

Abstract
The RpoE σ factor is essential for the viability of Escherichia coli. RpoE regulates extracytoplasmic functions including lipopolysaccharide (LPS) translocation and some of its non-stoichiometric modifications. Transcription of the rpoE gene is positively autoregulated by EσE and by unknown mechanisms that control the expression of its distally located promoter(s). Mapping of 5′ ends of rpoE mRNA identified five new transcriptional initiation sites (P1 to P5) located distal to EσE-regulated promoter. These promoters are activated in response to unique signals. Of these P2, P3, and P4 defined major promoters, recognized by RpoN, RpoD, and RpoS σ factors, respectively. Isolation of trans-acting factors, in vitro transcriptional and gel retardation assays revealed that the RpoN-recognized P2 promoter is positively regulated by a QseE/F two-component system and NtrC activator, whereas the RpoD-regulated P3 promoter is positively regulated by a Rcs system in response to defects in LPS core biosynthesis, overproduction of certain lipoproteins, and the global regulator CRP. Strains synthesizing Kdo2-LA LPS caused up to 7-fold increase in the rpoEP3 activity, which was abrogated in Δ(waaC rcsB). Overexpression of a novel 73-nucleotide sRNA rirA (RfaH interacting RNA) generated by the processing of 5′ UTR of the waaQ mRNA induces the rpoEP3 promoter activity concomitant with a decrease in LPS content and defects in the O-antigen incorporation. In the presence of RNA polymerase, RirA binds LPS regulator RfaH known to prevent premature transcriptional termination of waaQ and rfb operons. RirA in excess could titrate out RfaH causing LPS defects and the activation of rpoE transcription.
Keywords: bacterial transcription, glycosyltransferase, lipopolysaccharide (LPS), RNA polymerase, transcription termination, LapB, RirA sRNA, RpoE, RpoS, heptosyltransferase
Introduction
The cell envelope of Gram-negative bacteria, including Escherichia coli, contains two distinct membranes, an inner (IM)3 and an outer (OM) membrane separated by the periplasm, a hydrophilic compartment that includes a layer of peptidoglycan. The OM is an asymmetric lipid bilayer with phospholipids forming the inner leaflet and LPS forming the outer leaflet. LPSs are highly heterogeneous in composition. However, they share a common architecture composed of a membrane-anchored phosphorylated and acylated β(1→6)-linked GlcN disaccharide, termed lipid A, to which a carbohydrate moiety of varying size is attached (1, 2).
During the analyses of signals that induce the RpoE-dependent extracytoplasmic stress response, we showed that strains synthesizing heptoseless LPS due to the absence of either GmhD (RfaD/HtrM) or WaaC exhibited a constitutive induction of RpoE (3–6). Such strains also display constitutive synthesis of exopolysaccharide that is regulated by the Rcs two-component system (5, 7, 8). Subsequently, we showed that strains synthesizing the minimal LPS structure composed of either Kdo2-lipid IVA or only free lipid IVA exhibited hyper-elevated levels of the RpoE activity (6). The activity of RpoE is highly induced when the LPS assembly is severely compromised or when there is an imbalance between LPS and phospholipids (9). Lack of many LPS core biosynthetic genes also leads to defects in survival at high temperatures (5, 6, 10).
The RpoE synthesis is positively regulated at the transcriptional level and negatively at the post-transcriptional level by the IM-anchored anti-σ factor RseA and the periplasmic sensor RseB (Fig. 1) (3, 11–16). There is an intricate link between the LPS assembly, its non-stoichiometric modifications, and the RpoE-mediated control exerted via its regulon members (17, 18). Elevated levels of RpoE with intact core biosynthetic enzymes lead to global alterations in the LPS composition. Under such conditions, LPS is primarily composed of glycoforms with a third Kdo, phosphoethanolamine (P-EtN) on the second Kdo and rhamnose on the third Kdo (Fig. 2) (19). The synthesis of such LPS glycoforms is controlled by the RpoE-regulated RybB and EptB (19). RpoE also positively regulates transcription of micA and slrA (micL) sRNAs (9, 20, 21). MicA negatively regulates the PhoP expression by direct translational inhibition of the phoP mRNA (22), thus further linking the PhoP/Q two-component system, known to regulate LPS non-stoichiometric alterations, with the RpoE control. The LPS synthesis is further regulated by the transcriptional factor RfaH. RfaH strongly inhibits the Rho-dependent termination of waaQ and rfb operons, which contain ops (operon polarity suppression) sites in their 5′ UTR and also binds to the ribosome to activate translation (23–25). Thus, the synthesis of LPS, like many key cellular components, is regulated by various transcriptional factors including two-component systems and σ factors.
FIGURE 1.

Transcriptional regulation of the rpoE gene. Schematic drawing of previously identified transcriptional start sites of the rpoE gene (3, 16) (A) and those identified in this work (B). Major regulators and factors controlling the rpoE transcription are indicated.
FIGURE 2.

Proposed LPS structure of major glycoforms observed in E. coli K-12. Schematic drawing of LPS glycoforms I (A) and VII (B) with the indicated genes whose products are involved in the LPS core biosynthesis and non-stoichiometric modifications.
Interestingly, some two-component systems that regulate LPS modifications, like BasS/R and envelope stress responsive Rcs phosphorelay systems, are also linked with quorum sensing Qse systems by phosphorylation of the response regulator by a non-cognate kinase (26–28). Similarly, at the transcriptional level, the synthesis of different σ factors is interlinked due to the presence of multiple promoters adding additional layers of regulation. For example, the gene encoding the housekeeping σ factor RpoD is transcribed from two promoters and one of them is the RpoH-regulated heat shock promoter (29). Similarly, the rpoH gene is transcribed from multiple promoters whose transcription is regulated by different σ factors (3, 30). The presence of multiple promoters endows them the ability to respond to different signals. Such a mode of complex transcriptional regulation allows linking of different stress responsive pathways.
Despite numerous strides in the post-transcriptional regulation of the rpoE gene, its transcriptional regulation has not been fully addresses. In E. coli, two promoters were initially identified, one distally located with unknown regulation and a second promoter recognized by σE itself (Fig. 1A) (3, 16). However, transcription from the upstream promoter(s) is sustained under several stress and non-stress conditions (3). The RpoE activity is also induced by stresses like exposure to high osmolarity, cold shock conditions, and entry into the stationary phase (31, 32). Some of these factors, like the absence of RpoS, do not alter the LPS composition (19) and hence how these diverse signals are sensed is unknown. Thus, several single-copy chromosomal rpoEP-lacZ promoter fusions, devoid of DNA sequence covering the promoter recognized by EσE, were constructed to identify trans-acting factors that regulate rpoE transcription, followed by mapping of transcription start sites and verification by in vitro run-off assays. This analysis revealed five new transcription start sites that are utilized by different regulatory factors in response to distinct signals (Fig. 1B).
Results
LPS Defects and Signals Regulating RpoS, RpoN, and CRP Alter the Transcriptional Activity of the rpoE Gene
As the regulation of the rpoE promoter region distal to its EσE-transcribed promoter is unknown, a single-copy lacZ promoter fusion was constructed that contains the 529-bp region upstream of this autoregulated promoter. For simplicity, such a fusion derivative is referred to as rpoEP-lacZ. Strains with this promoter fusion (MC4100 derivative SR4245 and SR7917-based on BW25113) served as a host for saturated random transposon mutagenesis to identify mutations that alter the activity of this transcriptional region. The rationale of using two strains rules out any strain specificity. It is worth mentioning that the host strain BW25113 is also the parental strain for the Keio collection of gene knockouts (33) and hence its usage. Mapping and sequencing of Tn10 mutations conferring a Lac up phenotype revealed that the majority of them had insertion in LPS biosynthetic/LPS regulatory genes and a few in other loci (supplemental Table S1). A similar spectrum of mutants was obtained in SR4245 and SR7917. Among these, 12 Tn10 insertions were mapped to the waaC gene, three to the waaF gene, and one each was located in rfaH and waaQ genes. Furthermore, two Tn10 insertions were mapped to the rssB gene (both with Tn10 insertion at nucleotide position 240), one Tn10 insertion in the arcA gene (nucleotide position 72 within the coding region), and one at nucleotide position −78 upstream of the initiation codon of the ecfL gene. Mapping of Lac down mutations identified one mutation each in glnG (ntrC), glnA, and cya genes, indicating that their products could play positive regulatory roles. The introduction of defined non-polar deletion of various genes (33) confirmed the above results. This analysis identified additional genes that either down-regulate or induce transcription of rpoEP-lacZ. Thus, a ΔarcB mutation led to an increase in transcription, whereas ΔrpoS and Δcrp caused a decrease in the activity (Tables 1 and 2). All such deletion derivatives carrying the rpoEP-lacZ fusion in SR7917 were used for measurement of the β-galactosidase activity revealing involvement of several regulatory pathways (Tables 1 and 2).
TABLE 1.
Impact of non-polar deletion mutations or overexpression of various regulators that alter the activity of the rpoEP1-P5-lacZ transcriptional fusion
Values correspond to average of four independent samples from cultures grown in LB or in M9 minimal medium at A600 0.2 to 0.4.
| Function | Effect | Specificity | |
|---|---|---|---|
| Genotype | |||
| Wild-type SR7917 (rpoEP1-P5-lacZ) | 393 ± 26 | ||
| ΔwaaC | Heptosyltransferase I | 2630 ± 168 | rpoEP3 in Rcs-dependent manner, also induces the EσE-regulated rpoEP6 promoter that is only partially dependent on the Rcs system |
| ΔwaaF | Heptosyltransferase II | 1052 ± 65 | rpoEP3 |
| ΔwaaG | LPS glucosyltransferase I | 837 ± 46 | rpoEP3 |
| ΔwaaP | LPS core HepI kinase | 894 ± 51 | rpoEP3 |
| ΔrfaH | Transcriptional anti-terminator | 1505 ± 93 | rpoEP3 |
| Δcya | Adenylate cyclase | 230 ± 16 | rpoEP3 |
| Δcrp | Transcriptional dual regulator | 215 ± 18 | rpoEP3 |
| Δ(ecfLM) | RpoE regulon member, DedA family member | 870 ± 85 | rpoEP3 |
| ΔrssB | Regulator of RpoS | 531 ± 37 | rpoEP4 |
| ΔarcA | Transcriptional dual regulator | 831 ± 65 | rpoEP4 |
| ΔarcB | Sensory histidine kinase | 1120 ± 90 | rpoEP4 |
| ΔrpoS | RpoS σ factor | 193 ± 14 | rpoEP4 |
| ΔrpoN | RpoN σ factor | 302 ± 25 | rpoEP2, lack of RpoN in ΔwaaC, reduces rpoEP2 induction that is QseF-dependent |
| Δ(rpoN waaC) | 1290 ± 90 | ||
| ΔntrC | Transcriptional dual regulator | 312 ± 27 | rpoEP2 |
| Multicopy expression that alters the transcription | |||
| qseG | Part of qseE/F operon | 6920 ± 335 | rpoEP2 |
| yhdV and six other genes encoding lipoproteins | Lipoprotein | 1063 ± 68 | rpoEP3 (Rcs-dependent) |
| rirA | sRNA | 786 ± 52 | rpoEP3 |
| pcnB | Poly(A) polymerase I | 540 ± 38 | rpoEP4 |
| fliZ | DNA-binding transcriptional regulator | 249 ± 21 | rpoEP4 |
TABLE 2.
Supplementation of growth medium that alter the activity of the rpoEP1-P5-lacZ transcriptional fusion using strain SR7917
Cultures were grown in either LB medium adjusted to an A600 0.05, followed by challenge with specific reagent as indicated, or in nitrogen-limiting minimal medium containing 3 mm ammonium chloride as the nitrogen source. Values correspond to an average of four independent samples after a 30–60-min shift.
| Supplementation of growth medium that alter the transcription activity | ||
|---|---|---|
| Sucrose (0.25 m) | 1410 ± 110 | rpoEP4 |
| Glucose (0.5%) | 219 ± 15 | rpoEP3 |
| 25 mm Ammonium metavanadate | 1540 ± 90 | rpoEP2/P3 |
| 0.25 μg of Polymyxin B (A600 0.05) | 1503 ± 77 | rpoEP3 |
| 0.25 μg of Polymyxin B (A600 0.4) | 410 ± 29 | No induction |
| 3 mm Ammonium chloride | 575 ± 37 | rpoEP2 |
In a complementary approach, pools of plasmids with all ORFs of E. coli genome under control of the tightly regulated IPTG-inducible promoter (34) were introduced in SR4245 and SR7917 and screened for a Lac up or down phenotype in growth medium supplemented with 50 μm IPTG. This collection of cloned genes expresses only individual ORFs. Thus, one might miss genes whose products require the expression of whole operons or regulatory sRNAs. To overcome such limitations, a previously described complete genomic library in a medium-copy p15A vector (9, 35) was introduced to identify clones that exhibit a Lac up or down phenotype. These multicopy approaches identified qseG and several genes encoding lipoproteins (csgG, pgaB, spr, yhdV, yceB, yddW, and yghB), which, when overexpressed, increase the rpoEP-lacZ activity (Tables 1 and 2). Furthermore, DNA subcloning identified a novel sRNA located between waaQ and waaA ORFs, whose overexpression also induces the rpoEP-lacZ activity. Detailed analysis revealed that this sRNA is generated by the processing of 5′ UTR of the waaQ mRNA (see below). Additionally, overexpression of the pcnB gene encoding poly(A) polymerase I (PAP I) (36) also increases the rpoEP-lacZ activity (Tables 1 and 2). Among genes, whose induction repressed the rpoEP-lacZ activity, the fliZ gene was identified whose product could act as a negative regulator.
The quantification of the β-galactosidase activity of strains carrying the rpoEP-lacZ fusion under different growth conditions revealed that it is induced in the stationary growth phase, exposure to high osmolarity, limiting nitrogen conditions, upon challenge with either cationic antimicrobial peptide polymyxin B or ammonium metavanadate and repressed in glucose-supplemented medium (catabolite repression) (Tables 1 and 2). Ammonium metavanadate is a nonspecific phosphatase inhibitor (37) and hence can activate the expression of several two-component systems. The addition of ammonium metavanadate induces lipid A modifications (37, 38) and the PhoB/R-dependent incorporation of glucuronic acid (GlcUA) by WaaH in the inner core of LPS (39). Concerning the effect of polymyxin B, the rpoE transcriptional induction was observed when challenged at A600 of 0.05, but not when bacterial cultures were treated at or above an A600 of 0.4. Overall, these results revealed that transcription of the rpoE distal promoter region responds to diverse signals generated by defects in LPS, overproduction of certain lipoproteins, activators of RpoN and factors that influence the RpoS stability/activity. Such distinct signals could be sensed via multiple pathways. This could require recruitment of different σ factors, two-component systems, and hence potentially the presence of multiple promoters. As subsequent analysis indeed confirmed the presence of multiple promoters, multicopy libraries were once again introduced to identify genes whose overexpression modulates activity of specific rpoE promoters. Such additional approaches confirmed most of the above results and identified a specific mechanism of regulation of individual promoters recruiting different σ factors in each case (Tables 1 and 2).
Identification of 5′ Ends of the rpoE mRNA
Previous studies identified two promoters, out of them the initiation from the second promoter required EσE, whereas in vitro neither Eσ70 nor EσE could initiate transcription of the upstream promoter (Fig. 1) (3, 16). Thus, the transcriptional initiation from the upstream promoter region was examined using 5′ rapid amplification of cDNA ends. In parallel, total RNA extracted from the strain overexpressing the qseG gene was also used for mapping of transcription initiation site(s). This overexpressing strain was chosen, because the qseG was identified as a multicopy inducer of the rpoE transcription. These analyses revealed the utilization of five major new transcriptional initiation sites designated P1 (−381), P2 (−327), P3 (−327/−326), P4 (−218), and P5 (−153) (Fig. 3A). The frequency of usage of the P2 promoter (−327) was significantly pronounced when RNA was extracted from the strain overexpressing the qseG gene. However, when RNA from the ΔwaaC strain was used, both −327 and −326 initiation sites were identified in addition to the −218 site corresponding to the P4 promoter. Plasmids bearing the 5′ end of the P4 promoter were enriched when RNA was extracted from bacterial cultures harvested in the late stationary growth phase. Additional initiation sites located at −336, −321, and −319 were also identified (Fig. 3A). Of these, −321 and −319 could be secondary mRNA sites arising due to processing as the number of plasmid clones bearing these initiation sites was significantly reduced when mRNA was treated with calf intestinal phosphatase.
FIGURE 3.

Transcriptional regulation of different promoters of the rpoE gene. A, nucleotide sequence of the promoter region of the rpoE gene. Arrows in red indicate the position of transcription start sites. The shared TSS at −327 is utilized by RpoN and RpoD. The P4 start site represents the initiation site recognized by RpoS. The −10 and −35 elements of the RpoD (pink) and RpoS recognized promoter (green) and −12 and −24 elements of RpoN (blue) are indicated. The P6 initiation site corresponds to the EσE-regulated promoter. Nucleotides marked with boxes correspond to IHF, RcsB, NtrC, and CRP recognition sites. Three palindromic regions marked with inverted arrows correspond to UAS1, UAS2, and UAS3 representing QseF-binding sites required for the P2 promoter. Nucleotides marked with asterisks (*) correspond to putative processing sites. B, the alignment of −12 and −24 regions of the rpoEP2 promoter with well characterized RpoN-regulated promoters. C, the alignment of −10 and extended −10 elements of the rpoEP4 promoter with well known RpoS-regulated promoters.
Examination of the DNA sequence upstream of the P2 TSS showed that it contains at −12 (GC) and −24 (GG) motifs characteristic of RpoN recognition sites (Fig. 3A) and a very good match was observed with the promoter region of the RpoN-regulated genes (Fig. 3B). Transcription initiation sites −327 or −326 also define the P3 promoter. It is likely that the −326 site is generated by the processing of mRNA, because the overlapping rpoEP3 initiation site corresponding to the −327-nt position contains a high degree of similarity to consensus −10 and −35 elements of housekeeping promoters and the extended −10 promoter. Of interest is the presence of conserved −7T and −11A nucleotide residues that are critical in the −10 promoter element (40). Examination of the DNA sequence upstream of the −327 site also revealed the presence of consensus regions for binding to CRP and RcsB activators for the P3 promoter (Fig. 3A). The initiation site corresponding to the rpoEP1 promoter located at nucleotide position −381 overlaps with the promoter region of the divergently transcribed nadB gene. The DNA sequence in the −10 and −35 regions upstream of the initiation site corresponding to the P4 promoter suggests consensus sequences that could be recognized by either Eσ70 or EσS. The frequency of plasmids containing the 5′ end of the P5 promoter was the lowest of all. Next, DNA sequences covering five individual promoters were cloned in promoter probe vectors and transferred in a single-copy on the chromosome for further analyses (supplemental Table S1). Among these five promoters, rpoEP1 and rpoEP5 were not followed as no specific growth conditions/regulatory factors could be identified that alter their activity. Taken together, these data reveal the existence of multiple functional promoters that regulate transcription of the rpoE gene.
In Vitro Transcriptional Run-off Assays
Regulation of the P4 Promoter Is RpoS-dependent
As the 5′ rapid amplification of cDNA ends analysis revealed the existence of multiple transcriptional initiation sites that could utilize different σ factors, in vitro run-off assays were performed to validate these results/predictions. One template contained 45 bp downstream of the −218 transcription initiation site corresponding to the P4 promoter. Examination of its −10 and −35 regions suggests the similarity with promoters recognized by Eσ70 and EσS. Thus, run-off assays were undertaken using the RNAP core supplemented with either σ70 or σS (Fig. 4A, lanes 2 and 3). Only EσS was able to initiate transcription from the P4 promoter and gave the transcript size as expected of 45 nt (Fig. 4A). Supplementation of Eσ70 with RcsB did not result in any transcription initiation from this promoter (Fig. 4A, lane 4). The −10 and extended −10 regions of the P4 promoter indeed contain a high degree of similarity to the counterpart promoter regions of well known EσS-transcribed genes like dps (Fig. 3C). Consistent with these in vitro experiments with the rpoEP4 promoter revealing EσS-dependent transcriptional initiation, genetic data also support such results (see below) and allow us to conclude that this promoter is indeed regulated by RpoS.
FIGURE 4.
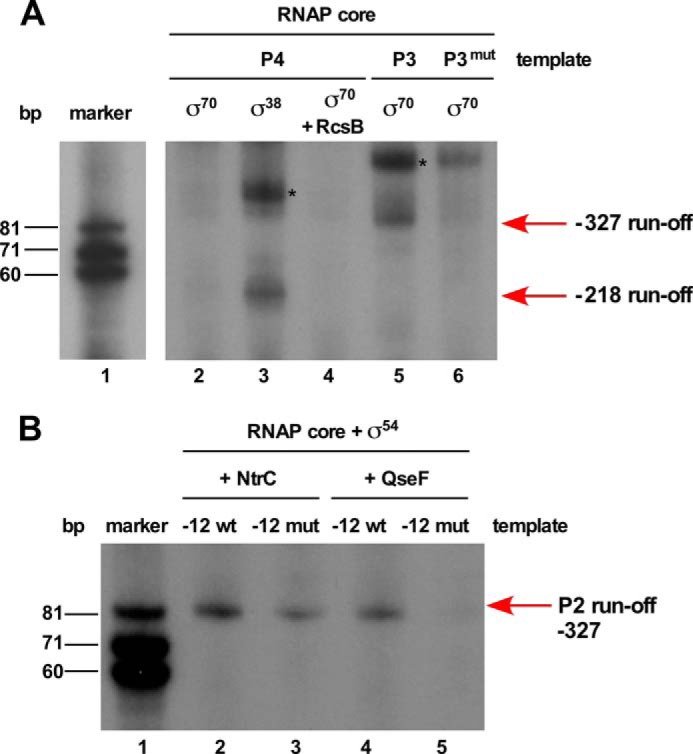
In vitro transcription run-off assays showing selective recruitment of different promoters of the rpoE gene with various forms of RNA polymerase. A, RpoS (σ38) and RpoD complexed with the RNAP core initiate transcription from −218 TSS and TSS at −327, respectively. Lane 1 corresponds to the size standard. For lanes 2–4, a DNA template of 105 bp was used. Lane 2 shows the incubation reaction with Eσ70, lane 3 the incubation with Eσ38 resulting in the synthesis of expected 45-nt product marked with the arrow. Lane 4 was incubated with Eσ70 in the presence of phosphorylated RcsB. Lanes 5 and 6 show RNA transcripts synthesized from DNA template of 170 bp. An expected size of 85-nt RNA product was observed when Eσ70 was incubated with the wild-type DNA template (lane 5), whereas only a weak signal was visible when Eσ70 was used with template with mutation at −7T(C) and −11A(G) in the −10 element (lane 6). Bands marked with an asterisk (*) symbol in lanes 3 and 5 indicate nonspecific end-to-end transcription reaction products. B, RpoN (σ54) complexed with the core RNAP in the presence of either NtrC or QseF can initiate transcription from the rpoEP2 promoter using the −327 TSS. Lane 1 corresponds to size standard, lanes 2 and 4 corresponds to reactions with the wild-type DNA template in the presence of either NtrC or QseF. Lanes 3 and 5 correspond to the RNA transcript produced with DNA template containing mutations at −12 (GC to AT) and −24 (GG to GA).
P2 and P3 Promoters are Regulated by RpoN and RpoD Using the Same Initiation Start Site
DNA sequence examination of the promoter region corresponding to the −327 TSS suggested that transcription initiation could be mediated by RNA polymerase containing either RpoN or housekeeping σ factor RpoD or subjected to a dual usage. EσN cannot transcribe on its own, because it requires ATP-dependent activators like NtrC belonging to the AAA family (41). NtrC is known to act as an enhancer-like protein for many RpoN-regulated promoters (41, 42). A consensus matching the RpoN-regulated promoter is located in the −12 and −24 regions and a recognition site for NtrC is also present (Fig. 3). Thus, in vitro run-off assays with reconstituted RNA polymerase, containing RpoN and either NtrC or QseF (GlrR) as activators, were performed. The rationale for using QseF is based on our results that showed that the QseE/F two-component system positively regulates the rpoEP2 promoter in response to overexpression of the qseG gene (see below). QseF, like NtrC, belongs to the family of AAA activators and contains a response regulator domain (42) with a key conserved aspartate residue and an RpoN-interaction domain including the GAFTGA motif (amino acid residues 211 to 216).
Thus, for in vitro run-off experiments, DNA template covering the P2 promoter region and 85 bp downstream of the initiation site located at −327 was used. In multiround transcription assays, the expected size of the transcript was observed when RpoN was supplemented with either NtrC or QseF (Fig. 4B, lanes 2 and 4). To validate recognition of the P2 promoter by RpoN, specific mutations in the −12 (GC to AT) and −24 (GG to GA) RpoN recognition sites were introduced in the template for in vitro run-off assays. Transcription reaction with the RNAP core containing RpoN + QseF using such a mutated DNA template resulted in a significant inhibition in the initiation of transcription, as compared with the same reaction with the wild-type DNA template (Fig. 4B, lane 5). RpoN + NtrC also recognized such a mutated template less efficiently as compared with the wild-type, although the reduction is not as severe as observed with the RpoN + QseF reaction (Fig. 4B, lane 3). These results thus confirm that RpoN recognizes the rpoEP2 promoter and QseF acts as the main activator/enhancer.
The initiation site corresponding to the −327 TSS has also a good similarity for the −10 and −35 promoter elements recognized by RpoD (σ70) (Fig. 3A). Accordingly, the wild-type DNA template used for in vitro run-off assay with RpoN also served as a template for reaction with Eσ70. This assay showed that Eσ70 without any activator efficiently initiates transcription (Fig. 4A, lane 5). The size of transcript was similar to that obtained with RpoN. Thus, it is quite likely the −327-nt residue defines the initiation site for P2 (RpoN-regulated) and P3 (RpoD-regulated) promoters. As the −10 element corresponding to the P3 promoter contains conserved −7T- and −11A-nt residues, they were mutated to −7C and −11G, respectively. Next, such a mutated DNA template was used in in vitro run-off assays with the RNAP core and RpoD. Unlike the wild-type template, the mutated template could not be recognized efficiently by the RNAP core and RpoD to initiate transcription (Fig. 4A, lane 6). This mutated P3 promoter was cloned in the promoter probe vector pRS551 (pGK4838) and transferred in a single-copy to the chromosome (SR19089). In vivo such a fusion did not respond to signals that activate the rpoEP3 promoter and exhibited reduced activity (see below). Thus, we can conclude that RpoN and RpoD recognize P2 and P3 promoters, respectively, and they share the same initiation start site located at −327 nt upstream of the rpoE translational start site.
QseF (GlrR) Positively Regulates the RpoN-recognized P2 Promoter
To understand the mechanism of transcriptional regulation of various rpoE promoters, genes whose overexpression induces the rpoE transcription were sought using multicopy libraries. Subsequently, such an overexpression-induced transcription was analyzed for specificity toward individual promoters. One of the genes, whose overexpression specifically induced the rpoEP2 promoter activity, identified yfhG (qseG), whose product could mediate signal transduction. The qseG gene encodes an OM α-helical protein and has been characterized in enterohemeorrhagic E. coli (EHEC) and Edwardsiella tarda (28, 43). The qseG gene is co-transcribed within the qseE (glrK) qseF (glrR) operon. The QseEFG system regulates key virulence factors and cross-talks with other regulatory systems, such as PhoP/Q and RcsB/C (28). QseG and its other operon members are involved in the formation of attaching pedestal and responding to phosphate and sulfate sources (28, 44). In E. coli K-12, the QseE/F two-component system positively regulates transcription of the glmY sRNA, located upstream of the qseE gene, in an RpoN-dependent manner and hence usage of Glr terminology in that study (42). The quantification of the β-galactosidase activity revealed approximately a 20-fold increase in the rpoEP2 promoter activity upon the overexpression of the qseG gene (Fig. 5A). This induction of the rpoEP2 promoter by QseG overproduction requires the QseF response regulator. This was shown by near abrogation of rpoEP2-lacZ induction upon qseG overexpression in a ΔqseF derivative (Fig. 5A).
FIGURE 5.
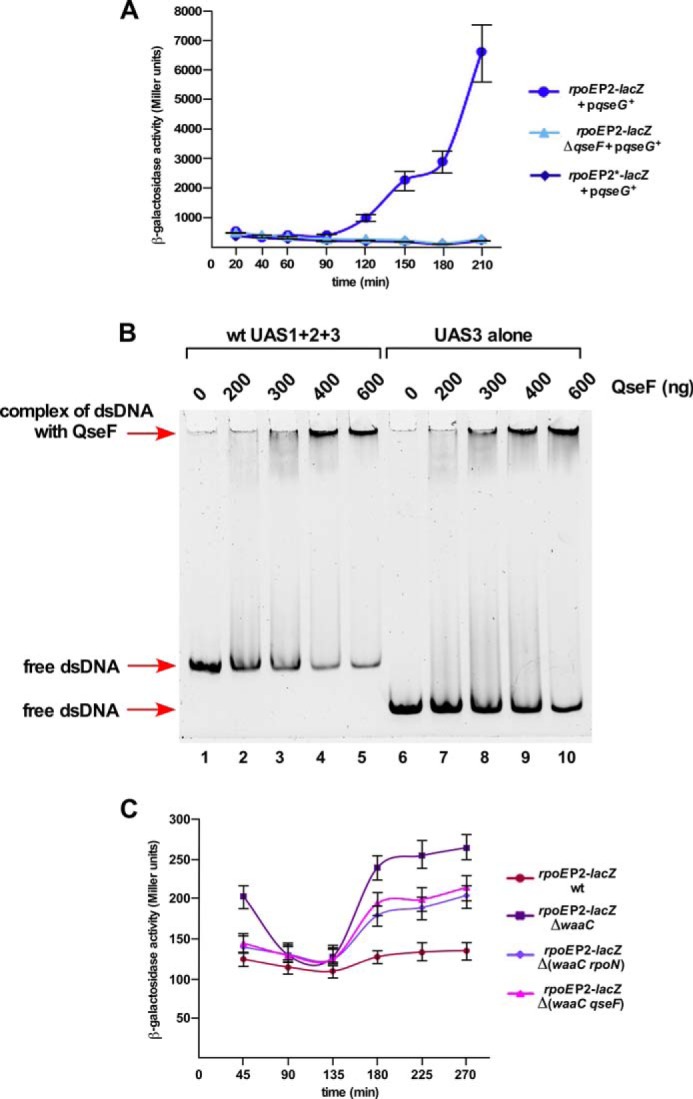
The positive regulation of the rpoEP2 promoter by the QseF activator. A, four independent cultures of wild-type and its ΔqseF derivative carrying either the wild-type rpoEP2 promoter fusion or with rpoEP2* promoter fusion with mutations in the −12 and −24 regions expressing the cloned qseG gene were analyzed for β-galactosidase activity after different growth intervals in the presence of 50 μm IPTG. B, the QseF-DNA interaction at the P2 promoter. Thirty-five ng of the wild-type DNA fragment covering QseF binding sites UAS1 + 2+3 (lanes 2–5) were incubated with increasing concentrations of QseF and analyzed by EMSA. A DNA probe containing only UAS3 was also incubated with QseF as indicated. Lanes 1 and 6 serve as a control with DNA alone. Samples were analyzed as described under “Experimental Procedures.” C, the RpoN-regulated rpoEP2 promoter also senses LPS defects. Isogenic cultures of SR19089 and its ΔwaaC, Δ(waaC rpoN), and Δ(waaC qseF) were analyzed for β-galactosidase activity after different growth intervals and averages of four independent derivatives are plotted.
As the in vitro run-off assay demonstrated that RpoN-dependent transcription from the P2 promoter requires QseF, this was also examined in vivo. Thus, the plasmid expressing the qseG gene was introduced in the isogenic strain carrying the mutated single-copy rpoEP2*-lacZ promoter fusion (−12 and −24 mutations). This rpoEP2*-lacZ derivative could not be induced upon qseG overexpression as compared with the wild-type (Fig. 5A). Hence, we can conclude that the RpoN-regulated rpoEP2 promoter can recruit QseF as an activator providing a link between RpoN and RpoE via a Qse two-component system.
QseF Binds to Upstream Elements of the rpoEP2 Promoter
The results presented in above sections established the presence of the active RpoN-dependent P2 promoter that recruits QseF as the activator. To further confirm the role of QseF in recognition of the rpoEP2 promoter, binding of purified QseF was analyzed by electrophoretic mobility shift assays (EMSAs). Two DNA fragments of 529 and 265 bp, containing 329 and 65 bp, respectively, upstream of the transcriptional start site for the P2 promoter, were used in EMSAs. Up to now, the only known promoter regulated by RpoN together with QseF is that of the glmY gene in E. coli K-12 (42). It was proposed, based on the alignment of the glmY promoter region, that the QseF-binding site located upstream of about 100 bp (up to three sites) contains a conserved palindromic sequence of TGTCN10GACA. Examination of the DNA sequence revealed the presence of three such sequence elements upstream of the P2 TSS with a similar palindromic sequence and similar distance between two half-sites. These sequence elements are designated UAS1, UAS2, and UAS3 (upstream activator sequence) (Fig. 3A). The UAS1 is located −100 nt upstream of the P2 TSS. A similar palindromic sequence is also present immediately downstream of the P2 TSS (Fig. 3A). A DNA fragment of 529 bp contains all three UAS elements, whereas the 265-bp fragment contains only UAS3. These DNA fragments were incubated with increasing concentrations of QseF and their complexes analyzed by a native polyacrylamide gel electrophoresis. As shown in Fig. 5B, the longer DNA fragment containing UAS1 to UAS3 was shifted much more efficiently as compared with the shorter fragment containing only UAS3. Thus, we can conclude that QseF binds DNA located upstream of the rpoEP2 TSS and based on in vitro assays acts as an activator for RpoN-dependent rpoEP2 transcription.
Modulators of RpoS Regulate the rpoEP4 Promoter
To identify signals and regulators of individual promoters, various mutations in different genes that caused activation of the rpoE transcription (Tables 1 and 2) were introduced into strain SR18874 carrying the rpoEP4-lacZ fusion. Thus, a ΔrpoS mutation resulted in ∼60% reduction as compared with the wild-type (Fig. 6). Furthermore, introduction of the ΔrssB caused more than 35% increase in the rpoEP4 promoter activity consistent with the isolation of Tn10 mutations in the rssB gene (Fig. 6). RssB acts as an adapter protein in the pathway of the ClpXP-dependent proteolysis of RpoS and hence rssB mutants exhibit elevated RpoS levels (45, 46). Thus, the isolation of Tn10 mutations in the rssB gene that increase rpoEP4 promoter activity and the reduction of P4 activity in ΔrpoS is overall consistent with results from in vitro run-off assays establishing that this promoter is uniquely transcribed by EσS.
FIGURE 6.
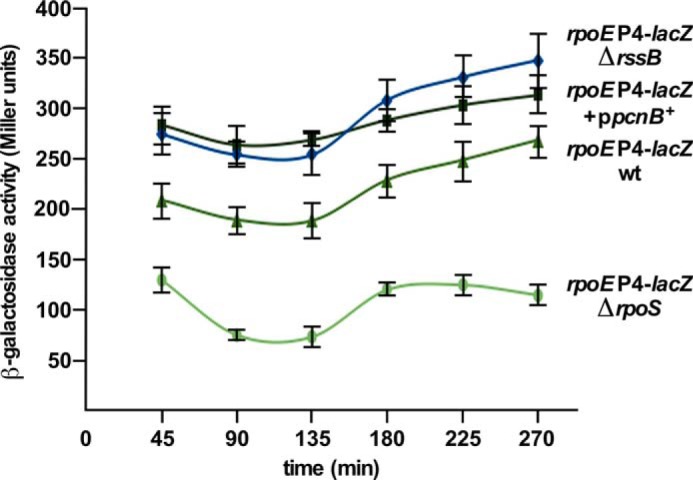
The rpoEP4 promoter is regulated positively by RpoS and responds to its modulators. Cultures of SR18874 carrying the rpoEP4 promoter fusion, its ΔrpoS or ΔrssB derivatives, and derivative carrying the cloned pcnB gene were grown in LB medium at 30 °C either in the presence of 50 μm IPTG for strain with plasmid or without IPTG for plasmid-free derivatives. Samples from four independent cultures at different growth intervals in each case were analyzed for the β-galactosidase activity.
Among various growth conditions that were identified to induce the rpoE transcription, the exposure to increased osmolarity (addition of 0.25 m sucrose or 0.5 m NaCl) also caused a 2–3-fold induction in rpoEP4 promoter activity (Tables 1 and 2). An increase in osmolarity increases RprA-dependent RpoS translation (47). Thus, in vivo the P4 promoter behaves in a typical RpoS-dependent manner.
Next, cloned genes, whose overexpression altered the rpoE transcription, were introduced in SR18874. Among these, overexpression of the pcnB gene caused an ∼40% increase in the rpoEP4 activity (Fig. 6). However, a mild induction of the fliZ gene repressed the P4 promoter activity by 40% (Tables 1 and 2). The pcnB gene encodes PAP I and in its absence the RssB-dependent proteolysis of RpoS is enhanced (36). The negative effect on the activity of the rpoEP4 promoter upon fliZ overexpression can be rationalized, because FliZ behaves as a repressor of RpoS by acting as its antagonist due to overlapping DNA-binding properties (48). Thus, overall these results support our conclusions that the rpoEP4 promoter is regulated by RpoS and conditions that either stabilize RpoS (PAP I) or destabilize it (RssB) alter the activity of the rpoEP4 promoter in accordance with its positive regulation by RpoS.
Defects in Early Steps of the LPS Core Biosynthesis Induce the rpoEP3 Transcription
As the majority of trans-acting Tn10 mutations that increased the rpoEP-lacZ activity mapped to the waaC gene and some other genes in the waa locus (supplemental Table S1) suggested that defects in LPS activate the rpoE transcription from distally located promoter(s). The second largest number of Tn10 insertion mutations were mapped to the waaF gene. The waaC gene encodes heptosyltransferase I transferring l-glycero-α-d-manno-heptopyranose (Hep) to Kdo. The second Hep transfer to the HepI requires heptosyltransferase II WaaF (1, 2). To avoid any effect of polarity, non-polar deletion derivatives of various waa genes were constructed and transduced into strains carrying the lacZ fusion to the P2, P3, and P4 promoter to identify which of these promoters is specifically induced when LPS synthesis is defective. Such isogenic strains were used to measure the β-galactosidase activity and also verified for their LPS composition.
As the P2 and P3 promoter use the same TSS, two different promoter fusions were constructed in a single-copy to distinguish between modes of activation of these promoters. One such construct SR18987 contains mutations in RpoN recognition sites for the P2 promoter in −12 and −24 regions, but with intact −10 and −35 elements for the P3 promoter recognized by RpoD. Another strain, SR19089, carries the mutation in the −10 element of the RpoD-regulated P3 promoter with mutated −7- and −11-nt residues. Thus, SR18987 reflects mainly the activity of the P3 promoter, whereas the promoter fusion in SR19089 shows the RpoN-dependent P2 promoter activity. Into these strains, different null alleles of various waa genes were introduced and quantified for the promoter-specific transcriptional activity. Such an analysis revealed up to a 7-fold increase in the rpoEP3-lacZ promoter activity with ΔwaaC as compared with its basal activity in the wild-type (Fig. 7A). However, the ΔwaaC derivative of rpoEP2-lacZ fusion exhibited only 70% to a 2-fold increase upon entry into the stationary phase (Fig. 5C). No induction of the RpoS-regulated P4 promoter was observed when the ΔwaaC mutation was introduced. Next, impact of the individual non-polar deletion of other genes within the waa locus on the rpoEP3-lacZ activity was addressed. A ΔwaaF derivative exhibited a nearly 2–3-fold increase in the rpoEP3-lacZ activity (Fig. 7A). Similarly, ΔwaaG, ΔwaaP, and ΔwaaO mutants exhibited a more than 2-fold increase in the rpoEP3 promoter activity (Fig. 7A). WaaG glycosyltransferase mediates the incorporation of GlcI, whereas WaaP is required for the phosphorylation of HepI (49, 50). Induction of the P3 promoter in ΔwaaG and ΔwaaP reflects the importance of phosphorylation in OM integrity (50). Deletion derivatives of waaQ, waaS, waaY, and waaZ did not result in any major induction of the rpoEP3 promoter or other rpoE promoters (data not shown). The above results allow us to conclude that severe LPS defects in the most conserved part of the inner core and the lack of GlcI induce the activity of the RpoD-dependent P3 promoter.
FIGURE 7.

The Eσ70-recognized rpoEP3 promoter is positively regulated by the Rcs two-component system in response to LPS defects in an RcsB-dependent manner. A, cultures of SR18987 carrying the rpoEP3 promoter fusion and its derivatives with non-polar deletion in various waa genes were grown in LB medium at 30 °C and analyzed for β-galactosidase activity after different intervals. Four independent derivatives in each case were analyzed and averaged data are presented after a 250-min incubation. B, cultures of SR18987, its ΔwaaC and Δ(waaC rcsB) derivatives were grown in LB medium at 30 °C and analyzed for β-galactosidase activity after different intervals of growth. Averages from four cultures in each case are plotted. C, the RcsB-DNA interaction at the P3 promoter. Thirty-five ng of a 280-bp wild-type DNA fragment that includes a putative RcsB recognition site was incubated with increasing concentrations of phosphorylated RcsB and analyzed by EMSA on a 4% native gel. D, specificity of RcsB-DNA interaction at the P3 promoter. Three DNA probes included a 81-bp DNA fragment with the wild-type RcsB sequence (lanes 2 and 3), a second 78-bp DNA fragment lacking conserved CAT trinucleotide residues of RcsB consensus (lanes 5 and 6), and a third 71-bp probe lacking nucleotides CATGGTTTGG of RcsB consensus (lanes 8 and 9) were incubated with 120 and 180 ng of RcsB for each probe, respectively. Lanes 1, 4, and 7 serve as control with DNA alone. After incubation the reaction mixtures were analyzed on a 6% native gel.
waaC and waaF Mutants Incorporate P-EtN on the Second Kdo Rather Than in the Lipid A
To validate the authenticity of different waa mutants and impact on LPS modifications, LPS was obtained from isogenic strains grown under conditions that induce lipid A and inner core modifications (6, 19, 39). As LPS defects due to mutations like ΔwaaC and ΔwaaF caused a significant induction of the rpoE transcription, this should be reflected in the modification of Kdo by the EptB-dependent incorporation of the P-EtN residue. Mass spectrometric analysis of LPS of the ΔwaaC strain revealed the mass peak at 2,237.3 Da (Fig. 8A). This is in agreement with the predicted composition of the Kdo2-lipid Ahexa 1,4′-bisphosphate. The spectra of LPS obtained from a ΔwaaF derivative contains the mass peak at 2,429.4 Da reflecting the incorporation of one heptose with a predicted composition of Kdo2-lipid Ahexa + Hep1 (Fig. 8B). Additional mass peaks in the spectra of ΔwaaC at 2,360.3 and 2,491.4 Da correspond to the addition of P-EtN and an additional 4-amino-4-deoxy-l-arabinose (l-Ara4N) residue, respectively (Fig. 8). Similarly, mass peaks at 2,560.5 and 2,683.5 Da present in the spectra of LPS of ΔwaaF can be explained to arise due to the incorporation of l-Ara4N and an additional P-EtN residue, respectively (Fig. 8). The mass spectrometric analysis of LPS obtained from the ΔwaaG strain revealed mass peaks at 2,621.5 and 2,824.4 Da. These mass peaks correspond to the predicted structure composed of LAhexa + Kdo2 + Hep2 and LAhexa + Kdo2 + Hep2 + P + P-EtN1, respectively (Fig. 8C), similar to previously described structural analysis of LPS from the waaG mutant in the E. coli strain with the R1 core type (49) reflecting partial phosphorylation of HepI. Additional mass peaks at 3,078.5 Da can be attributed to the mass composition of LAhexa + Kdo2 + Hep2 + P + P-EtN2 + l-Ara4N1. The mass spectrometric analysis of LPS of a ΔwaaP derivative revealed the presence of pentaacylated (LApenta + Kdo2 + Hep2 + Hex, the mass peak at 2,573.3 Da) and derivatives of hexaacylated species (2,783.5 and 3,037.6 Da). These mass peaks can be explained to be composed of LAhexa + Kdo2 + Hep2 + Hex1 and LAhexa + Kdo2 + Hep2 + Hex1 + P-EtN1 + l-Ara4N1, respectively. Further mass peaks at 3,199.6 and 3,361.7 Da correspond to the addition of one and two Hex residues, respectively (Fig. 8D). No mass peaks corresponding to phosphorylated derivatives of Hep residues are present, consistent with the lack of WaaP kinase due to the ΔwaaP mutation. The presence of two P-EtN residues reveals modification of Kdo as well as lipid A by P-EtN in ΔwaaP.
FIGURE 8.

The LPS composition of derivatives with specific mutations causing LPS truncation leading to the induction of the rpoEP3 transcription. Charge deconvoluted ES FT-ICR mass spectra in the negative ion mode of native LPS obtained from isogenic deletion derivatives of the wild-type strain carrying the ΔwaaC mutation (A), ΔwaaF (B), ΔwaaG (C), and ΔwaaP (D) grown in phosphate-limiting medium at 30 °C. Mass numbers refer to monoisotopic peaks. The mass peaks corresponding to additional substitutions with P-EtN and/or l-Ara4N are indicated.
The analysis of lipid A revealed the incorporation of l-Ara4N represented by the mass peak at 1,928.3 Da in the spectra of all mutants (Fig. 9). In E. coli K-12, the most preferred position of l-Ara4N in lipid A is at the 4′-phosphate (51). Interestingly, ΔwaaC and ΔwaaF derivatives did not reveal any P-EtN incorporation in the lipid A part. Thus, the mass peak at 2,360.3 Da in ΔwaaC and at 2,552.4 Da in ΔwaaF corresponding to the incorporation of P-EtN is predicted to have P-EtN on the Kdo, which is EptB-dependent (Fig. 8, A and B). These results are consistent with previous detailed fragmentation analysis of LPS obtained from various derivatives of waaC mutants (6). The EptB-dependent P-EtN modification of Kdo in ΔwaaC, ΔwaaF, and ΔwaaP derivatives is consistent with the activation of the rpoE transcription due to LPS defects and the RpoE-dependent regulation of the eptB gene. These structural alterations of LPS upon the hyper-induction of the rpoE transcription in ΔwaaC and ΔwaaF derivatives indicate the preferential modification of Kdo by P-EtN at the expense of its incorporation in the lipid A part.
FIGURE 9.

Defects in the lipid A biogenesis of Δwaa mutants exhibiting increased rpoEP3 transcriptional activity. Charge deconvoluted ESI FT-ICR mass spectra of isogenic ΔwaaC (A), ΔwaaF (B), ΔwaaG (C), and ΔwaaP (D) in the negative ion mode depicting the lipid A composition and its modifications from the LPS obtained from cultures grown in phosphate-limiting medium at 30 °C. Part of the negative ion mass spectra of the native LPS after unspecific fragmentation, leading to the cleavage of the labile lipid A-Kdo linkage, is presented. The mass peaks corresponding to the hexaacylated lipid A part and substitutions with P-EtN and/or l-Ara4N are indicated.
The Transcriptional Induction of the rpoE Gene Due to LPS Defects Requires Qse and Rcs Two-component Systems
Results presented above established that defects in LPS biosynthesis induce transcription of rpoEP2/P3 promoters. To address the molecular mechanism of the activation of the rpoE transcription when LPS is defective, panels of double deletion strains lacking various regulators in the ΔwaaC background were analyzed. Among these, the Δ(waaC rpoN) derivative showed ∼40–45% reduction in the rpoEP2-lacZ activity as compared with the isogenic ΔwaaC mutant (Fig. 5C). These results of RpoN dependence could require recruitment of the QseF activator based on the in vitro run-off assays. Consistent with these data, activation of the rpoEP2-lacZ fusion in a Δ(waaC qseF) derivative was reduced by more than 30% as compared with the parental ΔwaaC strain (Fig. 5C).
Severe defects in LPS are known to induce the Rcs two-component system leading to overproduction of colanic acid (7). In the Rcs two-component system, RcsB acts as the major response regulator (8). Thus, a ΔrcsB mutation was transduced into ΔwaaC derivatives carrying rpoEP3-lacZ fusion. Strikingly, activation of rpoEP3-lacZ fusion in a Δ(waaC rcsB) derivative is nearly abrogated as compared with highly elevated levels of the rpoEP3-lacZ activity in ΔwaaC (Fig. 7B). These data suggest that Rcs and Qse systems regulate the rpoE transcription in response to LPS defects with the Rcs system playing the major role.
In Vitro RcsB Binds in the P3 Promoter Region
To establish the role of RcsB in regulation of the rpoEP3 promoter, the ability of RcsB to bind DNA containing the region upstream of this promoter was tested by EMSA. A 280-bp DNA fragment was incubated with increasing concentrations of RcsB. Indeed, such a DNA fragment could efficiently shift in the presence of RcsB (Fig. 7C). Examination of DNA sequence upstream of the P3 promoter revealed that it contains a DNA sequence element with similarity to the RcsB box proximal to the −35 region of the P3 promoter (Fig. 3A). The specificity of RcsB binding to the RcsB box was examined using a smaller 81-bp DNA fragment with the intact RcsB box, a 78-bp fragment lacking three conserved nucleotides marked as asterisks and a 71-bp fragment lacking RcsB box. The 78-bp fragment with a deletion of 3 bp was shifted much less efficiently and this mobility shift was further reduced with the 71-bp lacking the conserved RcsB box (Fig. 7D). Thus, these results of RcsB binding upstream of the P3 promoter region support in vivo results of involvement of the Rcs two-component system in regulating the rpoE transcription from its P3 promoter.
Defects in the LPS Assembly Also Induce the rpoEP3 Promoter
Previously, we showed that the balanced biosynthesis of LPS requires the essential LapB protein and in its absence transcription of the rpoE gene is significantly induced (9). ΔlapB mutants synthesize excess LPS with a significant proportion of LPS comprised of precursor forms and also have a dysfunctional Lpt translocation system (9). Thus, ΔlapB derivatives were constructed under permissive growth conditions in strains carrying rpoEP1-P5-lacZ and rpoEP3-lacZ (with mutated RpoN recognition site) fusions in the wild-type background and ΔrcsB. The Δ(lapA lapB) derivative exhibited more than a 5-fold increase in the rpoEP1-P5-lacZ activity and a 3-fold induction of the rpoEP3-lacZ activity when the Rcs system is intact (Fig. 10, A and B). However, this activation in the absence of Lap proteins was significantly diminished in strains lacking RcsB (Fig. 10A). Thus, these results demonstrate that transcriptional activation of the rpoE gene caused by severe defects in the LPS synthesis is driven from the rpoEP3 promoter and this signal activation requires the RcsB response regulator.
FIGURE 10.
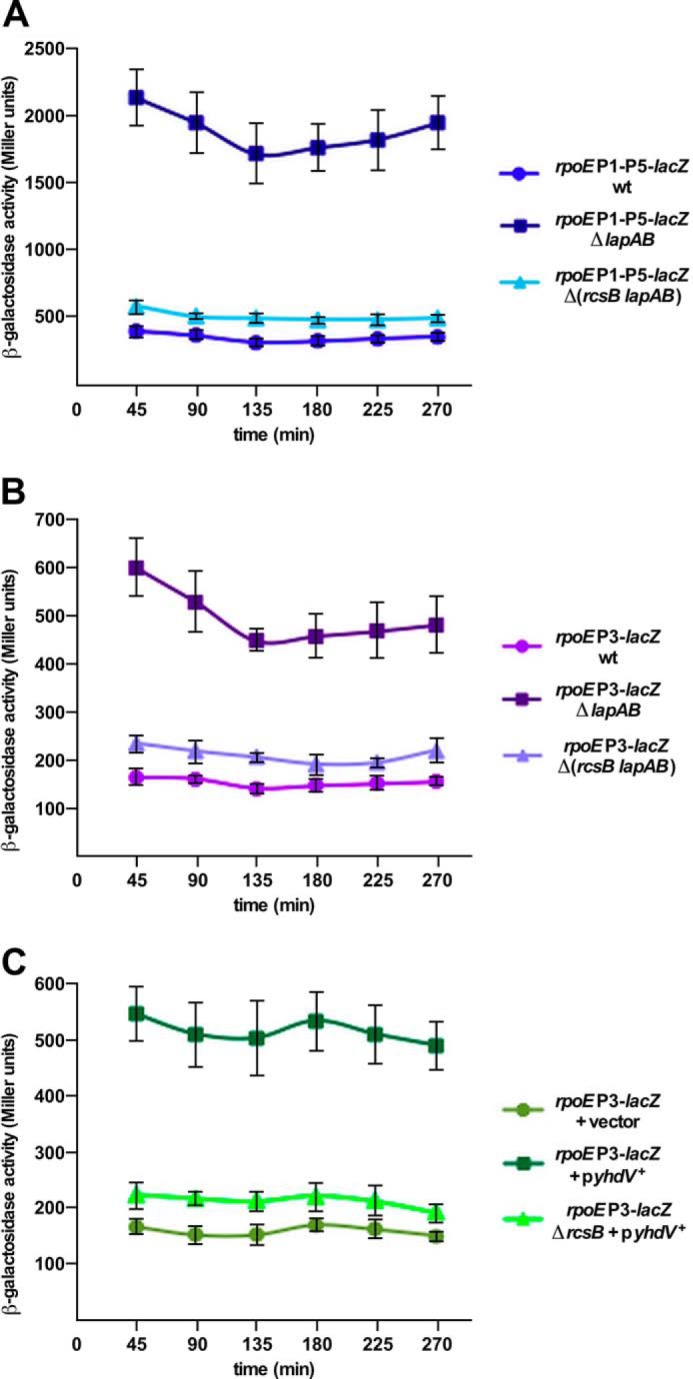
The absence of LapAB proteins or overexpression of lipoprotein encoded by the yhdV gene induces the rpoEP3 transcription. A, overnight cultures of SR7917 (carrying chromosomal rpoEP1-P5-lacZ fusion), its Δ(lapA lapB) and Δ(rcsB lapA lapB) derivatives, and B, SR18987 (rpoEP3-lacZ), its isogenic Δ(lapA lapB) and Δ(rcsB lapA lapB) derivatives, were grown in M9 medium at 30 °C and analyzed for β-galactosidase activity after different intervals. Data averaged from four independent samples are presented for panels A and B. C, cultures of SR18987 carrying rpoEP3-lacZ fusion with empty vector, its derivative with the cloned yhdV gene, and its ΔrcsB derivative with the yhdV gene on the plasmid were analyzed for β-galactosidase activity after the addition of 50 μm IPTG. Data from four replicates are plotted.
Multicopy Inducers of the rpoEP3 Promoter Identify a Novel RfaH-interacting sRNA RirA
One of the approaches, using a multicopy plasmid library, led to the identification of a novel sRNA, whose overexpression induced the rpoEP1-P5-lacZ activity. Subcloning identified a minimal clone carrying DNA overlapping the 5′ UTR of the waaQ mRNA. Further analysis revealed ∼2-fold induction of the rpoEP3 promoter when this sRNA was expressed in a medium-copy plasmid from its own promoter (Fig. 11B). Mapping of the 5′ and 3′ ends revealed that this sRNA shares its 5′ end with the known 5′ end of the waaQ mRNA and is a 73-nt sRNA (Fig. 11, A and C). This sRNA could arise due to processing of the 5′ end of the waaQ mRNA. Examination of its nucleotide sequence revealed that it contains the conserved JUMPstart and the ops site (GGCGGTAG) located between nt 40 and 60 recognized by the RfaH transcriptional factor (Fig. 11A). Based on biochemical evidence and the observed interaction with RfaH, this sRNA is designated RirA (RfaH interacting RNA). The −10 promoter region of the rirA gene contains similarity to RpoD-recognized promoters (Fig. 11A). It also contains TGA nucleotides next to the −10 element resembling the extended −10 promoter.
FIGURE 11.

A novel sRNA rirA in multicopy induces the rpoE transcriptional activity and controls LPS biosynthesis. A, nucleotide sequence of the gene encoding the rirA sRNA and its promoter region. The arrow indicates the TSS of the rirA sRNA. The −10 and extended −10 promoter elements, the conserved JUMPstart, and ops sites are depicted. B, either overexpression of the rirA gene or the absence of RfaH induces the rpoEP3 promoter activity. Cultures of SR18987 with the vector pRS551, its ΔrfaH derivative, or when the rirA gene is expressed from its own promoter in pRS551 were grown in LB medium at 30 °C and analyzed for the β-galactosidase activity after different growth intervals. Average of four independent samples are presented. C, model of RirA generated using M-fold.
RfaH is an operon-specific transcription factor belonging to the NusG family of proteins (25). RfaH is known to regulate the expression of genes transcribed as long operons encoding the LPS core, capsule, O-antigen biosynthesis, hemolysin, and conjugation system (23–25). RfaH functions by binding to the ops site located in the 5′ region of non-template DNA of these operons and prevents premature transcriptional termination by Rho. This is achieved by RfaH simultaneous contacts with RNA polymerase and coupling with translating ribosome by interaction with the S10 protein (25). We wondered if the rirA mRNA would bind RfaH. This was addressed by EMSA wherein RfaH was incubated with RirA alone and in the presence of the RNAP core. RfaH could efficiently gel shift RirA in the presence of RNAP, but not when incubated with RfaH alone (Fig. 12A). As RirA contains the conserved ops site, we tested if the interaction between RirA and RfaH requires the core ops element. Hence, gel shift assays were performed using synthetic wild-type RirA or RirA* with mutated ops site RNAs. EMSA experiments revealed that RirA with the mutated ops site does not bind RfaH (Fig. 12B). Thus, we can conclude that RirA indeed interacts with RfaH in the presence of RNA polymerase. Furthermore, this interaction is dependent on the presence of the ops site within RirA.
FIGURE 12.

RirA sRNA binds to RfaH and its overexpression causes defects in the LPS synthesis. A, interaction of RirA with RfaH in the presence or absence of RNAP. Fifty ng of RirA were incubated at 37 °C with 150, 300, and 450 ng of RfaH in the presence of 50 μg of RNA polymerase core (lanes 2–4) or in the absence of RNAP (lanes 5–7). Lane 1 corresponds to RirA alone. Samples were analyzed on a 6% native polyacrylamide gel. Arrows indicate the position of complex and free RirA. B, the replacement of the 8-nt ops site by 8 A nt in RirA abolishes interaction with RfaH + RNAP core. Fifty ng of wild-type RirA (lanes 2–4) and RirA with mutated ops site (lanes 5–7) were analyzed for the complex formation with RfaH in the presence of RNAP and resolved by native gel electrophoresis. C, a portion of whole cell lysate obtained from the wild-type E. coli K-12 strain BW25113 with vector pRS551 (lane 1), its derivative expressing the wild-type rirA gene in pRS551 (lane 2), and rirA with 8-nt ops site replaced by 8 A residues rirA* (lane 3) were applied on a 16.5% SDS-Tricine gel and LPS was revealed after silver staining. The arrow indicates the position of the LPS core. D, a portion of whole cell lysates after proteinase K treatment obtained from isogenic strains with pMF19 and vector pRS551 alone (lane 1) and its derivative expressing the rirA gene from its own promoter in pRS551 with pMF19 (lane 2) were resolved on a 14% SDS-Tricine gel and LPS was revealed after silver staining. Arrows indicate the position of the lipid A-core, lipid A-core + 1 O-antigen unit, and lipid A-core + polymeric O-antigen. E, isogenic cultures carrying vector pRS551 alone (lane 1) or expressing the wild-type rirA gene (lane 2) in the strain with chromosomal waaO, rfbB, or waaC genes epitoped at the C-terminal end with 3× FLAG were grown in LB medium at 30 °C up to an A600 of 0.2. An equivalent amount of total protein was analyzed on 12% SDS-PAGE, followed by immunoblotting using anti-FLAG monoclonal antibody.
Because overexpression of the rirA gene induces the rpoEP3 activity, a ΔrfaH mutant was also tested for influence on the rpoHP3 activity. A 4-fold induction of rpoEP3 promoter activity was observed (Fig. 11B). These results are consistent with isolation of a rfaH::Tn10 mutation conferring an increase in the rpoE transcriptional activity (supplemental Table S1). To address the basis of induction of the rpoEP3 by RirA, the effect of the rirA overexpression on the LPS content was examined. Whole cell lysates were prepared from isogenic strains with and without overexpression of RirA. The overexpression of RirA caused a reduction in the total amount of LPS (Fig. 12C). As the conserved ops site is also located in front of the O-antigen gene cluster, the wild-type E. coli K-12 strain BW25113 was transformed with plasmid pMF19 (expressing the wbbL gene (52)) to restore O-antigen biosynthesis. Next, this strain was transformed with the compatible vector alone or its derivative carrying the rirA gene and examined for the LPS profile. A quite robust reduction in the O-antigen was observed, when the rirA gene is present on the plasmid (Fig. 12D). It should be noted that the cloned rirA gene was expressed from its own promoter. These results suggest that RirA in excess could cause titration of RfaH and limit its availability to bind in vivo ops sites. This could lead to decreased expression of LPS core biosynthetic genes transcribed from the waaQ operon and other operons with the ops site in the 5′ UTR like O-antigen biosynthetic genes. Defects in the O-antigen presence upon the rirA overexpression can be due to an additive effect of reduction in the rfb operon expression and reduced number of sites for O-antigen incorporation.
To further understand the molecular basis of reduction of LPS upon RirA overexpression, the expression of RfaH-regulated waaQ and rfb operons was monitored. Western blot analysis revealed that the WaaO-FLAG and RfbB-FLAG amounts are reduced, whereas as a control WaaC-FLAG levels were unaltered (Fig. 12E). The waaO gene is transcribed as a part of the waaQ operon and the rfbB gene also contains the ops site in the 5′ UTR. The waaC gene is transcribed as a part of the gmhD waaF waaC operon lacking the ops site. Taken together, these results establish that RirA sRNA acts as a novel sRNA that interacts with RfaH and its excess reduces LPS amounts by the potential sequestration model that limits the availability of RfaH.
The overexpression of Genes Encoding Lipoproteins Induces the rpoEP3 Promoter
The most prominent class of multicopy inducers of the rpoEP1-P5 transcription identified genes encoding lipoproteins (Tables 1 and 2). From these, impact of overexpression of the yhdV gene was analyzed, because it was repeatedly isolated as inducer of the rpoEP1-P5-lacZ or specifically the rpoEP3-lacZ transcriptional activity. Measurement of the rpoEP3-lacZ activity revealed a nearly 3.4-fold increase (Fig. 10C). Most of the genes encoding lipoproteins identified in this work encode proteins of low abundance like YhdV, YddW, and YghB, and Spr is in the bottom 25% range (based on pax-db.org), making their isolation physiologically relevant. We reasoned that the increased synthesis of lipoprotein could cause limitation of the Lol system required for sorting of lipoproteins. Defective lipoprotein is known to induce the Rcs pathway (53). Thus, we tested if the mechanism of activation of the rpoEP3 promoter was also Rcs-dependent. Accordingly, expression of the yhdV gene was induced in the wild-type and its ΔrcsB derivative. This analysis revealed a reduction in the activation of the rpoEP3 promoter by more than 50% (Fig. 10C). A similar dependence was observed for the lipoprotein-induced rpoEP3 activity for the presence of RcsF (data not shown). Hence, these results establish that the Rcs two-component system senses imbalances in the OM due to either LPS defects or overexpression of lipoproteins causing activation of the rpoEP3 transcription. However, these results do not show if the overexpression of every lipoprotein will induce the rpoEP3 transcription.
The rpoEP3 Promoter Is Subjected to Catabolite Repression
During the quantification of transcription, we repeatedly observed that the activity of the rpoEP1-P5 promoter is higher when glycerol is used as the sole carbon source as compared with glucose-supplemented medium (Tables 1 and 2). After the identification of multiple promoters regulating the rpoE transcription, this effect was observed more specifically for the rpoEP3 promoter. Its activity is also severely repressed upon the addition of glucose to LA medium, like well known CRP-cAMP-regulated genes. It is well established that in the presence of glucose cAMP levels are low, resulting in reduced or lack of expression of promoters that require CRP binding (54). These observations are consistent with the identification of a Tn10 insertion in the cya gene (supplemental Table S1) and an observed 50% reduction in the activity of the rpoEP3 promoter in Δcrp (Fig. 13A). Thus, direct binding of the CRP protein in the presence of cAMP was examined by EMSA using 118-bp DNA fragments that contain nucleotides upstream of the P3 TSS. This DNA template is efficiently shifted in the presence of activated CRP (Fig. 13B) consistent with the presence of DNA sequences that match the CRP-binding site (Fig. 3A). Overall, these results establish cAMP-CRP-mediated control of rpoEP3 transcription.
FIGURE 13.
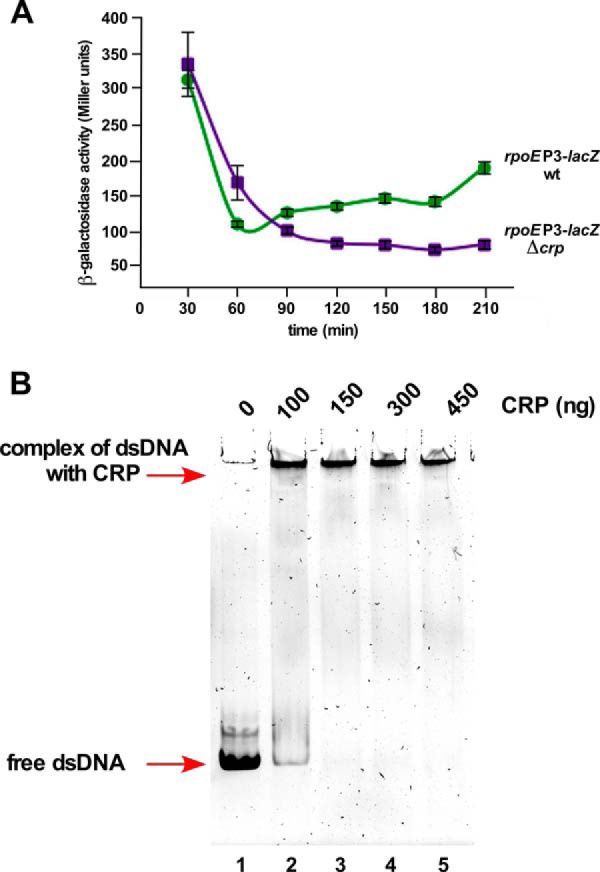
The rpoEP3 promoter is positively regulated by the CRP activator protein. A, cultures of the wild-type strain carrying the rpoEP3 promoter fusion and its four independent Δcrp transductants were grown in LB medium at 30 °C and analyzed for β-galactosidase activity after different intervals. B, interaction of CRP with DNA containing the P3 promoter and upstream DNA region. A 118-bp DNA fragment covering the P3 promoter region was incubated with increasing concentrations of CRP activated by cAMP (lanes 2–5) and complexes were resolved on a 6% native gel. Lane 1 serves as a negative control with DNA alone. Arrows show the position of free dsDNA and CRP-DNA complex.
The RpoE-regulated ecfLM Operon Influences the rpoE Transcription
During the mutagenesis screen for Tn10 insertion(s) that in trans alter the rpoE transcription, one mutation mapped to the ecfL gene (supplemental Table S1). ecfL and ecfM genes are transcribed as an operon with a RpoE-dependent transcription (31). Non-polar deletion of either the ecfL gene alone or the ecfM gene alone does not alter the transcriptional activity of any rpoE promoters. However, a total deletion of the ecfLM operon does increase the rpoEP3 activity by a 2–3-fold (Fig. 14). EcfL belongs to the family of conserved DedA IM proteins (31). Consistent with our data, it has been recently reported that multiple deletion combinations of ecfL orthologs, including ecfL, cause an increase in the rpoE transcriptional activity (55). EcfM, also called MzrA (56), could act as a connector protein in cross-talk among two-component systems and needs more studies.
FIGURE 14.
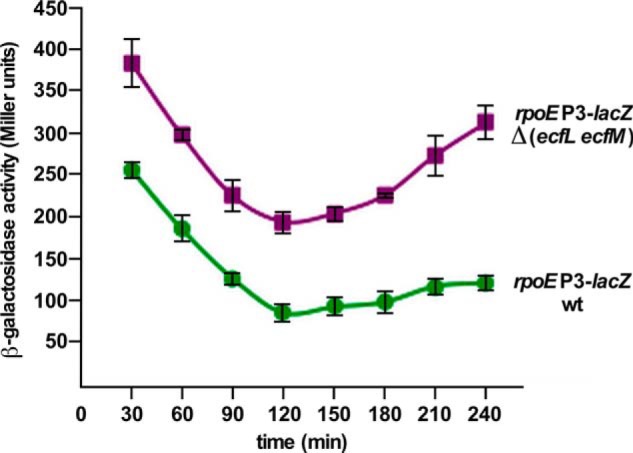
The rpoEP3 promoter activity is induced in the absence of ecfLM genes. Isogenic cultures of the wild-type and its Δ(ecfL ecfM) derivative carrying the rpoEP3 promoter fusion were grown in LB medium at 37 °C and analyzed for the β-galactosidase activity.
Discussion
In this work, we addressed the transcriptional regulation of the rpoE gene, encoding the extracytoplasmic function σ factor. Several members of its regulon have dedicated functions, particularly whose products are involved in the assembly of OM, including components of LPS translocation, OM protein maturation, and some sRNAs that maintain homeostasis of OM components (31). Despite the wealth of knowledge about the function of RpoE and its negative regulation by the anti-σ factor RseA, transcriptional regulation of the rpoE gene has not been fully understood. Previously, we and others showed that transcription of the rpoE gene is autoregulated by EσE from one of its promoters (3, 16). However, the regulation of transcription from its distal promoter remained elusive for more than two decades.
The construction of various transcriptional fusions and the identification of trans-acting factors that modulate the rpoE transcription revealed that the distal promoter region responds to several divergent stimuli. Factors that alter the rpoE transcription from the distal promoter region include activation by (i) severe defects in LPS, (ii) the addition of ammonium metavanadate, (iii) entry into the stationary phase, (iv) a shift to high osmolarity, (v) growth phase-dependent challenge with polymyxin B, (vi) a shift to nitrogen-limiting growth conditions, and (vii) overexpression of certain genes encoding lipoproteins. However, transcription is repressed in glucose-supplemented medium. Significantly, no single transposon insertion or any defined deletion could be identified that totally abolished transcription from this promoter region. These results suggested that either an essential transcriptional factor regulates expression of the rpoE gene from the upstream region or multiple transcriptional factors are involved in mediating the transcriptional control.
To address possibilities of the existence of multiple regulatory controls, 5′ ends of the rpoE mRNA were mapped. This led to the identification of at least five new transcriptional start sites located upstream of the EσE-transcribed promoter. The presence of multiple authentic promoters was confirmed by in vitro run-off assays and the construction of the single-copy promoter fusion to individual promoters. Of five promoters, P2 and P3 promoters share the same TSS located at −327. The P2 promoter shares homology to RpoN-regulated promoters with the presence of conserved −12 and −24 RpoN recognition sites. Interestingly, the P2 promoter is strongly activated by QseF, which also contains extensive homology to the NtrC family of activators (42). QseF activation of the rpoEP2 promoter occurs via RpoN and requires RpoN-recognition motifs (−12 and −24) as shown by mutagenesis and in vitro run-off assays. A role for QseF in regulating the rpoEP2 promoter was first observed by a nearly 20-fold induction upon overexpression of the OM protein QseG. The qseG gene is co-transcribed with qseE and qseF genes. QseE and QseF constitute a two-component system that positively regulates the expression of the promoter of the glmY sRNA in an RpoN-dependent manner (42). GlmY regulates the synthesis of GlmS in response to amounts of glucosamine 6-phosphate (GlcN6P). GlcN6P is the precursor for the synthesis of UDP-GlcNAc and hence, it constitutes the first rate-limiting step in LPS and peptidoglycan synthesis (17, 42). QseG interaction with the QseE kinase could trigger phosphorylation of the QseF response regulator. Indeed, activation of the rpoEP2 promoter by overexpression of qseG is solely dependent on the presence of QseF. Examination of the DNA sequence upstream of the rpoEP2 promoter identified three QseF-binding sites in the upstream region, which were verified by gel-retardation assays. A fourth QseF binding-like motif is also present in the downstream region of the rpoEP2 promoter.
The predicted −10 promoter element of rpoEP3 contains a consensus for RpoD-recognized promoters, with the presence of conserved −7T and −11A residues and an extended −10 TGC (Fig. 3). Mutation of −7T(C) and −11A(G) severely repressed the rpoEP3 promoter activity. This allowed us to establish that the −327 TSS is used by RpoD and RpoN. The −326 TSS could arise due to in vivo processing of the 5′ end or may be used in vivo under specific conditions. The RpoD-regulated P3 promoter is further subjected to catabolite repression and activated specifically when the LPS has truncation in the inner core region or the LPS assembly is dysfunctional.
As the rpoE gene is essential in E. coli under all growth conditions, the presence of multiple promoters regulated by RpoN (P2), RpoD (P3), and RpoS (P4) ensures that the basal level of transcription is sustained even when the OM integrity is not compromised (Fig. 15). Furthermore, each of these promoters fine-tunes transcription under specific conditions. Thus, the induction of the rpoE transcription upon entry into the stationary phase could recruit RpoS via the initiation from the P4 promoter. The P4 promoter utilization could integrate diverse signals that regulate the activity of RpoS. RpoS is induced not only in the stationary phase, but also upon challenge with high osmolarity, carbon starvation, oxidative stress, and in response to stringent growth conditions. We also observed that rpoEP4 is induced upon the shift to high osmolarity. In support of the RpoS-mediated transcription of the P4 promoter, our genetic analyses also identified factors that control the RpoS transcription/stability, like ArcA/B, RssB, and PcnB affecting the rpoEP4 activity.
FIGURE 15.

Co-integration of multiple signaling pathways and recruitment of different transcriptional factors in the regulation of the transcription of the rpoE gene in response to specific stimuli. A schematic drawing of the promoter region of the rpoE gene, depicting the organization of six promoters designated P1 to P6. The transcription from rpoEP6 is initiated by EσE and responds to OM protein defects via the RseA. P2 and P3 promoters utilize the same TSS. The rpoEP2 is recognized by σ54 and can recruit either NtrC or QseF as activators. The QseE/F system can be activated by QseG. QseE/F-regulated transcription of the rpoEP2 and glmY sRNA can co-integrate signals of cell envelope constituents like LPS and peptidoglycan synthesis to rpoE transcription. The rpoEP3 is recognized by σ70 and its transcription is specifically induced when LPS biosynthesis is compromised (lack of assembly protein LapB, titration of RfaH by RirA sRNA, or when the inner core of LPS is truncated). LPS defects could the transmit signal from the RcsF OM lipoprotein leading to the activation of the RcsB response regulator. The P2 promoter can also be induced in response to LPS defects albeit to a lower extent than the rpoEP3 promoter. The global regulator CRP protein in response to cAMP levels also positively regulates the rpoEP3 promoter. The rpoEP4 promoter is recognized by the stationary phase σ factor RpoS and is activated in response to diverse stresses like challenge with high osmolarity and factors that regulate transcription of the rpoS gene and the stability of σS.
The Mechanism of Sensing of LPS Alterations and the Overexpression of Lipoproteins
As the maximal number of mutations that induce transcription from the rpoE proximal promoter caused LPS defects, the mode of signal transduction was addressed. LPS defects significantly induce the RpoD-regulated rpoEP3 promoter that was dependent on the activation of the Rcs two-component system. Thus, the induction of this promoter was mitigated in the absence of either the OM protein RcsF or the response regulator RcsB when either were challenged with polymyxin B or when the LPS synthesis was defective. This mode of the rpoEP3 promoter activation via the Rcs system was demonstrated using strains with truncation of the LPS inner core or strains lacking LPS regulators like RfaH or LapA/B proteins. However, some signals due to LPS defects are also transmitted to the P2 promoter in the RpoN-QseF dependent manner. To distinguish between the activation of P2 and P3 promoters, mutations were introduced in either RpoN recognition sequences or in the −10 conserved element recognized by RpoD. Usage of such mutated promoter fusions confirmed that both P2 and P3 promoters can be activated when LPS is defective, with the main signal activation mediated by the Rcs system to the RpoD-regulated P3 promoter. Observed binding of RcsB and QseF upstream of the P2/P3 TSS further supports these conclusions.
The molecular basis of RpoE induction due to specific LPS defects and participation of any specific signal relay system, like Rcs or other two-component systems, has not been addressed. Two models have been proposed, suggesting sensing of acylation defects in lipid A and a trade-off between the lipopolysaccharide transporter LptA and RpoE negative regulator RseB (38, 57). However, as shown here, the Rcs system can independently sense LPS defects. Our data of signal transduction via multiple systems are compatible with additional modules like RseB and sensing involving additional pathways like the QseGEF system that can signal LPS defects. In support of such a model, we still observed a nearly 2-fold induction of the EσE-regulated promoter of the rpoE gene in Δ(waaC rcsB) and also involvement of RpoN-mediated Qse-dependent control. Utilization of multiple pathways could make the system to launch rapid response and make it more robust.
One of the novel findings from this work is the identification of a new 73-nt sRNA located in the 5′ UTR of waaQ mRNA. This sRNA shares the same start site as the waaQ mRNA and contains the ops site recognized by RfaH. This sRNA, designated RirA, binds RfaH in the presence of the RNAP core. The rirA gene was cloned, because its overexpression induces the rpoE transcription via the P3 promoter. Based on the LPS profile, a mild overexpression of the rirA sRNA caused severe reduction in the incorporation of O-antigen. When RirA was overexpressed in a wild-type E. coli K-12, which does not synthesize O-antigen, the overall LPS amount was reduced. RfaH regulates the expression of many long operons including waaQ and rfb operons by preventing transcription termination and enhancing translation of their mRNAs. RfaH is known to bind to ops sites and our model of reduction of the LPS amount and severe decrease in the O-antigen incorporation upon the rirA overexpression posits that an excess of RirA could titrate RfaH. This could make RfaH limiting and reduce the expression of operons that require RfaH, like rfb and waaQ operons. Thus, the induction of rpoE transcription upon rirA overexpression can be attributed to limiting amounts of LPS or an imbalance with other components of the OM. Hence, not only LPS defects, but also any imbalance (excess of LPS as with lapAB mutants or its reduction when RirA is in extra copies) are sensed by the rpoEP3-dependent transcriptional response. It is tempting to speculate that an accumulation of RirA sRNA under regulated mRNA processing of the waaQ UTR might constitute an internal checkpoint to balance the LPS synthesis.
Furthermore, it was noticed that overexpression of genes encoding some lipoproteins causes an induction of the rpoEP3 promoter. Mislocalization of lipoproteins has been shown to cause induction of the Rcs pathway (53). Consistent with the positive regulation of the rpoEP3 promoter by the Rcs system, its activation by overexpression of the lipoproteins is also Rcs-dependent. Among the genes encoding lipoproteins identified in this work that induce the rpoEP3 transcription, yhdV, yghG, spr, and yceB were previously shown to induce the transcription of the rpoE-regulated degP promoter (58). Some of the lipoproteins are known to show significant genetic interactions with several components of LPS biosynthesis (59). For example, yceB shows interactions with many genes whose products are involved in LPS and phospholipid biosynthesis. Thus, an excess of such lipoproteins could alter the delicate balance in the cell envelope leading to induction of rpoE transcription.
In summary, we have shown that in addition to autoregulation by RpoE, transcription of the rpoE gene is further regulated by RpoS, RpoD, and RpoN σ factors that specifically respond to different stimuli to sustain its transcription under all growth conditions (Fig. 15). Additional recruitment of envelope responsive two-component systems (QseE/F and Rcs) and global regulator CRP further modulate transcriptional of the rpoEP2 and P3 promoters linking several networks to regulate the expression of RpoE. This mode of interlinked regulatory control allows integration of several signals to rapidly respond to a variety of stress conditions and cell envelope defects including LPS alterations.
Experimental Procedures
Bacterial Strains, Plasmids, and Media
The bacterial strains and plasmids used in this study are described in supplemental Table S1. Luria-Bertani (LB) broth, M9 (Difco), and 121 phosphate-limiting minimal media were prepared as described (19, 60). For assays monitoring response to nitrogen concentrations, minimal medium was supplemented with either 3 or 10 mm NH4Cl. When required, media were supplemented with ampicillin (100 μg ml−1), kanamycin (50 μg ml−1), tetracycline (10 μg ml−1), spectinomycin (50 μg ml−1), or chloramphenicol (20 μg ml−1). The indicator dye 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was used at final concentrations of 20 or 40 μg ml−1 in the agar medium. Ammonium metavanadate was added at a final concentration of 25 μg ml−1 to LB or LA media. Polymyxin B (Sigma) was added to LB medium at concentrations ranging from 0.2 to 0.3 μg ml−1. Lactose-containing MacConkey agar (Difco) was supplemented with appropriate antibiotics when required.
The Isolation of Trans-acting Mutations
The MC4100-derived bacterial strain SR4245 and the BW25113 derivative SR7917 were used as parental strains to isolate Tn10 insertion mutants. Each of these strains carries an identical single-copy chromosomal transcriptional rpoEP-lacZ fusion at the λ attachment site as bacteriophage λ lysogen. Given the essentiality of the rpoE gene, a chromosomal replacement of the wild-type rpoE gene with such rpoEP-lacZ fusion was not attempted. More than 50,000 transposon insertion mutants were isolated on LA medium at 30 °C and screened for Lac up or down phenotypes as described previously (3, 61). Bacteriophage P1 or T4 lysates were prepared on individual Lac up candidates and transduced back into SR4245 or SR7197. The position of Tn10 was determined by the inverse PCR with nested primers and sequenced using the Tn10 primer as described previously (9).
The Identification of Trans-acting Factors Whose Overexpression Alters the rpoE Transcription
The complete genomic library of all predicted ORFs of E. coli cloned in pCA24N (33) was used to transform SR4245, SR7917 (carrying rpoEP1-P5-lacZ fusion), SR18874 (rpoEP4-lacZ), and SR18868 (rpoEP2/P3-lacZ). The plasmid carrying the lacZ gene was omitted to reduce the background noise. Lac up or Lac down transformants were isolated at 30 °C in the presence of IPTG (50 μm) based on X-Gal phenotype. DNA insert of all relevant plasmids that yielded reproducible results was sequenced. In parallel, previously described whole genomic libraries obtained from the wild-type E. coli K-12, cloned in medium-copy plasmids (9, 35), were used in similar screens to identify genes whose overexpression alters the rpoE transcriptional activity. The minimal genomic DNA fragment cloned in the same plasmid was obtained by standard subcloning.
Generation of Null Mutations and the Construction of Their Combinations
Non-polar antibiotic-free deletion mutations of various genes used in this study were constructed by using the λ Red recombinase/FLP-mediated recombination system as described previously (9, 39, 62). PCR products from such amplification reactions were electroporated into BW25113 containing the λ Red recombinase-encoding plasmid pKD46 (GK1942). Each deletion was verified by PCR amplification and sequencing of PCR products. Such deletions were transduced into BW25113 by bacteriophage T4-mediated transduction. Multiple null combinations were constructed as described previously, followed by the removal of aph or cat cassettes using the pCP20 plasmid and confirmed to be non-polar. The construction of deletion derivatives of waaC, waaO genes, and the EσE-regulated rpoE-lacZ fusion were previously described (3, 9, 19).
Cloning of Various Genes for Their Overexpression and Complementation Studies
For protein induction, the minimal coding sequences of rpoS, ntrC, qseF, rcsA, rcsB, and crp genes of E. coli were cloned in different expression vectors. The minimal coding region of each relevant gene was PCR-amplified, digested with specific restriction endonucleases, and cloned in expression vectors. rcsA and rcsB genes were cloned in pET22b (NdeI-XhoI), resulting in plasmids pGK4661 and pGK4662. In parallel, crp and rpoS genes with the C-terminal His6 tag were cloned in pET22b and pET24b (NdeI-XhoI), resulting in plasmids pSR16739 and pSR16691, respectively. These plasmids were verified to retain wild-type properties of the respective genes by confirming the complementation of strain Δcrp (GK2986) and ΔrpoS (GK2876). qseF and ntrC genes with the C-terminal His6 tag were cloned in pET28b (NdeI-XhoI) (pSR13036 and pSR18976, respectively). For the induction of RpoN and RfaH, the expression of corresponding genes was induced from clones in the expression vector pCA24N (34).
For LPS analysis and quantification of changes in the rpoE transcription, the rirA gene with its own promoter was subcloned into the pRS551 vector (pSR9446). The minimal coding region of rirA sRNA was PCR-amplified from the chromosomal DNA obtained from the wild-type strain BW25113 using specific oligonucleotides (supplemental Table S2).
RNA Purification and Mapping of 5′ Ends
For the identification of the transcription start site(s) of rpoE and rirA mRNAs, total RNA was extracted from wild-type strain BW25113 and its ΔwaaC derivative or in the presence of multicopy inducers of the rpoE transcription. Cultures were grown in LB medium at 30 °C until an absorbance of 0.2 at 600 nm (exponential phase) or after entry into the stationary phase. RNA was purified according to the manufacturer's protocol (Bioline UK) and digested with RQ1 DNase (Promega) to remove any chromosomal DNA, followed by phenol extraction and RNA precipitation.
The GeneRacer from Invitrogen was used to obtain 5′ ends of cDNA according to the manufacturer's protocol as described previously (9). RNA with and without treatment with calf intestinal phosphatase was used to identify primary and processed products according to the manufacturer's instructions. The cDNA was amplified by PCR using 2 pmol of reverse gene-specific primers (supplemental Table S1) and the GeneRacer 5′ primer. An additional round of nested PCR was employed, using GeneRacer-nested and gene-specific oligonucleotides (supplemental Table S2). PCR products from the second round nested PCR were purified and cloned into the pCR-4-TOPO vector. From each reaction, at least 10 independent plasmids were isolated and sequenced using the M13 forward primer (−21 universal).
In Vitro Run-off Assays
For the identification of the transcription initiation site with specific forms of RNA polymerase, DNA template (50 ng) was incubated with the core RNAP and the specific σ factor at a molar ratio of 4:1. Twenty μl of reaction was carried in buffer containing 40 mm Tris-HCl, pH 7.5, 150 mm KCl, 10 mm MgCl2, 1 mm DTT, 0.5 mm each of ATP, GTP, CTP, and 0.1 mm UTP plus 3.7 kBq of [α-32P]UTP. The reaction was allowed to proceed for 20 min at 37 °C, as described previously (3), and terminated by the addition of formamide stop solution. For in vitro assays with RpoN, NtrC, and QseF, the wild-type template and the template with mutations in the −12 and −24 elements were used. After the open complex formation in reactions with RpoN + NtrC and RpoN + QseF, 5 mm ATP was added and further incubated at 37 °C for 5 min before the addition of nucleotides. RNA samples (4 μl) labeled with [α-32P]UTP were electrophoresed as described previously (3).
Gel Retardation Assays
EMSAs were performed on PCR-generated probes containing various sizes of DNA. When required, mutations were introduced using specific oligonucleotides during PCR (supplemental Table S2). Binding reactions were performed by incubating 35 ng of various DNA probes with an increasing amount of purified RcsB or RcsA or CRP or QseF, using binding buffer (Invitrogen). Whenever required, RcsB and QseF were phosphorylated with acetyl-phosphate at 30 °C for 30 min. Similarly, the CRP-cAMP complex was generated by the incubation of purified CRP with 1 mm cAMP in buffer containing 10 mm Tris-HCl (pH 7.8 at 4 °C), 150 mm NaCl, and 3 mm Mg(OAc)2. Products were analyzed on 4 or 6% native acrylamide gel and visualized after staining with SYBR Green (Invitrogen).
β-Galactosidase Assays
To measure the activity of rpoE promoters, single-copy chromosomal promoter fusions to the lacZ gene were constructed and used to monitor their β-galactosidase activity. DNA sequences located upstream of the known RpoE-autoregulated promoter were amplified by PCR, using specific oligonucleotides (supplemental Table S2). After PCR amplification, gel-purified DNA was digested with EcoRI and BamHI, cloned into either pRS415 or pRS551 vector, and transferred to the chromosome in a single-copy by recombination with λRS45 (63). Lysogens were selected either on the basis of Lac+ phenotype or by directly selecting for Kan-resistant lysogens as described previously (3, 6, 9). Mutations in the −12 and −24 regions of the RpoN-regulated P2 promoter were introduced by PCR using mutagenic oligonucleotides (supplementalTableS2), cloned into pRS551 and transferred to the chromosome to generate single-copy promoter fusion (SR18987). Nucleotide changes corresponding to the −7T and −11A residues of the rpoEP3 promoter were constructed similarly with mutagenic oligonucleotides and cloned in pRS551. Such single-copy lysogens carrying lacZ fusions with mutated promoter regions were directly selected for Kan resistance (SR19089) and verified. Null alleles in different genes studied in this work were transduced into strains carrying various rpoE-lacZ promoter fusions in a single-copy on the chromosome. To measure the β-galactosidase activity, isogenic bacterial strains carrying promoter fusions were grown in LB or M9 medium with appropriate antibiotics at 30 °C, adjusted to an A600 of 0.05 and allowed to grow further. The β-galactosidase activity was measured in Miller units at different growth intervals from four independent cultures in each case as described previously (6, 9).
Protein Purification
The expression of His6-tagged RpoN, RfaH was induced in the E. coli BW25113 strain at an optical density of 0.1 at 600 nm in a 1-liter culture by the addition of 0.5 mm IPTG. For the purification of His6-tagged RpoS, CRP, RcsB, RcsA, NtrC, and QseF, their cognate genes cloned in pET expression vectors were induced in E. coli BL21(DE3) strain at 28 °C. After induction (4 h-5 h at 28 °C), cells were harvested by centrifugation at 12,000 rpm for 20 min. The pellet was resuspended in B-PER reagent (Pierce) and adjusted to contain 50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole (buffer A), supplemented with lysozyme to a final concentration of 200 μg ml−1. A mixture of protease inhibitors (Sigma) was added as per the manufacturer's instructions. The mixture was incubated on ice for 15 min with gentle mixing. To this lysate, 30 units of benzonase (Merck) was added and incubated with gentle mixing at 4 °C for another 15 min. The mixture was centrifuged at 45,000 × g for 30 min at 4 °C. Soluble proteins were applied over nickel-nitrilotriacetic acid beads (Qiagen), washed, and eluted with buffer A with a linear gradient (50 mm- 500 mm) of imidazole.
LPS Extraction
LPS was extracted by the phenol/chloroform/petroleum ether procedure (64) from cultures grown in phosphate-limiting medium and lyophilized. For the LPS analysis, lyophilized material was dispersed in water by sonication and resuspended at a concentration of 2 mg ml−1. For detection of the chemotype, an equivalent portion of whole cell lysate treated with proteinase K was applied to a 16.5% Tricine gel. To detect O-antigen, cultures of BW25113 carrying the vector alone (pRS551) or its derivative carrying the rirA sRNA expressed in pRS551, were transformed with the plasmid pMF19 (to restore the O-antigen biosynthesis). In these experiments, samples were applied on 14% Tricine gel. Gels were silver-stained for the LPS analysis.
Mass Spectrometry
Electrospray ionization-Fourier transform ion cyclotron (ESI-FT-ICR)-mass spectrometry was performed on intact LPS in the negative ion mode using an APEX QE (Bruker Daltonics) equipped with a 7-tesla actively shielded magnet and dual ESI-MALDI. LPS samples were dissolved at a concentration of ∼10 ng μl−1 and analyzed as described previously (6, 19). For the nonspecific fragmentation, the DC offset (collision voltage) of the quadruple interface was set from 5 to 30 V. Under these conditions, the labile linkage between lipid A and the core oligosaccharide is cleaved. Mass spectra were charge deconvoluted, and mass numbers given refer to the monoisotopic peaks.
Author Contributions
S. R. and G. K. designed the study and performed in vitro run-off assays and wrote the manuscript. G. K., A. S., P. W., and D. B. conducted genetic screens, quantified promoter strengths, and purified various proteins used in this study. G. K. purified LPS and B. L. performed MS analysis. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank D. Missiakas, G. Storz, M. Valvano, M. Yamada, and H. Brade for helpful suggestions and D. Clark and H. Mori for the gift of strains. We also acknowledge contributions made by Y. Yun, P. Sulima, A. Przeslakiewicz, J. Barski, D. Polak, P. Gorzelak, and M. Grygorewicz.
This work was supported by National Science Grants 2011/03/B/NZ1/02825 and 2013/11/B/NZ2/02641 (to S. R.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1 and S2.
- IM
- inner membrane
- OM
- outer membrane
- RNAP
- RNA polymerase
- TSS
- transcription start site
- Kdo
- 3-deoxy-α-d-manno-oct-2-ulosonic acid
- P-EtN
- phosphoethanolamine
- l-Ara4N
- 4-amino-4-deoxy-l-arabinose
- ESI-FT-ICR MS
- electrospray ionization Fourier transform-ion cyclotron mass spectrometry
- ops
- operon polarity suppression
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- nt
- nucleotide(s)
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- CRP
- cyclic AMP receptor protein
- sRNA
- small RNA.
References
- 1. Raetz C. R., and Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gronow S., Xia G., and Brade H. (2010) Glycosyltransferases involved in the biosynthesis of the inner core region of different lipopolysaccharides. Eur. J. Cell Biol. 89, 3–10 [DOI] [PubMed] [Google Scholar]
- 3. Raina S., Missiakas D., and Georgopoulos C. (1995) The rpoE gene encoding the σE (σ24) heat shock σ factor of Escherichia coli. EMBO J. 14, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Missiakas D., Betton J. M., and Raina S. (1996) New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21, 871–884 [DOI] [PubMed] [Google Scholar]
- 5. Raina S., and Georgopoulos C. (1991) The htrM gene, whose product is essential for Escherichia coli viability only at elevated temperatures, is identical to the rfaD gene. Nucleic Acids Res. 19, 3811–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klein G., Lindner B., Brabetz W., Brade H., and Raina S. (2009) Escherichia coli K-12 suppressor-free mutants lacking early glycosyltransferases and late acyltransferases: minimal lipopolysaccharide structure and induction of envelope stress response. J. Biol. Chem. 284, 15369–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parker C. T., Kloser A. W., Schnaitman C. A., Stein M. A., Gottesman S., and Gibson B. W. (1992) Role of the rfaG and rfaP genes in determining the lipopolysaccharide core structure and cell surface properties of Escherichia coli K-12. J. Bacteriol. 174, 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Majdalani N., and Gottesman S. (2005) The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59, 379–405 [DOI] [PubMed] [Google Scholar]
- 9. Klein G., Kobylak N., Lindner B., Stupak A., and Raina S. (2014) Assembly of lipopolysaccharide in Escherichia coli requires the essential LapB heat shock protein. J. Biol. Chem. 289, 14829–14853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murata M., Fujimoto H., Nishimura K., Charoensuk K., Nagamitsu H., Raina S., Kosaka T., Oshima T., Ogasawara N., and Yamada M. (2011) Molecular strategy for survival at a critical high temperature in Escherichia coli. PLoS ONE 6, e20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Missiakas D., Mayer M. P., Lemaire M., Georgopoulos C., and Raina S. (1997) Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol. Microbiol. 24, 355–371 [DOI] [PubMed] [Google Scholar]
- 12. De Las Peñas A., Connolly L., and Gross C. A. (1997) The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Mol. Microbiol. 24, 373–385 [DOI] [PubMed] [Google Scholar]
- 13. Missiakas D., and Raina S. (1998) The extracytoplasmic function σ factors: role and regulation. Mol. Microbiol. 28, 1059–1066 [DOI] [PubMed] [Google Scholar]
- 14. Ades S. E. (2008) Regulation by destruction: design of the σE envelope stress response. Curr. Opin. Microbiol. 11, 535–540 [DOI] [PubMed] [Google Scholar]
- 15. Noor R., Murata M., Nagamitsu H., Klein G., Raina S., and Yamada M. (2009) Dissection of σE-dependent cell lysis in Escherichia coli: roles of RpoE regulators RseA, RseB and periplasmic folding catalysts PpiD. Genes Cells 14, 885–899 [DOI] [PubMed] [Google Scholar]
- 16. Rouvière P. E., De Las Peñas A., Mecsas J., Lu C. Z., Rudd K. E., and Gross C. A. (1995) rpoE, the gene encoding the second heat-shock σ factor, σE, in Escherichia coli. EMBO J. 14, 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein G., and Raina S. (2015) Regulated control of the assembly and diversity of LPS by noncoding sRNAs. Biomed. Res. Int. 2015, 153561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Needham B. D., and Trent M. S. (2013) Fortifying the barrier: the impact of lipid A remodeling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klein G., Lindner B., Brade H., and Raina S. (2011) Molecular basis of lipopolysaccharide heterogeneity in Escherichia coli: envelope stress-responsive regulators control the incorporation of glycoforms with a third 3-deoxy-α-d-manno-oct-2-ulosonic acid and rhamnose. J. Biol. Chem. 286, 42787–42807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johansen J., Rasmussen A. A., Overgaard M., and Valentin-Hansen P. (2006) Conserved small non-coding RNAs that belong to the σE regulon: role in down-regulation of outer membrane proteins. J. Mol. Biol. 364, 1–8 [DOI] [PubMed] [Google Scholar]
- 21. Guo M. S., Updegrove T. B., Gogol E. B., Shabalina S. A., Gross C. A., and Storz G. (2014) MicL, a new σE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev. 28, 1620–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coornaert A., Lu A., Mandin P., Springer M., Gottesman S., and Guillier M. (2010) MicA sRNA links the PhoP regulon to cell envelope stress. Mol. Microbiol. 76, 467–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creeger E. S., Schulte T., and Rothfield L. I. (1984) Regulation of membrane glycosyltransferases by the sfrB and rfaH genes of Escherichia coli and Salmonella typhimurium. J. Biol. Chem. 259, 3064–3069 [PubMed] [Google Scholar]
- 24. Marolda C. L., and Valvano M. A. (1998) Promoter region of the Escherichia coli O7-specific lipopolysaccharide gene cluster: structural and functional characterization of an upstream untranslated mRNA sequence. J. Bacteriol. 180, 3070–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burmann B. M., Knauer S. H., Sevostyanova A., Schweimer K., Mooney R. A., Landick R., Artsimovitch I., and Rösch P. (2012) An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell 150, 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto K., Hirao K., Oshima T., Aiba H., Utsumi R., and Ishihama A. (2005) Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280, 1448–1456 [DOI] [PubMed] [Google Scholar]
- 27. Guckes K. R., Kostakioti M., Breland E. J., Gu A. P., Shaffer C. L., Martinez C. R. 3rd, Hultgren S. J., and Hadjifrangiskou M. (2013) Strong cross-system interactions drive the activation of the QseB response regulator in the absence of its cognate sensor. Proc. Natl. Acad. Sci. U.S.A. 110, 16592–16597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reading N. C., Rasko D., Torres A. G., and Sperandio V. (2010) A transcriptome study of the QseEF two-component system and the QseG membrane protein in enterohaemorrhagic Escherichia coli O157:H7. Microbiology 156, 1167–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor W. E., Straus D. B., Grossman A. D., Burton Z. F., Gross C. A., and Burgess R. R. (1984) Transcription from a heat-inducible promoter causes heat shock regulation of the σ subunit of E. coli RNA polymerase. Cell 38, 371–381 [DOI] [PubMed] [Google Scholar]
- 30. Janaszak A., Nadratowska-Wesołowska B., Konopa G., and Taylor A. (2009) The P1 promoter of the Escherichia coli rpoH gene is utilized by σ70-RNAP or σS-RNAP depending on growth phase. FEMS Microbiol. Lett. 291, 65–72 [DOI] [PubMed] [Google Scholar]
- 31. Dartigalongue C., Missiakas D., and Raina S. (2001) Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 276, 20866–20875 [DOI] [PubMed] [Google Scholar]
- 32. Bianchi A. A., and Baneyx F. (1999) Hyperosmotic shock induces the σ32 and σE stress regulons of Escherichia coli. Mol. Microbiol. 34, 1029–1038 [DOI] [PubMed] [Google Scholar]
- 33. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., and Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., and Mori H. (2005) Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299 [DOI] [PubMed] [Google Scholar]
- 35. Missiakas D., Georgopoulos C., and Raina S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 7084–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Santos J. M., Freire P., Mesquita F. S., Mika F., Hengge R., and Arraiano C. M. (2006) Poly(A)-polymerase I links transcription with mRNA degradation via σS proteolysis. Mol. Microbiol. 60, 177–188 [DOI] [PubMed] [Google Scholar]
- 37. Zhou Z., Lin S., Cotter R. J., and Raetz C. R. (1999) Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-l-arabinose, phosphoethanolamine and palmitate. J. Biol. Chem. 274, 18503–18514 [DOI] [PubMed] [Google Scholar]
- 38. Tam C., and Missiakas D. (2005) Changes in lipopolysaccharide structure induce the σE-dependent response of Escherichia coli. Mol. Microbiol. 55, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 39. Klein G., Müller-Loennies S., Lindner B., Kobylak N., Brade H., and Raina S. (2013) Molecular and structural basis of inner core lipopolysaccharide alterations in Escherichia coli: incorporation of glucuronic acid and phosphoethanolamine in the heptose region. J. Biol. Chem. 288, 8111–8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feklistov A., and Darst S. A. (2011) Structural basis for promoter-10 element recognition by the bacterial RNA polymerase σ subunit. Cell 147, 1257–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Syed A., and Gralla J. D. (1997) Isolation and properties of enhancer-bypass mutants of σ54. Mol. Microbiol. 23, 987–995 [DOI] [PubMed] [Google Scholar]
- 42. Reichenbach B., Göpel Y., and Görke B. (2009) Dual control by perfectly overlapping σ54- and σ70-promoters adjusts small RNA GlmY expression to different environmental signals. Mol. Microbiol. 74, 1054–1070 [DOI] [PubMed] [Google Scholar]
- 43. Xiao J., Chen T., Yang M., Zhang Y., and Wang Q. (2012) Identification of qseGF genetic locus and its roles in controlling hemolytic activity and invasion in fish pathogen Edwardsiella tarda. Lett. Appl. Microbiol. 55, 91–98 [DOI] [PubMed] [Google Scholar]
- 44. Reading N. C., Rasko D. A., Torres A. G., and Sperandio V. (2009) The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 5889–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muffler A., Fischer D., Altuvia S., Storz G., and Hengge-Aronis R. (1996) The response regulator RssB controls stability of the σS subunit of RNA polymerase in Escherichia coli. EMBO J. 15, 1333–1339 [PMC free article] [PubMed] [Google Scholar]
- 46. Pratt L. A., and Silhavy T. J. (1996) The response regulator SprE controls the stability of RpoS. Proc. Natl. Acad. Sci. U.S.A. 93, 2488–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Madhugiri R., Basineni S. R., and Klug G. (2010) Turn-over of the small non-coding RNA RprA in E. coli is influenced by osmolarity. Mol. Genet. Genomics 284, 307–318 [DOI] [PubMed] [Google Scholar]
- 48. Pesavento C., and Hengge R. (2012) The global repressor FliZ antagonizes gene expression by σS-containing RNA polymerase due to overlapping DNA binding specificity. Nucleic Acids Res. 40, 4783–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yethon J. A., Vinogradov E., Perry M. B., and Whitfield C. (2000) Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J. Bacteriol. 182, 5620–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Whitfield C., and Trent M. S. (2014) Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 83, 99–128 [DOI] [PubMed] [Google Scholar]
- 51. Zhou Z., Ribeiro A. A., and Raetz C. R. (2000) High-resolution NMR spectroscopy of lipid A molecules containing 4-amino-4-deoxy-l-arabinose and phosphoethanolamine substituents. Different attachment sites on lipid A molecules from NH4VO3-treated Escherichia coli versus kdsA mutants of Salmonella typhimurium. J. Biol. Chem. 275, 13542–13551 [DOI] [PubMed] [Google Scholar]
- 52. Feldman M. F., Marolda C. L., Monteiro M. A., Perry M. B., Parodi A. J., and Valvano M. A. (1999) The activity of a putative polyisoprenol-linked sugar translocase (Wzx) involved in Escherichia coli O antigen assembly is independent of the chemical structure of the O repeat. J. Biol. Chem. 274, 35129–35138 [DOI] [PubMed] [Google Scholar]
- 53. Tao K., Narita S., and Tokuda H. (2012) Defective lipoprotein sorting induces lolA expression through the Rcs stress response phosphorelay system. J. Bacteriol. 194, 3643–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Browning D. F., and Busby S. J. (2004) The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65 [DOI] [PubMed] [Google Scholar]
- 55. Sikdar R., Simmons A. R., and Doerrler W. T. (2013) Multiple envelope stress response pathways are activated in an Escherichia coli strain with mutations in two members of the DedA membrane protein family. J. Bacteriol. 195, 12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gerken H., Charlson E. S., Cicirelli E. M., Kenney L. J., and Misra R. (2009) MzrA: a novel modulator of the EnvZ/OmpR two-component regulon. Mol. Microbiol. 72, 1408–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lima S., Guo M. S., Chaba R., Gross C. A., and Sauer R. T. (2013) Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 340, 837–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miyadai H., Tanaka-Masuda K., Matsuyama S., and Tokuda H. (2004) Effects of lipoprotein overproduction on the induction of DegP (HtrA) involved in quality control in the Escherichia coli periplasm. J. Biol. Chem. 279, 39807–39813 [DOI] [PubMed] [Google Scholar]
- 59. Babu M., Díaz-Mejía J. J., Vlasblom J., Gagarinova A., Phanse S., Graham C., Yousif F., Ding H., Xiong X., Nazarians-Armavil A., Alamgir M., Ali M., Pogoutse O., Pe'er A., Arnold R., et al. (2011) Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 7, e1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Torriani A. (1960) Influence of inorganic phosphate in the formation of phosphatases by Escherichia coli. Biochim. Biophys. Acta 38, 460–469 [DOI] [PubMed] [Google Scholar]
- 61. Klein G., Dartigalongue C., and Raina S. (2003) Phosphorylation-mediated regulation of heat shock response in Escherichia coli. Mol. Microbiol. 48, 269–285 [DOI] [PubMed] [Google Scholar]
- 62. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Simons R. W., Houman F., and Kleckner N. (1987) Improved single and multicopy lac-based cloning vectors for protein and operon fusion. Gene 53, 85–96 [DOI] [PubMed] [Google Scholar]
- 64. Galanos C., Lüderitz O., and Westphal O. (1969) A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 9, 245–249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.