced endothelial barrier disruption through RhoA independently of receptor-activated Ca2+ signaling (23). Likewise, the stabilizing effects of S1P on barrier function occur via Rac-mediated pathways and are independent of STIM1, Orai1, and Ca2+ signaling. Collectively, the data in the literature support that regulation of endothelial barrier disruption and stabilization is likely mediated by the balance between the activities of Rho and Rac/Cdc42.
The “increased cytosolic Ca2+-induced endothelial contraction” hypothesis stipulates that Ca2+ entry upon GPCR ligation activates MLCK, which in turn phosphorylates MLC to cause endothelial contraction and disruption of barrier function (3, 18, 21). Earlier studies that proposed a role for MLCK in controlling endothelial barrier function downstream GPCR agonists relied on nonspecific approaches such as the use of MLCK inhibitory peptides or MLCK pharmacological inhibitors, namely ML7 and ML9 (46–48). Subsequently, ML9 was shown to inhibit STIM1 aggregation and movement upon store depletion in an MLCK-independent manner (40). Work in our laboratory showed that effective knockdown of endothelial MLCK failed to alter the disruption of endothelial barrier function in response to thrombin, ruling out the requirement of MLCK in GPCR-mediated acute control of barrier function (23).
Other Ca2+-sensitive enzymes, including Ca2+/calmodulin-dependent kinase II (CaMKII) and PKC, were proposed to regulate the cell-cell junctional VE-cadherin·catenin complex disassembly in endothelial cells in response to thrombin (49–55). For instance, the endothelial CaMKIIδ6 isoform was shown to play a role in thrombin-mediated endothelial barrier disruption. However, CaMKIIδ6 contribution was through RhoA/ROCK-dependent mechanism and was apparent only at low concentrations of thrombin (2.5 nm) where the Ca2+ signal activated by thrombin was negligible, but not at higher concentrations of thrombin (56). Therefore, increased cytosolic Ca2+ and MLCK are not absolutely required for triggering the acute and rapid disruption in barrier function in response to GPCR agonists; RhoA activation appears necessary and sufficient. However, we cannot obviously rule out subtle regulatory roles for cytosolic Ca2+ and MLCK (or other Ca2+-activated enzymes) during the signaling processes impacting on cytoskeletal rearrangements such as endothelial permeability or migration. In most in vivo studies, Ca2+ signals and endothelial permeability to agonists, measured for instance on mesenteric venules of anesthetized rats, are merely correlative and are based on the use of pharmacological compounds with questioned specificity (57–59). However, a regulatory role for cytosolic Ca2+ rise and subcellular activation of Ca2+-activated enzymes in endothelial barrier function might be more significant under in vivo conditions that are more complex and where subtle rearrangements of the barrier at specific sites are sufficient to allow passage of cells and solutes. We are arguing herein that the idea of acute and rapid changes in endothelial barrier function being triggered by a Ca2+ signal in a manner analogous to contraction of smooth muscle is not supported by the evidence and therefore the model of “Ca2+-induced endothelial contraction” should be abandoned.
Experimental Procedures
Reagents
siRNA sequences siSTIM1 (siSTIM1 #1, AAGGGAAGACCUCAAUUACCAUU; siSTIM1 #2, GGUGGUGUCUAUCGUUAUUUU), siOrai1 (siOrai1 #1, CUGUCCUCUAAGAGAAUAAUU; siOrai1 #2, CGUGCACAAUCUCAACUCGUU), and non-targeting siControl (UGGUUUACAUGUCGACUAAUU) were from Dharmacon Scientific. Anti-Orai1 (rabbit) antibody was from Sigma (HPA016583), used at a dilution of 1:1000. Anti-STIM1 (mouse, raised using STIM1 amino acids 25–139) antibody is from BD Transduction Laboratories, used at a dilution of 1:250. Anti-β-actin is from Sigma and used at a dilution of 1:10,000. Anti-GAPDH (mouse) is from Sigma (catalog no. G8795), used at a dilution of 1:3000, and anti-HSP 70/HSC 70 (W27) (mouse) from Santa Cruz Biotechnology (catalog no. sc-24) was used at a dilution of 1:2000. Anti-rabbit IgG-HRP and anti-mouse IgG-HRP are from Calbiochem used at 1:10,000. Anti-VE-Cadherin (goat) antibody used for immunofluorescence was purchased from Santa Cruz Biotechnology and used at 1:200. Secondary Alexa Fluor 594®-labeled anti-goat antibody (rabbit) is from Molecular Probes, used at 1:250. Alexa Fluor 488® phalloidin is from Molecular Probes. BCA protein assay kit, Super Signal West Pico, West Femto, and Restore Western blot stripping buffer were from Pierce. All other chemicals were purchased from Fisher Scientific unless specified otherwise.
Cell Culture
HDMECs were isolated from human neonatal foreskin and purified by CD31 antibody-coated magnetic beads (DYNAL®) as described elsewhere (60). Endothelial origin was tested by VE-cadherin and lectin staining. The cells were cultured in EBM2 basal medium (Lonza; catalog no. CC-3156) supplemented with EGM2-MV bullet kit for microvascular endothelial cells (Lonza; catalog no. CC-4147), 2 mm l-glutamine (Thermo Fisher Scientific, HyCloneTM; catalog no. SH30034.01) and antibiotic-antimycotic (Gibco®, Life Technologies; catalog no. 15240-062). The cells were subcultured every 3–4 days when at 90% confluence by trypsination (0.05% trypsin, 0.53 mm EDTA in HBSS from Corning, Cellgro®; catalog no. 25-052-CI) and used up to passage 12.
For impedance measurements, the cells were grown onto ECIS cultureware arrays from Applied BioPhysics Inc. (Troy, NY). If not specified otherwise electrode type 8W10E+ PET were used. The other array types used were 96w20idf PET and 2W4 × 10E PC. Before use, arrays were treated with sterile filtered 10 mm cysteine in water for 10 min to generate stable electrode impedance. Afterward electrodes were rinsed three times with sterile water and coated with 300 μl of 0.2% gelatin in PBS (8.0 g/liter NaCl, 0.2 g/liter KCl, 1.42 g/liter Na2HPO4, and 0.24 g/liter KH2PO4 pH 7.4) for 10 min (gelatin was purchased from Acros Organics, gelatin for analysis, granular; catalog no. AC410875000, cas 9000-70-8). HDMEC were seeded onto the ECIS cultureware at a density of 100,000 cells/cm2 in 400 μl of complete medium. To allow uniform settling of cells at the bottom of the wells, the arrays were left for ∼10 min on the bench. The cells were maintained in culture for 2–4 days (depending on the agonist studied; see below) in a humidified incubator with 5% CO2 at 37 °C. Every 24 h, half of the medium was exchanged by removing 200 μl of old medium and adding back 200 μl of fresh medium. Exchanging only 50% of medium instead of a full exchange reduces mechanical stress on the cell layer. Without medium changes, the cell layers established lower baseline resistances and higher variability in baseline values, as well as in kinetics of response to agonists. 6–24 h before the experiment complete medium was exchanged with low serum (0.3%) medium.
siRNA Transfections
For specific knockdown of STIM1 and Orai1, we used siRNA sequences (see “Reagents”) that have been established and tested for knockdown efficiency as previously described (11, 23). Transfections with siRNA sequences were performed with the Amaxa 4D-Nucleofector® (Lonza, Amaxa Biosystems) using transfection solution P5 and the program EH100, following the manufacturer's instructions. 0.5 μg of GFP plasmid construct (Amaxa) was co-transfected with siRNA to control transfection efficiency, which typically exceeded 80%. Protein knockdown efficiency was quantified by Western blot.
For impedance measurements, transfected cells were directly seeded onto the gelatin-coated electrodes for thrombin or histamine experiments. For S1P experiments where 2-day-old cell layers were used, the cells were first grown for 2 days on regular culture flasks, before seeding for impedance measurements.
Impedance Measurements
Impedance measurements were performed using the electric cell-substrate impedance sensing (ECIS) Zθ machine (Applied BioPhysics Inc., Troy, NY) with an array holder station for two 8-well arrays of type 8W10E+ (or 2W4 × 1E PC) and an alternative 96-well plate holder for 96W20idf type electrode arrays (26). The measurements were performed in a humidified cell culture incubator unless stated otherwise. Barrier function of HDMEC cell layers was measured at 4000 Hz when using 8W10E+ electrodes or at 2000 Hz when using 96W20idf electrode arrays, which were the respective frequencies with maximum dynamic range between the cell-free and cell-covered electrodes (26). Single frequency time collect mode allowed detecting HDMEC response to GPCR agonists with a time resolution of 8 s for 16 wells or 48 s for 96 wells, respectively. For data presentation the resistance, the real part of the impedance data, which is most indicative of paracellular currents via cell-cell junctions was normalized to the last value before agonist addition (normalized resistance) and plotted versus time. For measurements of the complete attachment, growth and cell layer maturation process electrodes were pretreated with 10 mm cysteine solution 1 day before cell seeding. After 10 min of incubation with 150 μl of cysteine solution, arrays were rinsed three times with water and incubated in HDMEC medium overnight to support establishment of stable electrode-electrolyte interface. HDMEC attachment, spreading, and cell layer maturation was measured with the multiple frequency time collect mode. A baseline of the cell-free array with 400 μl of medium in each well was measured over at least 1 h. Data acquisition was paused for inoculation with single-cell suspension (100,000 cells/cm2) and for medium changes.
Monitoring of HDMEC Response to GPCR Agonists
Thromin (catalog no. T464), histamine (catalog no. 53300) and S1P (catalog no. A8806) were purchased from Sigma-Aldrich. Thrombin stock solutions in MilliQ water (100 μm) were stored at −80 °C. Immediately before use in an experiment, 100 nmol/liter working solution (for a final concentration of 5 nmol/liter, 20 μl/well) was prepared in prewarmed 0.3% serum medium (50 ml of EBM-2 150 μl of FBS, 500 μl of 100 × antibiotics-antimycotics, and 500 μl of 100 × l-glutamine solution). 1 mol/liter histamine stock solutions in MilliQ water were stored at −20 °C. Before use, a 200 μmol/liter working 20× solution was prepared in serum-free medium (for a final concentration of 10 μmol/liter and addition of 20 μl/well). The solution was prewarmed to 37 °C for ∼5 min before addition to the cells. S1P was solubilized in methanol, aliquoted, dried under nitrogen, and stored at −20 °C. For use in stimulation experiments, 20 μm S1P (20 ×) working solution was made in HBSS (Fisher Scientific; catalog no. 14025-092) with 0.03 mg/ml fatty acid-free BSA (from bovine plasma, Sigma-Aldrich; catalog no. A8806). Solubilization was supported by brief sonication for 5 min at 37 °C.
For experiments with thrombin, HDMEC cell layers were used after 4 days in culture. The cells were changed to serum-free medium 12–24 h before start of the experiment. Cell layer quality was checked by phase contrast microscopy and by either recording a single full impedance spectrum or collecting data for 10–30 min at multiple frequency time mode. The resistance at 4000 Hz should be in the range of 2500–4500 Ω for HDMEC grown on 8W10E+ electrodes for 4 d. On 96W20idf the resistance at 2000 Hz should be above 1000 Ω; if these basal resistances were not attained, the experiment was aborted. A baseline was recorded for at least 10 min. Thrombin was added as a 20× stock solution. Although the measurement was running, 20 μl of old medium were removed from the initial 400 μl of each well. 20 μl of 20× thrombin solution (100 nmol/liter) were then added to the remaining 380 μl to give a final concentration of 5 nmol/liter. Cell responses was monitored for at least 1 h after addition of thrombin. For experiments with histamine, HDMEC cell layers were used after 3–4 days in culture. The cells were changed to serum-free medium 6–12 h before start of the experiment. Histamine was added as a 20× stock solution of 200 μmol/liter histamine in low serum medium, following the procedure as described for thrombin. For S1P experiments, HDMEC were used after 2 days in culture, where cell layers were confluent, but cell-cell junctions were not fully mature (26). The cells were changed to serum-free medium 6 h before stimulation with 1 μm S1P. The experiment was conducted as described above for thrombin and histamine, using a 20× stock solution. For ECIS experiments using preincubation with Gd3+ and corresponding controls, a HEPES-based medium was used (130 mm NaCl, 3 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 5 mm glucose, and 20 mm Na-HEPES, pH 7.4) as previously described (23).
Calcium Imaging
For measuring changes in intracellular calcium, HDMECs were grown on gelatin-coated 30-mm round coverslips as previously described (61–63). The coverslips with cells were mounted in a Teflon chamber and incubated with 4 μm Fura-2/AM in culture medium for 35 min at 37 °C protected from light. Afterward, the cells were gently washed three times with HEPES-buffered saline (140 mm NaCl, 1.13 mm MgCl2, 4.7 mm KCl, 2 mm CaCl2, 10 mm d-glucose, and 10 mm HEPES, pH 7.4) and let sit for 10 min at room temperature and protected from light. Data acquisition was performed using InCyt Im2 Fluorescence Imaging System and IntCyt Im2 software (Intracellular Imaging Inc., Cincinnati, OH). Fura-2-loaded cells were alternately illuminated with 340 and 380 nm. Fluorescence emission was collected at 510 nm. 340/380 ratio images were obtained on pixel by pixel basis. Figures showing Ca2+ traces show averages from several cells per coverslip and are representative of several independent recordings of several cells as indicated in the respective figure legends. Thrombin and histamine solutions were made in HEPES-buffered saline, and S1P solutions additionally contained 0.03 mg/ml fatty acid-free BSA.
Immunofluorescence Staining
HDMECs were grown on gelatin-coated cover glasses for 4 days (initial seeding density 100,000 cells/cm2). The cell layers were stimulated with different agonists under the same conditions as used for ECIS experiments. At the indicated time after stimulation, cells were fixed with 4% (w/v) paraformaldehyde in PBS for 20 min. Fixed cells were rinsed with PBS three times and permeabilized with 0.02% Triton X-100 in PBS. Permeabilized cells were first treated with Image-iT® FX signal enhancer (Fisher Scientific; catalog no. I36933) for 30 min and afterward blocked with a mixture of 3% BSA and 5% serum in TBS for 1 h. The cell layers were rinsed two times before incubated with goat anti-VE-cadherin primary antibody (1:200) in TBS for 2 h at room temperature. Secondary antibody (Alexa Fluor 594®-labeled rabbit anti-goat antibody from Molecular Probes) incubation was for 1 h at room temperature together with Alexa Fluor 488® phalloidin and Hoechst 33342 incubation. The cells were rinsed three times and embedded with anti-fade mounting medium (Sigma-Aldrich). Images were taken on a Zeiss confocal microscope.
Western Blotting
The cells were transfected with respective siRNAs as described above. After 4 days the cells were lysed in radioimmune precipitation assay lysis buffer (Sigma-Aldrich) with 10% protease inhibitor mixture and 10% phosphatase inhibitor mixture (both from Roche Diagnostics). Suspensions were subjected to a 30 s sonication on ice. Cell debris was removed by 1.5 min centrifugation at 14,000 rpm and 4 °C. Protein concentration in the supernatant was determined using BCA assay (Pierce) and SpectraMax® Paradigm® multimode detection platform (Molecular Devices), and equal amounts of proteins were loaded in each well.
Lysates (50 μg/well) were subjected to SDS-PAGE using the Precision PlusTM protein dual color standard (Bio-Rad) as reference (with 10, 15, 20, 25, 37, 50, 75, 100, 150, and 250 kDa) and transferred to immunoblot PVDF membranes (Bio-Rad). The membranes were blocked (5% nonfat milk/TBS/Tween-20) at 4 °C overnight, incubated with respective antibodies, washed, and subsequently incubated with HRP-linked secondary antibody in 2% nonfat milk/TBS/Tween-20 for 2 h. Bound antibody was detected by enhanced chemiluminescence with Super Signal West Pico or Femto reagents (Pierce). Signal intensity was measured using a Bio-Rad ChemiDoc MP imaging system (Bio-Rad). The membranes were then stripped and reprobed with anti-β-actin, anti-GAPDH, or anti-HSP 70/HSC 70 antibody to verify equal protein loading. Quantitative densitometry analysis was performed using ImageJ software.
Statistical Analysis
Experimental data are presented as means ± S.D. Only for data sets with very high N numbers were means ± S.E. used, and the values are indicated as such in the figure legends. To test for statistical significance, data sets were subjected to ANOVA analysis. Levels of significance were indicated with asterisks as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.005. A p value of <0.05 was considered statistically significant. ANOVA analysis and Pearson's correlation calculations were performed with OriginLab Pro software. Density plots were generated with R software using Kernel density smoothing for histograms. All data were plotted using Origin Pro software.
Author Contributions
J. A. S. and M. T. designed research, analyzed data, and drafted the manuscript. J. A. S. performed most of research. X. Z., W. Z., and M. G. performed part of the experiments and analyzed data. C. R. analyzed data statistically and provided reagents, arrays, technical assistance, and critical input on the manuscript. K. M. provided reagents, technical assistance, and critical input on overall research. All authors discussed and approved the manuscript.
This work was supported by American Heart Association Grant 14GRNT18880008, National Institutes of Health Grants R01HL097111 and R01HL123364 (to M. T.) and R01HL095566 (to K. M.), and in part by a postdoctoral fellowship from Applied Biophysics Inc. (to J. A. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- VE-cadherin
- vascular endothelial cadherin
- GPCR
- G protein-coupled receptors
- Thr
- thrombin
- His
- histamine
- S1P
- sphingosine-1-phosphate
- STIM1
- stromal interacting molecule-1
- HDMEC
- human dermal microvascular endothelial cells
- ECIS
- electric cell-substrate impedance sensing
- 2-APB
- 2-aminoethoxydiphenyl borate
- BTP2
- N-[4-[3,5-bis(trifluoromethyl)pyrazol-1-yl]phenyl]-4-methylthiadiazole-5-carboxamide
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester
- SOCE
- store-operated Ca2+ entry
- ROCK
- Rho kinase
- IP3
- inositol 1,4,5-trisphosphate
- ER
- endoplasmic reticulum
- MLC
- myosin light chain
- MLCK
- myosin light chain kinase
- siNT
- non-targeting siRNA
- ANOVA
- analysis of variance
- CaMKII
- Ca2+/calmodulin-dependent kinase II
- HBSS
- Hank's balanced salt solution.
References
- 1. Amado-Azevedo J., Valent E. T., and Van Nieuw Amerongen G. P. (2014) Regulation of the endothelial barrier function: a filum granum of cellular forces, Rho-GTPase signaling and microenvironment. Cell Tissue Res. 355, 557–576 [DOI] [PubMed] [Google Scholar]
- 2. Beckers C. M., van Hinsbergh V. W., and van Nieuw Amerongen G. P. (2010) Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb. Haemost. 103, 40–55 [DOI] [PubMed] [Google Scholar]
- 3. Mehta D., and Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 4. van Nieuw Amerongen G. P., Musters R. J., Eringa E. C., Sipkema P., and van Hinsbergh V. W. (2008) Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/Rho kinase-myosin phosphatase axis. Am. J. Physiol. Cell Physiol. 294, C1234–C1241 [DOI] [PubMed] [Google Scholar]
- 5. Terry S., Nie M., Matter K., and Balda M. S. (2010) Rho signaling and tight junction functions. Physiology 25, 16–26 [DOI] [PubMed] [Google Scholar]
- 6. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., and Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 7. Garcia J. G., Liu F., Verin A. D., Birukova A., Dechert M. A., Gerthoffer W. T., Bamberg J. R., and English D. (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 108, 689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolodney M. S., and Wysolmerski R. B. (1992) Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J. Cell Biol. 117, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Putney J. W., Jr. (1986) A model for receptor-regulated calcium entry. Cell Calcium 7, 1–12 [DOI] [PubMed] [Google Scholar]
- 10. Trebak M. (2009) STIM1/Orai1, ICRAC, and endothelial SOC. Circ. Res. 104, e56–e57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., and Trebak M. (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou M. H., Zheng H., Si H., Jin Y., Peng J. M., He L., Zhou Y., Muñoz-Garay C., Zawieja D. C., Kuo L., Peng X., and Zhang S. L. (2014) Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 289, 29446–29456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Potier M., and Trebak M. (2008) New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Arch. 457, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moccia F., Tanzi F., and Munaron L. (2014) Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 14, 457–480 [DOI] [PubMed] [Google Scholar]
- 15. Fahrner M., Derler I., Jardin I., and Romanin C. (2013) The STIM1/Orai signaling machinery. Channels 7, 330–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosado J. A., Diez R., Smani T., and Jardín I. (2015) STIM and Orai1 variants in store-operated calcium entry. Front. Pharmacol. 6, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Putney J. W. (2011) Origins of the concept of store-operated calcium entry. Front. Biosci. (Schol. Ed.) 3, 980–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cioffi D. L., and Stevens T. (2006) Regulation of endothelial cell barrier function by store-operated calcium entry. Microcirculation 13, 709–723 [DOI] [PubMed] [Google Scholar]
- 19. Cioffi D. L., Lowe K., Alvarez D. F., Barry C., and Stevens T. (2009) TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid. Redox Signal. 11, 765–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moore T. M., Norwood N. R., Creighton J. R., Babal P., Brough G. H., Shasby D. M., and Stevens T. (2000) Receptor-dependent activation of store-operated calcium entry increases endothelial cell permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L691–L698 [DOI] [PubMed] [Google Scholar]
- 21. Shen Q., Rigor R. R., Pivetti C. D., Wu M. H., and Yuan S. Y. (2010) Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc. Res. 87, 272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mehta D., Konstantoulaki M., Ahmmed G. U., and Malik A. B. (2005) Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J. Biol. Chem. 280, 17320–17328 [DOI] [PubMed] [Google Scholar]
- 23. Shinde A. V., Motiani R. K., Zhang X., Abdullaev I. F., Adam A. P., González-Cobos J. C., Zhang W., Matrougui K., Vincent P. A., and Trebak M. (2013) STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci. Signal. 6, ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J., Cubbon R. M., Wilson L. A., Amer M. S., McKeown L., Hou B., Majeed Y., Tumova S., Seymour V. A., Taylor H., Stacey M., O'Regan D., Foster R., Porter K. E., Kearney M. T., et al. (2011) Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 108, 1190–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Antigny F., Jousset H., König S., and Frieden M. (2011) Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 49, 115–127 [DOI] [PubMed] [Google Scholar]
- 26. Stolwijk J. A., Matrougui K., Renken C. W., and Trebak M. (2015) Impedance analysis of GPCR-mediated changes in endothelial barrier function: overview and fundamental considerations for stable and reproducible measurements. Pflugers Arch. 467, 2193–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trebak M. (2012) STIM/Orai signalling complexes in vascular smooth muscle. J. Physiol. 590, 4201–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prakriya M., and Lewis R. S. (2015) Store-operated calcium channels. Physiol. Rev. 95, 1383–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takemura H., Hughes A. R., Thastrup O., and Putney J. W. Jr. (1989) Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells: evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J. Biol. Chem. 264, 12266–12271 [PubMed] [Google Scholar]
- 30. Trebak M., St J Bird G., McKay R. R., Birnbaumer L., and Putney J. W. Jr. (2003) Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J. Biol. Chem. 278, 16244–16252 [DOI] [PubMed] [Google Scholar]
- 31. Trebak M., Bird G. S., McKay R. R., and Putney J. W. Jr. (2002) Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J. Biol. Chem. 277, 21617–21623 [DOI] [PubMed] [Google Scholar]
- 32. He L. P., Hewavitharana T., Soboloff J., Spassova M. A., and Gill D. L. (2005) A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole derivative, BTP2. J. Biol. Chem. 280, 10997–11006 [DOI] [PubMed] [Google Scholar]
- 33. Law M., Morales J. L., Mottram L. F., Iyer A., Peterson B. R., and August A. (2011) Structural requirements for the inhibition of calcium mobilization and mast cell activation by the pyrazole derivative BTP2. Int. J. Biochem. Cell Biol. 43, 1228–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohga K., Takezawa R., Arakida Y., Shimizu Y., and Ishikawa J. (2008) Characterization of YM-58483/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo. Int. Immunopharmacol. 8, 1787–1792 [DOI] [PubMed] [Google Scholar]
- 35. Zitt C., Strauss B., Schwarz E. C., Spaeth N., Rast G., Hatzelmann A., and Hoth M. (2004) Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 279, 12427–12437 [DOI] [PubMed] [Google Scholar]
- 36. DeHaven W. I., Smyth J. T., Boyles R. R., Bird G. S., and Putney J. W. Jr. (2008) Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 283, 19265–19273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bootman M. D., Collins T. J., Mackenzie L., Roderick H. L., Berridge M. J., and Peppiatt C. M. (2002) 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16, 1145–1150 [DOI] [PubMed] [Google Scholar]
- 38. Prakriya M., and Lewis R. S. (2001) Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 536, 3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takezawa R., Cheng H., Beck A., Ishikawa J., Launay P., Kubota H., Kinet J. P., Fleig A., Yamada T., and Penner R. (2006) A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 69, 1413–1420 [DOI] [PubMed] [Google Scholar]
- 40. Smyth J. T., Dehaven W. I., Bird G. S., and Putney J. W. Jr. (2008) Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J. Cell Sci. 121, 762–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Motiani R. K., Abdullaev I. F., and Trebak M. (2010) A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 285, 19173–19183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Motiani R. K., Hyzinski-García M. C., Zhang X., Henkel M. M., Abdullaev I. F., Kuo Y. H., Matrougui K., Mongin A. A., and Trebak M. (2013) STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 465, 1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Motiani R. K., Zhang X., Harmon K. E., Keller R. S., Matrougui K., Bennett J. A., and Trebak M. (2013) Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 27, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McLaughlin J. N., Shen L., Holinstat M., Brooks J. D., Dibenedetto E., and Hamm H. E. (2005) Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J. Biol. Chem. 280, 25048–25059 [DOI] [PubMed] [Google Scholar]
- 45. Mikelis C. M., Simaan M., Ando K., Fukuhara S., Sakurai A., Amornphimoltham P., Masedunskas A., Weigert R., Chavakis T., Adams R. H., Offermanns S., Mochizuki N., Zheng Y., and Gutkind J. S. (2015) RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 6, 6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yuan S. Y., Wu M. H., Ustinova E. E., Guo M., Tinsley J. H., De Lanerolle P., and Xu W. (2002) Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ. Res. 90, 1214–1221 [DOI] [PubMed] [Google Scholar]
- 47. Tinsley J. H., De Lanerolle P., Wilson E., Ma W., and Yuan S. Y. (2000) Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am. J. Physiol. Cell Physiol. 279, C1285–C1289 [DOI] [PubMed] [Google Scholar]
- 48. Norwood N., Moore T. M., Dean D. A., Bhattacharjee R., Li M., and Stevens T. (2000) Store-operated calcium entry and increased endothelial cell permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L815–L824 [DOI] [PubMed] [Google Scholar]
- 49. Borbiev T., Verin A. D., Birukova A., Liu F., Crow M. T., and Garcia J. G. (2003) Role of CaM kinase II and ERK activation in thrombin-induced endothelial cell barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L43–54 [DOI] [PubMed] [Google Scholar]
- 50. Borbiev T., Verin A. D., Shi S., Liu F., and Garcia J. G. (2001) Regulation of endothelial cell barrier function by calcium/calmodulin-dependent protein kinase II. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L983–990 [DOI] [PubMed] [Google Scholar]
- 51. Sandoval R., Malik A. B., Naqvi T., Mehta D., and Tiruppathi C. (2001) Requirement for Ca2+ signaling in the mechanism of thrombin-induced increase in endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L239–L247 [DOI] [PubMed] [Google Scholar]
- 52. Sandoval R., Malik A. B., Minshall R. D., Kouklis P., Ellis C. A., and Tiruppathi C. (2001) Ca2+ signalling and PKCα activate increased endothelial permeability by disassembly of VE-cadherin junctions. J. Physiol. 533, 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mehta D., Rahman A., and Malik A. B. (2001) Protein kinase C-α signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J. Biol. Chem. 276, 22614–22620 [DOI] [PubMed] [Google Scholar]
- 54. Konstantoulaki M., Kouklis P., and Malik A. B. (2003) Protein kinase C modifications of VE-cadherin, p120, and β-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L434–442 [DOI] [PubMed] [Google Scholar]
- 55. Janmey P. A. (1994) Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu. Rev. Physiol. 56, 169–191 [DOI] [PubMed] [Google Scholar]
- 56. Wang Z., Ginnan R., Abdullaev I. F., Trebak M., Vincent P. A., and Singer H. A. (2010) Calcium/Calmodulin-dependent protein kinase II delta 6 (CaMKIIδ6) and RhoA involvement in thrombin-induced endothelial barrier dysfunction. J. Biol. Chem. 285, 21303–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu L., and He P. (2005) Platelet-activating factor increases endothelial [Ca2+]i and NO production in individually perfused intact microvessels. Am. J. Physiol. Heart Circ. Physiol. 288, H2869–H2877 [DOI] [PubMed] [Google Scholar]
- 58. Yuan D., Xu S., and He P. (2014) Enhanced permeability responses to inflammation in streptozotocin-induced diabetic rat venules: Rho-mediated alterations of actin cytoskeleton and VE-cadherin. Am. J. Physiol. Heart Circ. Physiol. 307, H44–H53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou X., and He P. (2011) Temporal and spatial correlation of platelet-activating factor-induced increases in endothelial [Ca2+]i, nitric oxide, and gap formation in intact venules. Am. J. Physiol. Heart Circ. Physiol. 301, H1788–H1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferreri D. M., Minnear F. L., Yin T., Kowalczyk A. P., and Vincent P. A. (2008) N-cadherin levels in endothelial cells are regulated by monolayer maturity and p120 availability. Cell Commun. Adhes. 15, 333–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kassan M., Zhang W., Aissa K. A., Stolwijk J., Trebak M., and Matrougui K. (2015) Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflugers Arch. 467, 1195–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bisaillon J. M., Motiani R. K., Gonzalez-Cobos J. C., Potier M., Halligan K. E., Alzawahra W. F., Barroso M., Singer H. A., Jourd'heuil D., and Trebak M. (2010) Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 298, C993–C1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. González-Cobos J. C., Zhang X., Zhang W., Ruhle B., Motiani R. K., Schindl R., Muik M., Spinelli A. M., Bisaillon J. M., Shinde A. V., Fahrner M., Singer H. A., Matrougui K., Barroso M., Romanin C., et al. (2013) Store-independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia. Circ. Res. 112, 1013–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]