

Abstract
The pleiotropic cytokine interleukin 2 (IL2) disrupts the blood-brain barrier and alters brain microcirculation, underlying vascular leak syndrome that complicates cancer immunotherapy with IL2. The microvascular effects of IL2 also play a role in the development of multiple sclerosis and other chronic neurological disorders. The mechanism of IL2-induced disruption of brain microcirculation has not been determined previously. We found that both human and murine brain microvascular endothelial cells express constituents of the IL2 receptor complex. Then we established that signaling through this receptor complex leads to activation of the transcription factor, nuclear factor κB, resulting in expression of proinflammatory interleukin 6 and monocyte chemoattractant protein 1. We also discovered that IL2 induces disruption of adherens junctions, concomitant with cytoskeletal reorganization, ultimately leading to increased endothelial cell permeability. IL2-induced phosphorylation of vascular endothelial cadherin (VE-cadherin), a constituent of adherens junctions, leads to dissociation of its stabilizing adaptor partners, p120-catenin and β-catenin. Increased phosphorylation of VE-cadherin was also accompanied by a reduction of Src homology 2 domain-containing protein-tyrosine phosphatase 2, known to maintain vascular barrier function. These results unravel the mechanism of deleterious effects induced by IL2 on brain microvascular endothelial cells and may inform the development of new measures to improve IL2 cancer immunotherapy, as well as treatments for autoimmune diseases affecting the central nervous system.
Keywords: brain, cancer therapy, endothelial cell, inflammation, permeability, interleukin 2
Introduction
The blood-brain barrier (BBB)3 is formed by endothelial cells that line the microcirculation and are part of the “neurovascular unit” composed of brain microvascular endothelial cells (BMECs), astrocytes, and neurons (1). BMECs play an essential role in these functional units, even prompting neurological changes following endothelial activation. For example, BMECs are stimulated by inflammatory mediator interleukin 1β, causing fever and lethargy (2). Likewise, IL2, the mainstay of the first effective cancer immunotherapy, induces neuropsychiatric changes in patients through a hitherto undefined mechanism (3). This therapy consists of IL2-induced activation and proliferation of tumor-infiltrating lymphocytes that lead to remission of renal cell carcinoma and malignant melanoma (4). However, these beneficial effects of IL2 are curtailed by the development of brain edema, a prominent manifestation of vascular leak syndrome (VLS). Brain edema subsides upon cessation of IL2 immunotherapy, indicating its direct role in this serious complication. Thus, IL2-induced vascular permeability in the central nervous system and other organs represents an undesirable off-target effect (5). Moreover, IL2 plays a significant role in several chronic neurological autoimmune diseases, including multiple sclerosis, neuromyelitis optica, and neuropsychiatric systemic lupus erythematosus (6–8). Disruption of the BBB is a critical step in development of these neurological conditions. However, the mechanism of adverse action of IL2 on the barrier function of brain microvascular endothelial cells remains unknown.
The maintenance of BBB function is largely vested in BMEC intercellular junctions, a complex network of adhesive proteins organized into adherens junctions (AJs) and tight junctions (9). AJs are ubiquitously expressed in the vascular tree (10) and are of particular importance in microvascular circulation, which provides the largest surface area of exchange between blood and tissue (11). Furthermore, postcapillary venules comprise the main endothelial surface for activation and emigration of immune cells that mediate inflammatory reactions (12). Formation of AJs is dependent on the homotypic interaction of cell surface transmembrane proteins termed cadherins whose cytoplasmic tails associate with intracellular binding partners termed catenins. Thus, the stability of AJs depends on complex formation between vascular endothelial cadherin (VE-cadherin), p120-catenin (p120), β-catenin, and plakoglobin (9). In contrast, destabilization of AJs is due to a loss of p120 and β-catenin binding, which causes the internalization of VE-cadherin (13–15). This process is induced by increased phosphorylation of VE-cadherin accompanied by a decrease in Src homology domain-containing protein-tyrosine phosphatase 2 (SHP2), which maintains vascular barrier function (15). Ultimately, disruption of AJs leads to leakiness of endothelial cell monolayers (9, 15–17).
In general, activation of endothelial cells results in expression of cell adhesion molecules along with proinflammatory cytokines and chemokines, enhancing an autoregulatory feed-forward loop that leads to further recruitment of immune cells and escalation of the inflammatory response (18). Genes encoding these mediators of inflammation are controlled by nuclear factor κB (NFκB), master regulator of immunity and inflammation. Increased expression of interleukin 6 (IL6), macrophage chemoattractant protein 1 (MCP1/CCL2), intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 among others has been linked to the NFκB pathway (19, 20). In lymphocytes, IL2-evoked signaling mediated by its high affinity trimeric receptor (IL2Rαβγ) has been previously coupled to activation of the NFκB pathway (21).
Here we analyze the mechanism of IL2-induced activation of human and murine brain microvascular endothelial cells and the resulting disruption of BMEC barrier function. We report that IL2 induces activation of the NFκB pathway, leading to increased expression of proinflammatory mediators. In parallel, IL2 causes disruption of AJs through an increase in VE-cadherin phosphorylation. As a consequence, microvascular endothelial monolayer permeability was increased. Whereas IL2-induced disruption of the BBB changes the central nervous system milieu and leads to subsequent brain edema, our study may contribute to the development of new strategies for protecting brain function from this deleterious process.
Experimental Procedures
Cell Culture
The human BMEC line hCMEC/D3 was provided through the courtesy of Professor Babette Weksler (Weill Cornell Medical College, New York, NY) (22). hCMEC/D3 cells were cultured at 37 °C in 5% CO2 in EBM-2 (Lonza) supplemented with 5% heat-inactivated fetal bovine serum (HI-FBS), 1% penicillin/streptomycin (Mediatech), 1.4 μm hydrocortisone (Sigma), 5 μg/ml ascorbic acid (Fisher Scientific), chemically defined lipid concentrate (Invitrogen) diluted 1:100, 10 mm HEPES (Mediatech), and 1 ng/ml human basic FGF (Sigma). Cells used for experiments were between passages 20 and 30. The murine brain endothelioma cell line bEnd.3 (23) was purchased from American Type Culture Collection and cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium (Mediatech) supplemented with 10% HI-FBS and 1% penicillin/streptomycin. Murine embryonic fibroblasts (MEFs) and the human embryonic kidney (HEK) cell line were cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% HI-FBS and 1% penicillin/streptomycin.
Quantitative Real Time PCR Analysis
Total RNA was isolated for quantitative real time PCR (qPCR) analysis using the RNeasy Mini kit (Qiagen) following the manufacturer's instructions and reverse transcribed utilizing the iScript cDNA synthesis kit (Bio-Rad). Targets were amplified by the CFX96 Touch real time PCR detection system (Bio-Rad) using the iQ SYBR Green Supermix kit (Bio-Rad) with specific primers listed in Table 1 for the indicated mRNAs. Results were analyzed by CFX Manager (Bio-Rad). Following normalization to GAPDH cDNA, relative levels were calculated using the 2−ΔΔCt method (24).
TABLE 1.
Oligonucleotide PCR primer sequences used in current study
h, human; m, murine.
| Gene | Primer sequences |
|---|---|
| h vWF | Forward, 5′-TGCTGACACCAGAAAAGTGC-3′ |
| Reverse, 5′-AGTCCCCAATGGACTCACAG-3′ | |
| m vWF | Forward, 5′-TTCATCCGGGACTTTGAGAC-3′ |
| Reverse, 5′-AGCCTTGGCAAAACTCTTCA-3′ | |
| h PECAM1 | Forward, 5′-GATAATTGCCATTCCCATGC-3′ |
| Reverse, 5′-GGGTTTGCCCTCTTTTTCTC-3′ | |
| m PECAM1 | Forward, 5′-ATGACCCAGCAACATTCACA-3′ |
| Reverse, 5′-CACAGAGCACCGAAGTACCA-3′ | |
| h IL2Rα | Forward, 5′-ATCAGTGCGTCCAGGGATAC-3′ |
| Reverse, 5′-GTGACGAGGCAGGAAGTCTC-3′ | |
| m IL2Rα | Forward, 5′-TACAAGAACGGCACCATCCTAA-3′ |
| Reverse, 5′-TTGCTGCTCCAGGAGTTTCC-3′ | |
| h IL2Rβ | Forward, 5′-GCTGATCAACTGCAGGAACA-3′ |
| Reverse, 5′-TGTCCCTCTCCAGCACTTCT-3′ | |
| m IL2Rβ | Forward, 5′-GGCTCTTCTTGGAGATGCTG-3′ |
| Reverse, 5′-GCCAGAAAAACAACCAAGGA-3′ | |
| h IL2Rγ | Forward, 5′-CCACTCTGTGGAAGTGCTCA-3′ |
| Reverse, 5′-AGGTTCTTCAGGGTGGGAAT-3′ | |
| m IL2Rγ | Forward, 5′-CTGGGGGAGTCATACTGTAGAGG-3′ |
| Reverse, 5′-AGGCTTCCGGCTTCAGAGAAT-3′ |
Immunoblotting and Immunoprecipitation Analysis
Antibodies against NFκB p65 (RelA), p120, VE-cadherin, SHP2 (Santa Cruz Biotechnology), β-catenin (BD Biosciences), phospho-β-catenin, IκBα, phospho-NFκB p65 (phospho-RelA) (Cell Signaling Technology), and phospho-VE-cadherin (ECM Biosciences) were used for immunoblotting analyses. Obtained values were normalized in reference to β-actin or TATA-binding protein detected with their cognate antibodies (Abcam) in cytosolic and nuclear extracts as indicated. Adherens junction protein complexes were immunoprecipitated from cell lysates with antibodies against p120 or VE-cadherin. Species-specific IgG (Santa Cruz Biotechnology) was used as a control. The lysates were precleared with protein A/G-agarose beads (Thermo) prior to addition of antibodies or control IgG. The immune complexes were then captured with protein A/G-agarose beads and analyzed by quantitative immunoblotting using antibodies against p120, VE-cadherin, and β-catenin with the LI-COR Odyssey infrared imaging system as described previously (25).
Cytokine/Chemokine Assays
Cytokines and chemokines in tissue culture medium supernatants were assayed by cytometric bead array (BD Biosciences) in the Vanderbilt Flow Cytometry Shared Resource according to the manufacturer's instructions as described previously (25). During time- and concentration-dependent titration experiments, supernatant samples were taken following stimulation as indicated. Cytokine and chemokine expression was also analyzed in medium from BMEC monolayers cultured in Transwell chambers for permeability experiments. Medium samples were taken from the upper chamber following stimulation as indicated but prior to addition of FITC-dextran tracer.
Immunofluorescence Staining and Fluorescence Microscopy
Human hCMEC/D3 or murine bEnd.3 cells grown to confluence on Transwell permeability supports were stimulated with species-specific IL2 (BD Biosciences, Prometheus) or TNFα (Invitrogen) as indicated. After stimulation, cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences), washed with PBS, and permeabilized with 0.1% Triton X-100 (Invitrogen). For immunostaining, cells were blocked with 5% normal goat serum (Jackson ImmunoResearch Laboratories) before overnight incubation at 4 °C with primary antibodies to NFκB p65 (Abcam) or VE-cadherin and p120 (Santa Cruz Biotechnology) followed by incubation with Alexa Fluor 488 (Invitrogen)-, Cy3-, or Cy5-labeled (Jackson ImmunoResearch Laboratories) secondary antibodies. F-actin polymerization was visualized in permeabilized cells with Alexa Fluor 488-labeled phalloidin (Cytoskeleton, Inc.). After staining, the semipermeable membranes were removed from the Transwell permeability supports and mounted on slides with ProLong Gold antifade reagent containing DAPI (Invitrogen) to counterstain nuclei. Images were captured and analyzed with MetaMorph software on an Axioplan widefield microscope in the Vanderbilt Cell Imaging Core facility using a ×63 oil immersion objective.
Endothelial Cell Permeability
Human hCMEC/D3 or murine bEnd.3 cells (1 × 105) were seeded onto 24-well Transwell semipermeable supports (Costar) precoated with type 1 collagen (Cultrex) and incubated until confluent. Upon confluence, as verified by microscopy, growth medium was exchanged for serum-depleted medium that comprised growth medium with a low concentration (0.5%) of HI-FBS. Following 24-h culture in low serum medium, cells were left unstimulated or stimulated for 24 h with species-specific 300 kilounits/ml IL2 or 30 ng/ml TNFα. In experiments utilizing neutralizing antibodies, species-specific murine anti-IL6 and anti-MCP1 (R&D Systems) and human anti-IL6 (R&D Systems) and anti-MCP1 (Sigma) were added concomitantly with IL2. Human anti-IL2Rβ antibody (R&D Systems) was added 30 min prior to IL2 stimulation. Antibody concentrations necessary for neutralization were calculated from the levels of cytokine/chemokine expression during titration of IL2. Monolayer permeability was assessed by detection of FITC-dextran in the lower chamber at various times after addition of 1 mg/ml 10-kDa FITC-dextran (Sigma) to the upper chamber.
Cellular Detachment and Apoptosis
Human and murine BMECs were cultured and treated as described previously for permeability experiments. Following 24-h stimulation, the supernatant was removed from the Transwell chambers, and cellular detachment/apoptosis was measured using the trypan blue exclusion assay.
Statistical Analyses
Data analysis and statistical calculations were performed using Prism (GraphPad). Cytokine and chemokine levels in cultured cell supernatants and Western blotting quantification of comparative controls were analyzed using an unpaired t test with Welch's correction or Student's t test as indicated. Western blotting quantifications of multiple time points were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Quantification of real time PCR results was calculated as a -fold change in transcripts compared with non-stimulated samples, and statistical differences were determined by Student's t test. For permeability experiments, p values shown represent comparison of the area under the curve calculated for each experiment and/or condition analyzed by unpaired t test with Welch's correction. In all experiments, a p value of <0.05 was considered significant.
Results
Characterization of BMECs and Their Expression of the IL2 Receptor Subunits
In an effort to ensure that the endothelial cell phenotype was maintained in this study, we analyzed two well known markers specific for endothelial cells, von Willebrand factor (vWF) and platelet endothelial cell adhesion molecule 1 (PECAM1) (26). As expected, both human hCMEC/D3 and murine bEnd.3 cells constitutively expressed high levels of vWF and PECAM1 transcripts compared with human and murine non-endothelial cells (HEK cells and MEFs) when analyzed by qPCR (Fig. 1A). Our qPCR results in both human hCMEC/D3 and murine bEnd.3 cells complement previous characterization in terms of protein expression of vWF and PECAM1 (22, 23, 27). Next, we assessed IL2 receptor subunit expression by qPCR and Western blotting to ascertain whether BMECs can respond to IL2. The IL2 receptor complex comprises the high affinity trimeric configuration (IL2Rαβγ) or the intermediate affinity dimer (IL2Rβγ) (28). We documented that IL2Rβ (CD122) and IL2Rγ (CD132) are expressed constitutively at both the transcript and protein levels. Conversely, these transcripts were barely detectable in control cell lines of human and murine origin (HEK cells and MEFs). IL2Rα (CD25) transcripts were only detected in BMECs, but not control cells, after agonist stimulation for 24 h and corroborated with detection of the protein by Western blotting (Fig. 1B). Without constitutive expression of CD25 and formation of the trimeric receptor complex, affinity for IL2 is reduced 10–100 times (29). Therefore, we based our experimental protocol on using sufficiently high concentrations of IL2 to stimulate naïve BMECs and overcome their lower signaling capacity via the intermediate affinity receptor complex (IL2Rβγ). As compared with human hCMEC/D3 cells, murine bEnd.3 cells displayed significantly higher transcript levels of IL2Rβ, the receptor subunit most attributed to mediating positive signal transduction (28, 30). No significant difference between human and murine cells was detected in transcript levels of inducible IL2Rα subunit or constitutive IL2Rγ subunit. However, a trend toward higher expression of IL2Rγ was observed in murine bEnd.3 cells. Thus, these results establish the expression of the IL2 intermediate affinity receptor complex on both human and murine BMECs.
FIGURE 1.
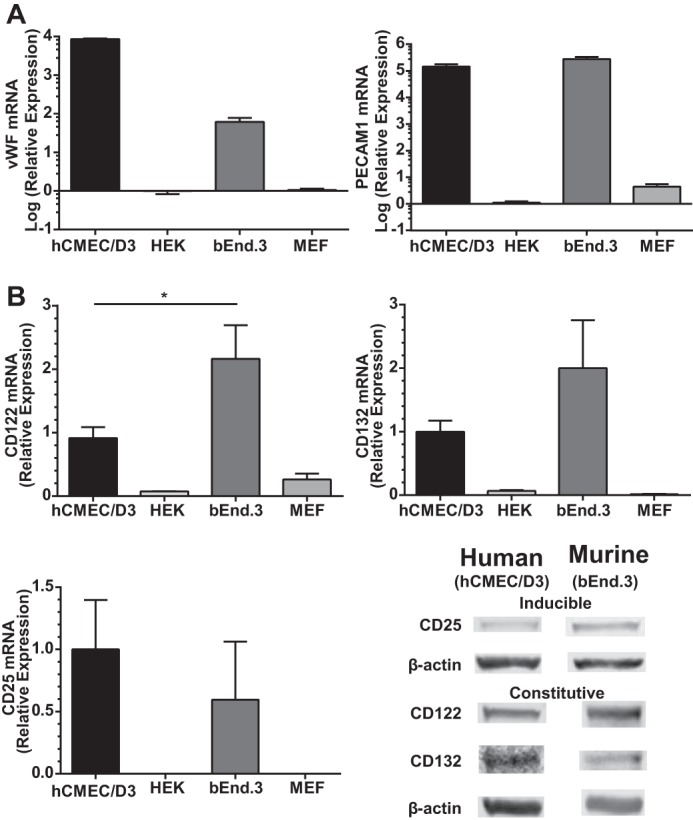
Expression of endothelial cell markers vWF, PECAM1, and IL2R subunits in human and murine BMECs. A, constitutive transcript levels of endothelial cell markers vWF and PECAM1 were assessed by qPCR. Following normalization to GAPDH, mRNA levels are presented as increased -fold change from species-specific non-endothelial cells: HEK 293 cells and MEFs. B, transcript levels of IL2R subunits were determined by qPCR, and protein expression was documented by immunoblotting. IL2Rβ (CD122) and IL2Rγ (CD132) were constitutively expressed in both human and murine BMECs. Transcript and protein levels for IL2Rα (CD25) were only detected following 24-h stimulation with species-specific 300 kilounits/ml IL2. The level of mRNA transcript is presented as relative expression comparing human and murine BMECs. Error bars represent mean ± S.E. of three independent experiments performed in triplicates. Statistical significance was determined by Student's t test (*, p < 0.05).
IL2-induced Activation of the NFκB Pathway
We hypothesized that IL2-induced activation of BMECs entails signaling through the NFκB pathway akin to IL15 (31). Therefore, we used three experimental approaches to analyze NFκB signaling in IL2-stimulated BMECs and compared the results with those evoked by a known endothelial agonist, lipopolysaccharide (LPS), a potent activator of the NFκB pathway (32). First, we determined that IκBα, the inhibitory protein responsible for sequestering NFκB p65 (RelA) in the cytoplasm, becomes degraded following IL2 stimulation in both human and murine BMECs (Fig. 2A). Due to the very rapid turnover kinetics of IκBα (33), we included the protein synthesis inhibitor cycloheximide to prevent reaccumulation of newly synthesized IκBα. Constitutive turnover was measured following addition of cycloheximide and compared with degradation in BMECs stimulated with agonist. Stimulation with IL2 caused time-dependent degradation of IκBα with a loss of 30–70% of its basal level, mimicking IκBα degradation following activation with LPS. Notably, the kinetics of IκBα degradation was faster in human hCMEC/D3 cells compared with murine bEnd.3 cells likely due to constitutively higher expression of physiologic suppressors of the NFκB signaling pathway such as A20 in murine cells (34). As a result of IκBα degradation, RelA is no longer sequestered in the cytoplasm, becomes phosphorylated, and is able to translocate into the nucleus. RelA phosphorylation enhances DNA binding and results in the activation of a number of proinflammatory genes (35). Accordingly, we detected an increase in phosphorylated RelA compared with total RelA in the nucleus (Fig. 2B). Both human and murine BMECs showed a time-dependent response to IL2, resulting in a 50% increase in Tyr-536 phosphorylation of RelA in nuclear extracts of agonist-treated cells. Finally, we verified nuclear translocation of RelA using immunofluorescence. Indeed, as compared with the unstimulated control, we noted elevated levels of nuclear RelA in BMECs stimulated with IL2 or TNFα, another known inducer and activator of the NFκB pathway (Fig. 2C). Taken together, our multipronged approach demonstrates that IL2 activates the NFκB signaling pathway in human and murine BMECs.
FIGURE 2.
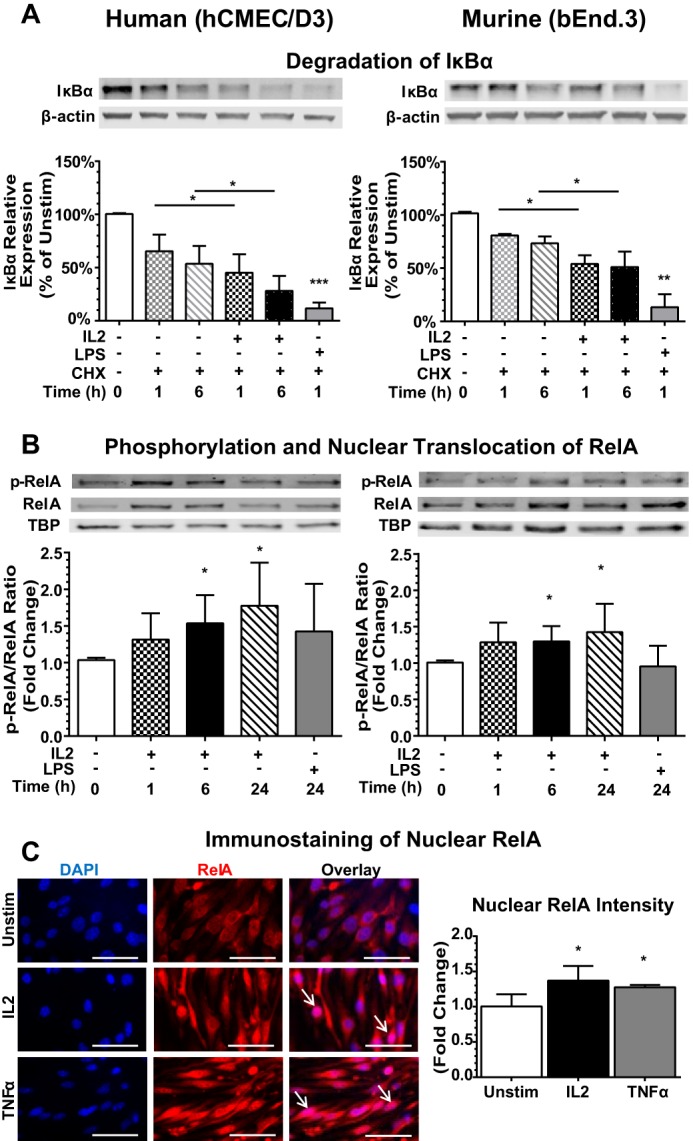
IL2 activates the NFκB pathway in human and murine BMECs. A, degradation of IκBα in BMECs. Human (hCMEC/D3) and murine (bEnd.3) BMECs were left unstimulated (Unstim) (0) or stimulated with species-specific 300 kilounits/ml IL2 or 1 μg/ml LPS, a positive control, for the indicated times after pretreatment for 30 min with the protein synthesis inhibitor cycloheximide (CHX) (10 μg/ml) to block de novo protein synthesis. Cytosolic extracts were isolated, separated by SDS-PAGE, and immunoblotted for the degradation of IκBα. Protein levels were quantified by immunoblotting, and values at each time point were normalized to the β-actin loading control. Error bars represent mean ± S.D. of at least four independent experiments. Statistical significance was determined using an unpaired t test with Welch's correction (*, p < 0.05; **, p < 0.005; ***, p < 0.0005). B, translocation of RelA to the nucleus. Human and murine BMECs were stimulated with 300 kilounits/ml species-specific IL2 or 1 μg/ml LPS for the indicated times, and nuclear extracts were isolated, separated by SDS-PAGE, and immunoblotted for the presence of total RelA and Tyr-536 phosphorylated RelA (p-RelA). TATA-binding protein (TBP) serves as a loading control for nuclear lysates. Quantification of signal is shown as -fold change compared with the unstimulated control. Error bars represent mean ± S.D. of at least four independent experiments. Statistical significance was determined using an unpaired t test with Welch's correction (*, p < 0.05). C, detection of RelA in the nucleus by immunofluorescence. Murine (bEnd.3) BMECs were analyzed following 24-h stimulation with 300 kilounits/ml murine IL2 or 30 ng/ml murine TNFα. Cells were immunostained with anti-RelA antibodies (red), and nuclei were counterstained with DAPI (blue). Arrows indicate strong nuclear staining of RelA. Images are representative of three independent experiments. Quantification was determined using MetaMorph imaging software. Statistical significance was determined using an unpaired t test with Welch's correction (*, p < 0.05). Magnification, ×63. Scale bars, 5 μm. Error bars represent mean ± S.D.
IL2 Induces Expression of Proinflammatory Cytokines and Chemokines
Several proinflammatory cytokines and chemokines have been previously implicated in endothelial activation and loss of barrier function, including IL6 and MCP1 (36–39). Both are regulated by the NFκB pathway (20, 40). Given that IL2 activated this pathway, we hypothesized that IL2 stimulation would also induce expression of these two proinflammatory mediators. Indeed, both pleiotropic cytokine IL6 and chemokine MCP1 were elicited by IL2 in a concentration-dependent manner in human and murine BMECs (Fig. 3). Thus, IL2 induces proinflammatory agonists that can also amplify signaling events involved in the potentiation of microvascular injury.
FIGURE 3.
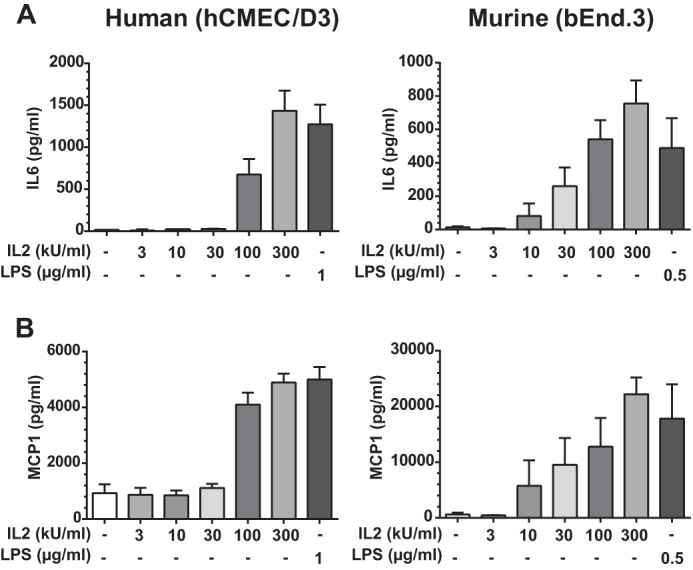
IL2-induced expression of proinflammatory IL6 and MCP1 in human and murine BMECs. IL6 (A) and MCP-1 (B) are expressed by activated BMECs in a dose-dependent manner. Protein levels of IL6 and MCP1 in culture medium were measured 24 h after stimulation of hCMEC/D3 and bEnd.3 cells with increasing concentrations of species-specific IL2 as indicated (kilounits (kU)) or 1 μg/ml LPS in hCMEC/D3 cells and 0.5 μg/ml LPS in bEnd.3 cells. Error bars represent mean ± S.D. from three independent experiments performed in duplicate or triplicate.
IL2-induced Permeability of the BMEC Monolayer
IL2 administered during cancer immunotherapy causes VLS, manifested by a rise in brain water content that is attributed to increased permeability of the brain microvascular endothelium (5). Therefore, we next analyzed the potential mechanism of IL2-induced disruption of endothelial barrier function in BMEC monolayers in Transwell chambers. We monitored the passage of FITC-labeled dextran across an intact monolayer. FITC-dextran was added to the upper Transwell chamber following 24-h stimulation of BMECs with species-specific IL2. The lower chamber supernatant was then sampled at the indicated time points. IL2 induced a significant increase in the movement of FITC-dextran across the IL2-stimulated BMEC monolayer compared with the non-stimulated control, indicating a gradual loss of endothelial barrier function and increased permeability (Figs. 4A and 5A). As IL2-induced permeability can potentially be enhanced by IL6 and/or MCP1, we utilized species-specific neutralizing antibodies against IL6 and MCP1. Neutralizing these two potential permeability agonists did not significantly alter an IL2-induced increase in permeability, although a trend toward lower permeability was observed. In contrast, IL2-induced permeability enhancement was counteracted in human hCMEC/D3 cells by species-specific IL2Rβ-blocking antibody (Fig. 4A). These results indicate that IL2 can directly increase endothelial monolayer permeability. A similar blocking experiment could not be performed in murine bEnd.3 cells due to a lack of species-specific antibody against IL2Rβ. Murine bEnd.3 cells demonstrated a greater change in endothelial permeability than human hCMEC/D3 cells most likely due to constitutively higher expression of IL2Rβ. Even though permeability increases in the hCMEC/D3 monolayer were not as robust, these results were significant and consistent. Endothelial monolayer confluence and integrity were checked by microscopy prior to initiating permeability assays. To this end, extending culture time from 24 to 72 h without agonist treatment in the Transwell system of both human hCMEC/D3 and murine bEnd.3 endothelial cells did not change their baseline permeability level for FITC-labeled dextran.
FIGURE 4.
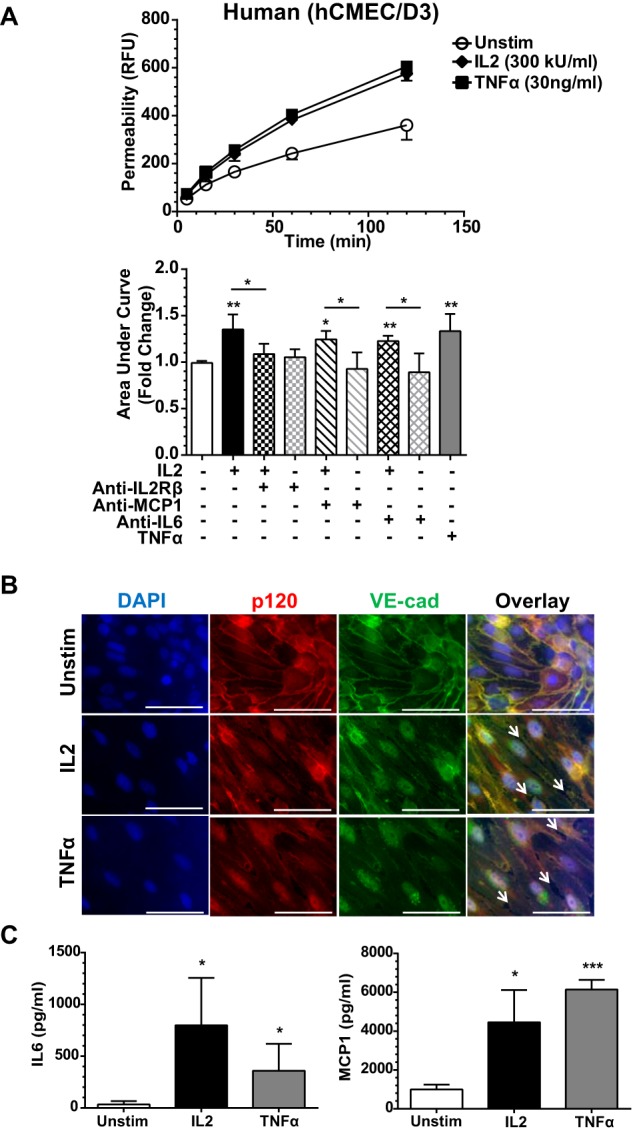
IL2-induced permeability and expression of proinflammatory mediators in human BMEC monolayer. A, human (hCMEC/D3) cells were grown to confluence on Transwell inserts and left unstimulated (Unstim) or stimulated with 300 kilounits (kU)/ml human IL2 or 30 ng/ml human TNFα for 24 h as indicated. Concurrent with stimulation, a portion of the Transwell inserts were treated with species-specific neutralizing antibodies against IL6 and MCP1 as indicated. Shown are representative permeability tracings (upper panel) and quantitative representation of all permeability tracings recorded at 120 min (bottom panel). To assess permeability, 10-kDa FITC-dextran was added to the top chamber of each insert, and fluorescence in the lower chamber was measured at the indicated times. Error bars represent mean ± S.D. of -fold change relative to the unstimulated control from five independent experiments performed in at least duplicate; p values shown were determined by an unpaired t test with Welch's correction of the area under the curves from each individual experiment (*, p < 0.05; **, p < 0.005). B, the loss of cell border adherens junctions documented by immunofluorescence of VE-cadherin (VE-cad) and p120 following agonist stimulation. hCMEC/D3 cells were grown to confluence and then left unstimulated or stimulated with 300 kilounits/ml human IL2 or 30 ng/ml human TNFα for 24 h. Cells were immunostained with anti-p120 antibodies (red) and anti-VE-cadherin antibodies (green). Nuclei were counterstained with DAPI (blue). Arrows indicate openings between neighboring cells and disruption of both proteins at the cell membrane in agonist-stimulated human BMECs. Magnification, X63; scale bars, 5 μm. C, agonist-induced expression of cytokine IL6 and chemokine MCP1 in hCMEC/D3 cell monolayer lining Transwell inserts. The hCMEC/D3 cell activation state was assessed prior to initiation of the permeability assay utilizing identical cells for multiple analyses. Cells were grown to confluence on Transwell inserts and left unstimulated or stimulated with 300 kilounits/ml human IL2 or 30 ng/ml human TNFα for 24 h as indicated. The supernatants were collected and analyzed for cytokine/chemokine production prior to addition of FITC-dextran for permeability analysis. Error bars represent mean ± S.D. of four independent experiments performed in technical duplicates or triplicates. Statistical significance was determined by unpaired t test with Welch's correction (*, p < 0.05; ***, p < 0.0005). RFU, relative fluorescence units.
FIGURE 5.
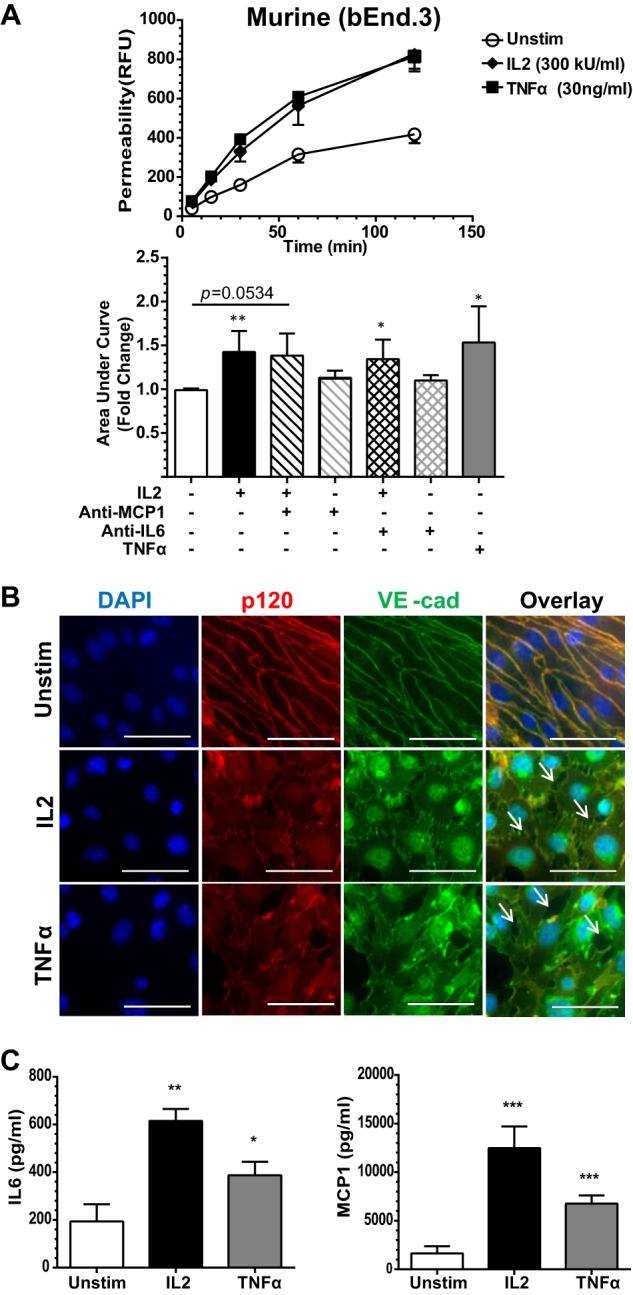
IL2-induced permeability and expression of proinflammatory mediators in murine BMEC monolayer. A, murine (bEnd.3) cells were grown to confluence on Transwell inserts and left unstimulated (Unstim) or stimulated for 24 h with 300 kilounits (kU)/ml murine IL2 or 30 ng/ml murine TNFα as indicated. Concurrent with stimulation, a portion of the Transwell inserts were treated with species-specific neutralizing antibodies against IL6 and MCP1 as indicated. Shown are representative permeability tracings (upper panel) and quantitative representation of all permeability tracings recorded at 120 min (bottom panel). To assess permeability, 10-kDa FITC-dextran was added to the top chamber of each insert, and fluorescence in the lower chamber was measured at the indicated times. Error bars represent mean ± S.D. of -fold change relative to the unstimulated control from at least three independent experiments performed in at least duplicate; p values shown were determined by unpaired t test with Welch's correction of the area under the curves from each individual experiment (*, p < 0.05; **, p < 0.005). B, the loss of cell border adherens junctions documented by immunofluorescence of VE-cadherin (VE-cad) and p120 following agonist stimulation. bEnd.3 cells were grown to confluence and then left unstimulated or stimulated with 300 kilounits/ml murine IL2 or 30 ng/ml murine TNFα for 24 h. Cells were immunostained with anti-p120 antibodies (red) and anti-VE-cadherin antibodies (green). Nuclei were counterstained with DAPI (blue). Arrows indicate openings between neighboring cells and disruption of both proteins at the cell membrane in agonist-stimulated murine BMECs. Magnification, X63; scale bars, 5 μm. C, agonist-induced expression of cytokine IL6 and chemokine MCP1 in bEnd.3 cell monolayer lining Transwell inserts. bEnd.3 cell activation state was assessed prior to initiation of the permeability assay utilizing identical cells for multiple analyses. Cells were grown to confluence on Transwell inserts and left unstimulated or stimulated with 300 kilounits/ml murine IL2 or 30 ng/ml murine TNFα for 24 h as indicated. The supernatants were collected and analyzed for cytokine/chemokine production prior to addition of FITC-dextran for permeability analysis. Error bars represent mean ± S.D. of at least three independent experiments performed in duplicates or triplicates. Statistical significance was determined by unpaired t test with Welch's correction (*, p < 0.05; **, p < 0.005; ***, p < 0.0005). RFU, relative fluorescence units.
We verified changes in endothelial barrier function by immunostaining the adherens junction protein complex that includes VE-cadherin and one of its adaptors, p120-catenin. In quiescent human and murine BMECs, immunostained VE-cadherin and p120 displayed a well defined, contiguous border between adjoining cells. This border was strikingly lost after treatment with IL2 or TNFα, a comparative positive control. Membrane-associated VE-cadherin·p120 complexes were disrupted with a concomitant reduction of these proteins along the periphery of the cells (Figs. 4B and 5B). We verified that endothelial cell activation correlated with the time course of increased permeability in human hCMEC/D3 and murine bEnd.3 cells by analyzing cytokine and chemokine expression in supernatant collected from the upper Transwell chamber prior to addition of FITC-dextran. In agreement with prior analysis in a different culture system (see Fig. 3), we documented significantly higher levels of IL6 and MCP1 following IL2 stimulation compared with the unstimulated control (Figs. 4C and 5C). However, within the time frame of our antibody-neutralizing permeability experiments, these two proinflammatory agonists evoked by IL2 did not significantly contribute to its effect on microvascular permeability (see above; Figs. 4A and 5A).
IL2-induced Disruption of the Adherens Junction
Disruption of the AJ protein complex is accompanied by changes in cell morphology and resulted in the opening of gaps between neighboring cells. These changes in endothelial monolayer integrity are coupled to cytoskeletal reorganization (25, 41, 42). Indeed, an increase in F-actin fiber bundles was prominently displayed in IL2-activated murine bEnd.3 cells (Fig. 6A). These cells displayed thicker and more prominent F-actin fiber bundles compared with thin F-actin fibers sparsely crossing the cell body in unstimulated controls. Moreover, agonist-induced cell loss due to detachment and apoptosis was documented (Fig. 6B). Agonist stimulation resulted in significantly higher levels of cellular detachment and apoptosis in human and murine BMECs. Detached circulating endothelial cells have been described in a number of human diseases as well as in animal models of vascular injury (43, 44).
FIGURE 6.
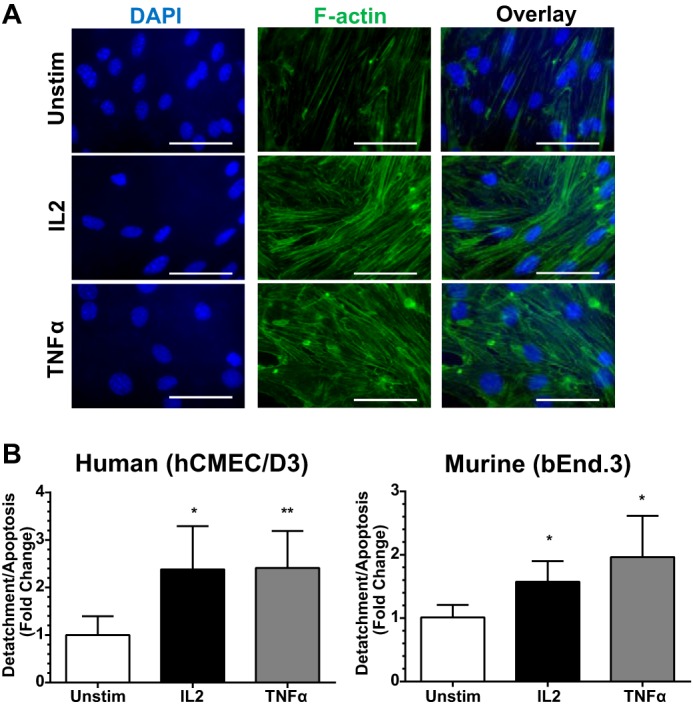
IL2-induced F-actin polymerization in BMECs and increased detachment following stimulation. A, F-actin polymerization changes in agonist-stimulated murine BMECs. bEnd.3 cells were grown to confluence and then left unstimulated (Unstim) or stimulated with 300 kilounits/ml murine IL2 or 30 ng/ml murine TNFα for 24 h. Cells were stained with Alexa Fluor 488-labeled phalloidin (green) to visualize F-actin. Nuclei were counterstained with DAPI (blue). Images are representative of at least three independent experiments. Magnification, ×63. Scale bars, 5 μm. B, increased BMEC detachment and apoptosis following agonist stimulation from Transwell inserts. Both human and murine BMECs were grown to confluence on Transwell permeability inserts and then left unstimulated or stimulated with species-specific IL2 (300 kilounits/ml) or TNFα (30 ng/ml) for 24 h. Supernatants were collected, and free floating endothelial cells detached from the Transwell membranes were counted. Error bars represent mean ± S.D. of at least three independent experiments performed in duplicates. Statistical significance was determined by unpaired t test with Welch's correction (*, p < 0.05; **, p < 0.005).
IL2 Changes the Composition of the AJ Complex
The striking disruption of the AJ protein complex following IL2 stimulation as demonstrated by immunostaining led us to analyze changes in AJ protein complex composition. In a series of co-immunoprecipitation experiments, we demonstrated that IL2-induced activation of BMECs results in a loss of interaction between VE-cadherin and p120 as reflected by their ratio (Fig. 7A). β-Catenin also binds to VE-cadherin, stabilizing the complex and allowing its interaction with F-actin. Loss of both p120 and β-catenin leads to destabilization of adherens junctions and endocytosis of VE-cadherin (12, 14, 42). Indeed, the VE-cadherin·β-catenin complex is also altered due to IL2 activation in both human and murine BMECs (Fig. 7B). Thus, IL2-induced dissociation of the AJ complex documented by co-immunoprecipitation explains the loss of VE-cadherin immunostaining at cellular borders seen in agonist-stimulated BMECs (Figs. 4B and 5B).
FIGURE 7.
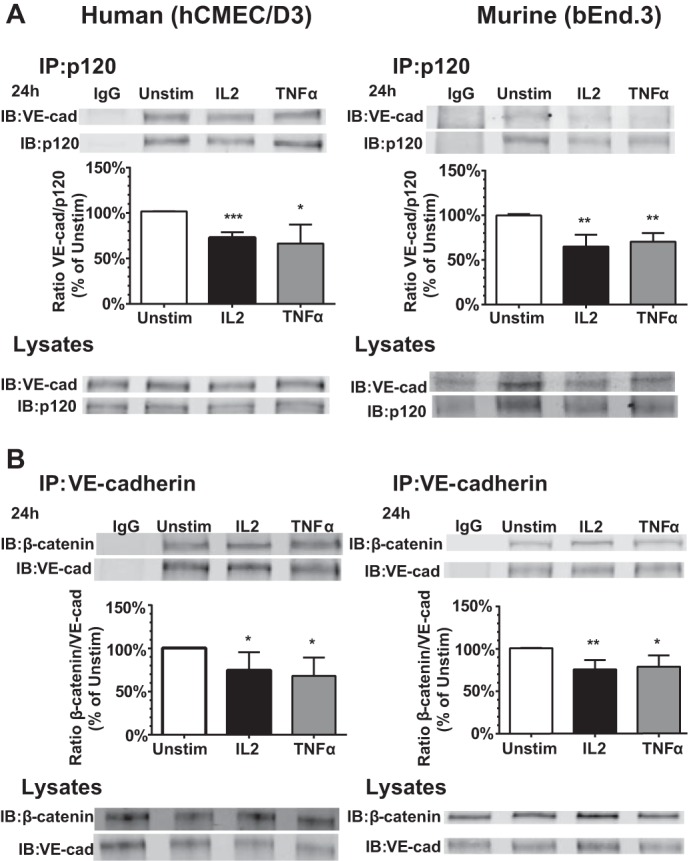
IL2-induced dissociation of the VE-cadherin·p120 and VE-cadherin·β-catenin complexes in human and murine BMECs. A, analysis of the VE-cadherin·p120 complex. Human (hCMEC/D3) and murine (bEnd.3) endothelial cells were grown to confluence and left unstimulated (Unstim) or stimulated with 300 kilounits/ml species-specific IL2 or 30 ng/ml species-specific TNFα for 24 h. Protein complexes in cell lysates precipitated with anti-p120 (IP) or treated with control IgG were quantified by immunoblotting (IB) with protein-specific antibodies. Cell lysates serve as input controls prior to immunoprecipitation. Error bars represent mean ± S.D. of at least three independent experiments. Statistical significance was determined by Student's t test (*, p < 0.05; **, p < 0.005; ***, p < 0.0005). B, analysis of the VE-cadherin·β-catenin complex. Human (hCMEC/D3) and murine (bEnd.3) endothelial cells were grown to confluence and left unstimulated or stimulated with 300 kilounits/ml species-specific IL2 or 30 ng/ml species-specific TNFα for 24 h. Protein complexes in cell lysates precipitated with anti-VE-cadherin (IP) or treated with control IgG were quantified by immunoblotting (IB) with protein-specific antibodies. Cell lysates serve as input controls prior to immunoprecipitation. Error bars represent mean ± S.D. of four independent experiments. Statistical significance was determined by Student's t test (*, p < 0.05; **, p < 0.005). VE-cad, VE-cadherin.
IL2-induced Phosphorylation of VE-cadherin
We next analyzed the mechanism by which VE-cadherin and its associated adaptor proteins (p120 and β-catenin) are dissociated upon IL2 stimulation. Post-translational modifications, such as phosphorylation events, contribute to destabilization of the AJ complex and loss of endothelial barrier function in various endothelial cell subsets (14, 16, 17, 45). Indeed, IL2 evoked a significant increase in Tyr-685 phosphorylation of VE-cadherin in these cells (Fig. 8A). This process was time-dependent with maximal phosphorylation levels reached between 6 and 24 h. Consistent with more robust activation and increased permeability recorded in murine bEnd.3 cells, we noted higher VE-cadherin phosphorylation than in human hCMEC/D3 cells. Thus, IL2-induced phosphorylation of VE-cadherin leads to the destabilization of the AJ complex most likely through dissociation of VE-cadherin from its adaptor proteins as documented above (Fig. 7, A and B).
FIGURE 8.
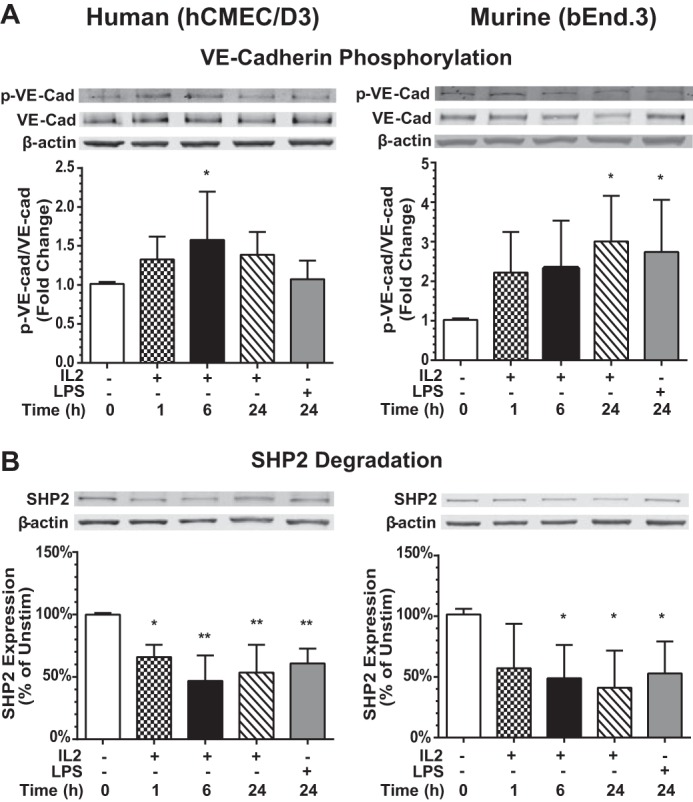
IL2-induced phosphorylation of VE-cadherin and degradation of SHP2 phosphatase in human and murine BMECs. A, analysis of VE-cadherin phosphorylation. Human (hCMEC/D3) and murine (bEnd.3) BMECs were left unstimulated or stimulated with 300 kilounits/ml species-specific IL2 or 1 μg/ml species-specific LPS for 24 h prior to isolation of cytosolic extracts. Samples were separated by SDS-PAGE and immunoblotted for the presence of total VE-cadherin (VE-cad) and Tyr-685 phosphorylated VE-cadherin (p-VE-cad). β-Actin is a loading control for cytosolic lysates. Quantification of signal is shown as a ratio of p-VE-cadherin to total VE-cadherin as -fold change compared with unstimulated control. Error bars represent mean ± S.D. of seven independent experiments. Statistical significance was determined using one-way ANOVA with Bonferroni correction for multiple comparisons for IL2 and an unpaired t test with Welch's correction for LPS (*, p < 0.05). B, analysis of SHP2 expression. Human (hCMEC/D3) and murine (bEnd.3) BMECs were left unstimulated (Unstim) or stimulated with 300 kilounits/ml species-specific IL2 or 1 μg/ml species-specific LPS for 24 h, and cytosolic extracts were isolated, separated by SDS-PAGE, and immunoblotted for the presence of SHP2. β-Actin is a loading control for cytosolic lysates. Normalized quantification of proteins is shown as a percent compared with the unstimulated control. Error bars represent mean ± S.D. of five independent experiments. Statistical significance was determined using one-way ANOVA with Bonferroni correction for multiple comparisons for IL2 and an unpaired t test with Welch's correction for LPS (*, p < 0.05; **, p < 0.005).
IL2-induced Loss of SHP2 Phosphatase
SHP2 phosphatase counteracts VE-cadherin phosphorylation, thereby contributing to the maintenance of lung microvascular endothelial barrier function (9, 15, 46). We found a significant decrease in expression of SHP2 phosphatase in IL2-stimulated BMECs that paralleled increased phosphorylation of VE-cadherin (Fig. 8B). SHP2 protein levels were reduced by 40–50% in both human and murine BMECs following IL2 stimulation. In contrast, observed transcript levels of SHP2 phosphatase remained at the same level or were increased.4 Thus, degradative loss of SHP2 phosphatase accompanies an increase in the phosphorylation state of AJ-associated proteins and destabilization of the complex.
Discussion
Taken together, our results decode the process through which IL2 activates BMECs and causes the loss of their barrier function. We found that human and murine BMECs constitutively express the intermediate affinity IL2 receptor comprising subunits β and γ (IL2Rβγ), whereas subunit α (CD25) is inducibly expressed following IL2 stimulation. We documented that IL2-induced signaling in BMECs involves transcription factor NFκB through (i) degradation of NFκB inhibitor IκBα, (ii) phosphorylation of NFκB p65 (RelA), and (iii) its nuclear translocation. Subsequently, we demonstrated that among NFκB-regulated genes in BMECs two pleiotropic mediators of inflammation, cytokine IL6 and chemokine MCP1 (CCL2), are expressed following IL2 stimulation. Both IL6 and MCP1 are known inducers of endothelial instability (19, 20). IL2-evoked NFκB activation in lymphocytes is well known (21) but to our knowledge has not been reported previously in non-immune cells.
Furthermore, we demonstrated that IL2 causes the loss of barrier function of BMECs. Their stimulation disrupts the interaction of AJ complex proteins by inducing phosphorylation of VE-cadherin along with a concomitant decline in SHP2 phosphatase. Thus, IL2 changes the phenotype of BMECs from basal physiologic quiescence to an activated state, contributing to an inflammatory milieu affecting brain neurovascular units that comprise the BBB (1). As schematically depicted in Fig. 9, IL2 interaction with its intermediate affinity receptor expressed on brain microvascular endothelial cells activates signaling mediated by the transcription factor NFκB, master regulator of inflammation. This feed-forward loop results in expression of proinflammatory mediators exemplified by IL6 and MCP1. In parallel, IL2 destabilizes adherens junctions through phosphorylation of VE-cadherin and dissociation of its complex with p120 and β-catenin. This process is enhanced by an IL2-induced decrease in SHP2 phosphatase expression and stress fiber formation. Increased microvascular permeability ensues, thereby explaining vascular leak syndrome and brain edema observed during IL2 cancer immunotherapy.
FIGURE 9.

Diagrammatic depiction of IL2-induced activation of brain microvascular endothelial cells. IL2 interacts with the constitutively expressed intermediate affinity IL2R complex (IL2Rβ and IL2Rγ), leading to degradation of IκBα and freeing NFκB to be translocated to the nucleus. Therein NFκB initiates transcription of its target genes, including two proinflammatory mediators, namely cytokine IL6 and chemokine MCP1, culminating in a feed-forward enhancement of the inflammatory process (left). In parallel, IL2 induces destabilization of adherens junctions through an increase in VE-cadherin (VE-cad) phosphorylation concomitant with a decrease in SHP2 phosphatase protein levels. An elevated VE-cadherin phosphorylation state results in diminished interaction between the constituents of the adherens junction complex, in particular p120 and β-catenin (β-cat). Furthermore, activation of the brain microvascular endothelial monolayer leads to increased F-actin stress fiber formation. Loss of adherens junction interaction between adjoining endothelial cells increases monolayer permeability (right). Thus, IL2 interaction with its cognate receptor on BMECs leads to a shift from a quiescent to an “activated” phenotype of BMECs and an increase in endothelial monolayer permeability.
As was initially reported (3), neuropsychiatric effects of IL2 are both dose- and time -dependent during IL2 cancer immunotherapy. The latency period in development of IL2 neurotoxicity points to a cumulative effect and/or increased availability of the inducible IL2Rα subunit (CD25). Therefore, in an effort to overcome this receptor limitation on naïve BMECs, we utilized a much higher agonist concentration range of IL2 in BMEC-based experiments.
Consistent with our study, expression of the IL2 receptor complex has been previously documented in pulmonary endothelial cells studied in a lung edema model (29, 41). Signaling through the IL2 receptor complex expressed on BMECs induces activation of the NFκB pathway similarly to the previously reported action of IL15, another member of the IL2 family. IL15 utilizes two subunits of IL2 receptor, IL2Rβ and IL2Rγ, in addition to the IL15-binding α subunit (31).
The structural and functional integrity of brain microvascular endothelium is of paramount importance for maintaining the physiologic function of the BBB (1, 47). IL2 stimulation of BMECs caused significant and consistent increases in endothelial monolayer permeability. VE-cadherin, the transmembrane protein responsible for homotypic interactions between adjacent endothelial cells, is stabilized by a group of cytoplasmic adaptor proteins, including p120 and β-catenin. We established that IL2 stimulation of BMECs disrupts this interaction of AJ proteins (see Fig. 7). Consistent with our study, VE-cadherin distribution in pulmonary microvascular endothelial cells was altered by blood serum obtained from patients treated with IL2 (41). Although our investigation was focused on AJs, it is important to note that tight junctions are also involved in restricting paracellular permeability in endothelial cells. It is likely that activation of BMECs also influences the barrier function of tight junctions (48).
Our study offers a deeper understanding of the mechanism of IL2-induced brain edema, which along with lung edema limits the effectiveness of IL2 cancer immunotherapy. This IL2-evoked mechanism includes Tyr-685 phosphorylation of VE-cadherin in BMECs (Fig. 8A) that is rapid and persists up to 24 h poststimulation, coinciding with the loss of AJs at endothelial cell borders. It is noteworthy that other VE-cadherin phosphorylation sites have been described in regulating AJ stability, and they contribute to destabilization of the AJ complex in response to agonists other than IL2 (9, 16, 17, 49). Thus, more than one phosphorylation site within VE-cadherin, along with other IL2-induced signaling events, may contribute to increased endothelial permeability.
We also discovered that IL2 stimulation resulted in the loss of SHP2 protein in BMECs (Fig. 8B). SHP2 is one of several phosphatases responsible for VE-cadherin dephosphorylation linked to maintenance of endothelial monolayer barrier function (9, 15, 46). The loss of SHP2 protein following IL2 stimulation enhances the phosphorylation status of VE-cadherin and/or its adaptor proteins and exacerbates the destabilization of AJs. Modification of protein-tyrosine phosphatases, including SHP2, as opposed to kinases has been proposed as the principal driver in modulating VE-cadherin phosphorylation levels (9).
In summary, our study provides new evidence that IL2 activates BMECs and leads to deterioration of their barrier function (see Fig. 9). A better understanding of IL2 action on brain microvascular endothelium, the cardinal component of the brain's neurovascular unit, may aid in the development of new measures to protect the central nervous system against IL2-induced VLS complicating high dose IL2 cancer immunotherapy and shed light on the role of IL2 in autoimmune inflammation associated with several brain diseases.
Author Contributions
L. S. W. designed and performed the experiments, analyzed the data, and prepared the manuscript. J. H. conceived the study and revised the manuscript. Both authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Professor Babette Weksler (Weil-Cornell Medical College, New York, NY) and Professor Pierre-Olivier Couraud and Dr. Nacho Romero (Institute Cochin, INSERM, Paris, France) for generously providing human BMEC line hCMEC/D3 and for helpful comments concerning the manuscript. We also thank Alyssa Hasty, Roger Colbran, David G. Harrison, Luc Van Kaer, and Ruth Ann Veach for valuable and constructive comments during the course of this study. Additionally, we thank Jozef Zienkiewicz for insightful discussions and suggestions throughout the course of experimentation and preparation of this manuscript. Fluorescence microscopy and analysis were performed in part through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by National Institutes of Health Grants CA68485, DK20593, DK58404, DK59637, and EY08126). Flow cytometry experiments were performed in the Vanderbilt Medical Center (VMC) Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (National Institutes of Health Grant P30 CA68485) and the Vanderbilt Digestive Disease Research Center (National Institutes of Health Grant DK058404).
This work was supported by United States Public Health Service National Institutes of Health Grants HL069452 and HL085833 (to J. H.), National Institutes of Health Ruth Kirschstein Institutional National Service Award T32 HL069765 (to L. S. W.), the Vanderbilt Immunotherapy Program, the Vanderbilt University Medical School Department of Medicine, and Vanderbilt Clinical and Translational Science Award UL1TR000445. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
L. S. Wylezinski and J. Hawiger, unpublished data.
- BBB
- blood brain barrier
- VE-cadherin
- vascular endothelial cadherin
- BMEC
- brain microvascular endothelial cell
- VLS
- vascular leak syndrome
- NFκB
- nuclear factor κB
- MCP1
- macrophage chemoattractant protein 1
- IL2R
- IL2 receptor
- AJ
- adherens junction
- p120
- p120-catenin
- SHP2
- Src homology 2 domain-containing protein-tyrosine phosphatase 2
- HI-FBS
- heat-inactivated fetal bovine serum
- MEF
- mouse embryonic fibroblast
- qPCR
- quantitative real time PCR
- vWF
- von Willebrand factor
- PECAM1
- platelet endothelial cell adhesion molecule 1
- ANOVA
- analysis of variance.
References
- 1. Abbott N. J., Rönnbäck L., and Hansson E. (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53 [DOI] [PubMed] [Google Scholar]
- 2. Ridder D. A., Lang M. F., Salinin S., Röderer J. P., Struss M., Maser-Gluth C., and Schwaninger M. (2011) TAK1 in brain endothelial cells mediates fever and lethargy. J. Exp. Med. 208, 2615–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Denicoff K. D., Rubinow D. R., Papa M. Z., Simpson C., Seipp C. A., Lotze M. T., Chang A. E., Rosenstein D., and Rosenberg S. A. (1987) The neuropsychiatric effects of treatment with interleukin-2 and lymphokine-activated killer cells. Ann. Intern. Med. 107, 293–300 [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg S. A. (2014) IL-2: the first effective immunotherapy for human cancer. J. Immunol. 192, 5451–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saris S. C., Patronas N. J., Rosenberg S. A., Alexander J. T., Frank J., Schwartzentruber D. J., Rubin J. T., Barba D., and Oldfield E. H. (1989) The effect of intravenous interleukin-2 on brain water content. J. Neurosurg. 71, 169–174 [DOI] [PubMed] [Google Scholar]
- 6. Ichinose K., Arima K., Ushigusa T., Nishino A., Nakashima Y., Suzuki T., Horai Y., Nakajima H., Kawashiri S. Y., Iwamoto N., Tamai M., Nakamura H., Origuchi T., Motomura M., and Kawakami A. (2015) Distinguishing the cerebrospinal fluid cytokine profile in neuropsychiatric systemic lupus erythematosus from other autoimmune neurological diseases. Clin. Immunol. 157, 114–120 [DOI] [PubMed] [Google Scholar]
- 7. Martins T. B., Rose J. W., Jaskowski T. D., Wilson A. R., Husebye D., Seraj H. S., and Hill H. R. (2011) Analysis of proinflammatory and anti-inflammatory cytokine serum concentrations in patients with multiple sclerosis by using a multiplexed immunoassay. Am. J. Clin. Pathol. 136, 696–704 [DOI] [PubMed] [Google Scholar]
- 8. Gallo P., Pagni S., Piccinno M. G., Giometto B., Argentiero V., Chiusole M., Bozza F., and Tavolato B. (1992) On the role of interleukin-2 (IL-2) in multiple sclerosis (MS). IL-2-mediated endothelial cell activation. Ital. J. Neurol. Sci. 13, 65–68 [PubMed] [Google Scholar]
- 9. Hatanaka K., Lanahan A. A., Murakami M., and Simons M. (2012) Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS One 7, e37600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lampugnani M. G., and Dejana E. (2007) Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 120, Suppl. 2, S1–S6 [DOI] [PubMed] [Google Scholar]
- 11. Hawiger J., Veach R. A., and Zienkiewicz J. (2015) New paradigms in sepsis: from prevention to protection of failing microcirculation. J. Thromb. Haemost. 13, 1743–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abu Taha A., and Schnittler H. J. (2014) Dynamics between actin and the VE-cadherin/catenin complex: novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adh. Migr. 8, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao K., Garner J., Buckley K. M., Vincent P. A., Chiasson C. M., Dejana E., Faundez V., and Kowalczyk A. P. (2005) p120-catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell 16, 5141–5151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Potter M. D., Barbero S., and Cheresh D. A. (2005) Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and β-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 280, 31906–31912 [DOI] [PubMed] [Google Scholar]
- 15. Ukropec J. A., Hollinger M. K., Salva S. M., and Woolkalis M. J. (2000) SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem. 275, 5983–5986 [DOI] [PubMed] [Google Scholar]
- 16. Orsenigo F., Giampietro C., Ferrari A., Corada M., Galaup A., Sigismund S., Ristagno G., Maddaluno L., Koh G. Y., Franco D., Kurtcuoglu V., Poulikakos D., Baluk P., McDonald D., Grazia Lampugnani M., et al. (2012) Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 3, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A. F., and Vestweber D. (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223–230 [DOI] [PubMed] [Google Scholar]
- 18. Greenwood J., Heasman S. J., Alvarez J. I., Prat A., Lyck R., and Engelhardt B. (2011) Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 37, 24–39 [DOI] [PubMed] [Google Scholar]
- 19. Kempe S., Kestler H., Lasar A., and Wirth T. (2005) NF-κB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 33, 5308–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Libermann T. A., and Baltimore D. (1990) Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol. Cell. Biol. 10, 2327–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arima N., Kuziel W. A., Grdina T. A., and Greene W. C. (1992) IL-2-induced signal transduction involves the activation of nuclear NF-κB expression. J. Immunol. 149, 83–91 [PubMed] [Google Scholar]
- 22. Weksler B. B., Subileau E. A., Perrière N., Charneau P., Holloway K., Leveque M., Tricoire-Leignel H., Nicotra A., Bourdoulous S., Turowski P., Male D. K., Roux F., Greenwood J., Romero I. A., and Couraud P. O. (2005) Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 19, 1872–1874 [DOI] [PubMed] [Google Scholar]
- 23. Montesano R., Pepper M. S., Möhle-Steinlein U., Risau W., Wagner E. F., and Orci L. (1990) Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell 62, 435–445 [DOI] [PubMed] [Google Scholar]
- 24. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 25. Qiao H., Liu Y., Veach R. A., Wylezinski L., and Hawiger J. (2014) The adaptor CRADD/RAIDD controls activation of endothelial cells by proinflammatory stimuli. J. Biol. Chem. 289, 21973–21983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Middleton J., Americh L., Gayon R., Julien D., Mansat M., Mansat P., Anract P., Cantagrel A., Cattan P., Reimund J. M., Aguilar L., Amalric F., and Girard J. P. (2005) A comparative study of endothelial cell markers expressed in chronically inflamed human tissues: MECA-79, Duffy antigen receptor for chemokines, von Willebrand factor, CD31, CD34, CD105 and CD146. J. Pathol. 206, 260–268 [DOI] [PubMed] [Google Scholar]
- 27. Sheibani N., and Frazier W. A. (1998) Down-regulation of platelet endothelial cell adhesion molecule-1 results in thrombospondin-1 expression and concerted regulation of endothelial cell phenotype. Mol. Biol. Cell 9, 701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Minami Y., Kono T., Miyazaki T., and Taniguchi T. (1993) The IL-2 receptor complex: its structure, function, and target genes. Annu. Rev. Immunol. 11, 245–268 [DOI] [PubMed] [Google Scholar]
- 29. Krieg C., Létourneau S., Pantaleo G., and Boyman O. (2010) Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 107, 11906–11911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruiz-Medina B. E., Ross J. A., and Kirken R. A. (2015) Interleukin-2 receptor β Thr-450 phosphorylation is a positive regulator for receptor complex stability and activation of signaling molecules. J. Biol. Chem. 290, 20972–20983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stone K. P., Kastin A. J., and Pan W. (2011) NFκB is an unexpected major mediator of interleukin-15 signaling in cerebral endothelia. Cell. Physiol. Biochem. 28, 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cordle S. R., Donald R., Read M. A., and Hawiger J. (1993) Lipopolysaccharide induces phosphorylation of MAD3 and activation of c-Rel and related NF-κB proteins in human monocytic THP-1 cells. J. Biol. Chem. 268, 11803–11810 [PubMed] [Google Scholar]
- 33. Donald R., Ballard D. W., and Hawiger J. (1995) Proteolytic processing of NF-κB/IκB in human monocytes. ATP-dependent induction by pro-inflammatory mediators. J. Biol. Chem. 270, 9–12 [DOI] [PubMed] [Google Scholar]
- 34. Cooper J. T., Stroka D. M., Brostjan C., Palmetshofer A., Bach F. H., and Ferran C. (1996) A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J. Biol. Chem. 271, 18068–18073 [DOI] [PubMed] [Google Scholar]
- 35. Wang D., Westerheide S. D., Hanson J. L., and Baldwin A. S. Jr. (2000) Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 275, 32592–32597 [DOI] [PubMed] [Google Scholar]
- 36. Tieu B. C., Lee C., Sun H., Lejeune W., Recinos A. 3rd, Ju X., Spratt H., Guo D. C., Milewicz D., Tilton R. G., and Brasier A. R. (2009) An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Investig. 119, 3637–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Desai T. R., Leeper N. J., Hynes K. L., and Gewertz B. L. (2002) Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 104, 118–123 [DOI] [PubMed] [Google Scholar]
- 38. Jee Y., Yoon W. K., Okura Y., Tanuma N., and Matsumoto Y. (2002) Upregulation of monocyte chemotactic protein-1 and CC chemokine receptor 2 in the central nervous system is closely associated with relapse of autoimmune encephalomyelitis in Lewis rats. J. Neuroimmunol. 128, 49–57 [DOI] [PubMed] [Google Scholar]
- 39. Stamatovic S. M., Keep R. F., Kunkel S. L., and Andjelkovic A. V. (2003) Potential role of MCP-1 in endothelial cell tight junction 'opening': signaling via Rho and Rho kinase. J. Cell Sci. 116, 4615–4628 [DOI] [PubMed] [Google Scholar]
- 40. Brasier A. R. (2010) The nuclear factor-κB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 86, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim D. W., Zloza A., Broucek J., Schenkel J. M., Ruby C., Samaha G., and Kaufman H. L. (2014) Interleukin-2 alters distribution of CD144 (VE-cadherin) in endothelial cells. J. Transl. Med. 12, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rajput C., Kini V., Smith M., Yazbeck P., Chavez A., Schmidt T., Zhang W., Knezevic N., Komarova Y., and Mehta D. (2013) Neural Wiskott-Aldrich syndrome protein (N-WASP)-mediated p120-catenin interaction with Arp2-actin complex stabilizes endothelial adherens junctions. J. Biol. Chem. 288, 4241–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dignat-George F., and Sampol J. (2000) Circulating endothelial cells in vascular disorders: new insights into an old concept. Eur. J. Haematol. 65, 215–220 [DOI] [PubMed] [Google Scholar]
- 44. Westlin W. F., and Gimbrone M. A. Jr. (1993) Neutrophil-mediated damage to human vascular endothelium. Role of cytokine activation. Am. J. Pathol. 142, 117–128 [PMC free article] [PubMed] [Google Scholar]
- 45. Chang S. F., Chen L. J., Lee P. L., Lee D. Y., Chien S., and Chiu J. J. (2014) Different modes of endothelial-smooth muscle cell interaction elicit differential β-catenin phosphorylations and endothelial functions. Proc. Natl. Acad. Sci. U.S.A. 111, 1855–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grinnell K. L., Casserly B., and Harrington E. O. (2010) Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L361–L370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lippmann E. S., Azarin S. M., Kay J. E., Nessler R. A., Wilson H. K., Al-Ahmad A., Palecek S. P., and Shusta E. V. (2012) Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., and Dejana E. (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 49. Adam A. P., Sharenko A. L., Pumiglia K., and Vincent P. A. (2010) Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J. Biol. Chem. 285, 7045–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]