

Abstract
Bile acid sequestrants are synthetic polymers that bind bile acids in the gut and are used to treat dyslipidemia and hyperphosphatemia. Recently, these agents have been reported to lower blood glucose and increase insulin sensitivity by altering bile acid signaling pathways. In this study, we assessed the efficacy of sevelamer in treating mice with non-alcoholic fatty liver disease (NAFLD). We also analyzed how sevelamer alters inflammation and bile acid signaling in NAFLD livers. Mice were fed a low-fat or Western diet for 12 weeks followed by a diet-plus-sevelamer regimen for 2 or 12 weeks. At the end of treatment, disease severity was assessed, hepatic leukocyte populations were examined, and expression of genes involved in farnesoid X receptor (FXR) signaling in the liver and intestine was analyzed. Sevelamer treatment significantly reduced liver steatosis and lobular inflammation. Sevelamer-treated NAFLD livers had notably fewer pro-inflammatory infiltrating macrophages and a significantly greater fraction of alternatively activated Kupffer cells compared with controls. Expression of genes involved in FXR signaling in the liver and intestine was significantly altered in mice with NAFLD as well as in those treated with sevelamer. In a mouse model of NAFLD, sevelamer improved disease and counteracted innate immune cell dysregulation in the liver. This study also revealed a dysregulation of FXR signaling in the liver and intestine of NAFLD mice that was counteracted by sevelamer treatment.
Keywords: bile acid, inflammation, innate immunity, macrophage, obesity, bile acid sequestrants, FXR, NAFLD, sevelamer, steatohepatitis
Introduction
The epidemic proportions of obesity in the United States have resulted in non-alcoholic fatty liver disease (NAFLD)2 becoming a serious healthcare burden. Recent estimates of nationwide disease prevalence have been as high as 37% with the mean around 30% (1, 2). This makes NAFLD the most common cause of chronic liver disease in the United States (3). The high prevalence of NAFLD is alarming because people with NAFLD and comorbidities such as type 2 diabetes mellitus, hypertension, and obesity are at serious risk of developing liver fibrosis and even cirrhosis (4–6). Given the risks and prevalence, it is no wonder that NAFLD is projected to be the leading indication for liver transplant by the year 2020 (7).
Bile acid sequestrants are non-absorbable, cationic polymers that bind bile acids as well as a host of molecules in the gut and prevent their reabsorption. They are approved by the Food and Drug Administration for the treatment of dyslipidemia and hyperphosphatemia (8–10). Beyond their ability to prevent bile acid and phosphate absorption in the gut, bile acid sequestrants have been reported to modulate fat and glucose metabolism by activating TGR5, a G protein-coupled receptor that binds bile acids (11–13). Recently, it has been discovered that bile acid sequestrants also improve insulin sensitivity and reduce obesity and liver steatosis in humans and mice (11, 14–16).
In recent years, convincing evidence has surfaced that farnesoid X receptor (FXR) plays an important role in modulating inflammation caused by metabolic stress (e.g. dyslipidemia, overfeeding). However, it is unclear exactly what that role is. There is evidence that FXR signaling can be beneficial or detrimental depending on the setting. In one study, Ldlr−/−Fxr−/− mice fed a high-fat diet developed more severe liver inflammation and hepatocyte damage compared with Ldlr−/− mice (17). However, in another study, overexpression of FXR sensitized mice to cholesterol-mediated liver damage (18). Further complicating the matter is the recent finding that FXR signaling may behave differently depending on the specific tissue in which it is expressed. FXR agonism in the liver has been shown to improve NAFLD inflammation (19), but, paradoxically, antagonism of small intestine-specific FXR also improves NAFLD (20).
In this study, we evaluated the effects that sevelamer has on NAFLD mouse livers. We assessed how sevelamer treatment affects immune cell infiltration and the phenotype of macrophages in the liver. Furthermore, we investigated how Western diet feeding and sevelamer affect FXR signaling in the liver and intestine. We report that sevelamer treatment improves NAFLD liver pathology and reduces inflammation and innate leukocyte dysregulation, shifting Kupffer cells toward a non-inflammatory, reparative phenotype. Furthermore, our results suggest that Western diet feeding overactivates FXR in the liver and intestine and that sevelamer counteracts this phenomenon.
Results
Western Diet Feeding Induces NAFLD Pathology and Pro-inflammatory Transcriptional Changes That Are Improved with Long-term Sevelamer Treatment
To better understand the effects of oral sevelamer on diet-induced steatohepatitis, we established a mouse model of obesity by feeding a group of C57BL/6 mice a Western diet (WD) for an extended period of time (a total of 24 weeks, see “Experimental Procedures”), whereas a control group was fed a low-fat (LF), low-sucrose diet. Western diet feeding caused a significant increase in serum alanine aminotransferase (ALT) (Fig. 1D) as well as severe increases in mean body weight (↑33%) and liver weight (↑261%) compared with LF mice (Fig. 1B). Furthermore, analysis of H&E-stained liver sections of WD mice revealed marked changes consistent with NAFLD (Fig. 1A). Liver steatosis and lobular inflammation scores were significantly increased in WD mice compared with controls (Fig. 1C). Sevelamer treatment in these long-term WD-fed mice (WDS) improved the histology and body and liver weight as well as liver steatosis and inflammation (Fig. 1, A–C).
FIGURE 1.

Sevelamer ameliorates NAFLD liver pathology in mice fed a Western diet. A, H&E-stained liver sections of LF, LFS, WD, and WDS mice. WD livers demonstrated macrovesicular steatosis (blue arrow), microvesicular steatosis (circled region), and prominent inflammatory foci (red arrow). B, average body and liver weights for each treatment group. C, average semiquantitative score of liver steatosis and lobular inflammation for each treatment group. D, average serum ALT per group. Each column represents the mean ± S.E. of 8–10 mice/group. *, p < 0.05.
Supporting the histologic analyses, transcriptional changes in pro-inflammatory and pro-fibrotic genes were also caused by Western diet feeding. As expected, WD mice had markedly higher hepatic expression of the pro-inflammatory cytokines interleukin 1 β (Il1b), tumor necrosis factor α (Tnf), chemokine (C-C motif) ligand 2 (Ccl2), and interleukin 6 (Il6; Fig. 2, A–D), all of which have been implicated in NAFLD pathogenesis (23–27). These cytokines and chemokines recruit and activate innate immune cells and drive pro-fibrotic processes. WD mice also had higher hepatic expression of the pro-fibrotic genes transforming growth factor β 1 (Tgfb1) and α smooth muscle actin (α-SMA; Acta2) compared with controls (LF mice; Fig. 2, E and F). Tgfb1 is a mediator of stellate cell activation in the liver, whereas α-SMA is expressed by activated stellate cells (28).
FIGURE 2.

Sevelamer treatment reduces hepatic expression of pro-inflammatory and pro-fibrotic genes. Quantitative RT-PCR performed on whole liver tissue taken from LF, LFS, WD, and WDS mice confirmed that sevelamer treatment in diet-induced obese mice had significant anti-inflammatory (A–D) and anti-fibrotic effects (E and F). Each column represents mean -fold expression ± S.E. of 5–7 mice/group. *, p < 0.05.
After 12 weeks of diet feeding, WD and LF mice were treated with sevelamer or no drug, creating four treatment groups: WD, WDS, LF diet, and low fat diet plus sevelamer (LFS). The treatment phase lasted an additional 12 weeks, after which we assessed the ability of sevelamer to improve disease. We found that sevelamer treatment in Western diet-fed mice significantly improved obesity and disease by multiple measures. On average, WDS mice had a 61% lower body weight and 76% lower liver weight than WD mice (Fig. 1B). In fact, the body and liver weights of WDS mice were nearly at control levels (LF mice). Pathology scoring revealed that liver steatosis and lobular inflammation were significantly reduced in WDS mice compared with WD mice (Fig. 1C). Although some steatosis persisted in the WDS treatment group, both macro- and microvesicular steatosis were significantly reduced (Fig. 1A). Consistent with the histology findings, average serum ALT was significantly lower in WDS mice compared with Western diet controls (Fig. 1D).
Gene expression analysis showed that WDS mice had much lower expression of pro-inflammatory and pro-fibrotic genes than their WD counterparts (Fig. 2, A–F). Indeed, it is worth noting that expression levels of Il1b, Tnf, Il6, Ccl2, Tgfb1, and Acta2 (α-SMA) in WDS mouse liver were not significantly higher than in LF mouse liver (Fig. 2, A–F).
Sevelamer Inhibits Activation of Liver and Intestinal FXR in NAFLD Mice
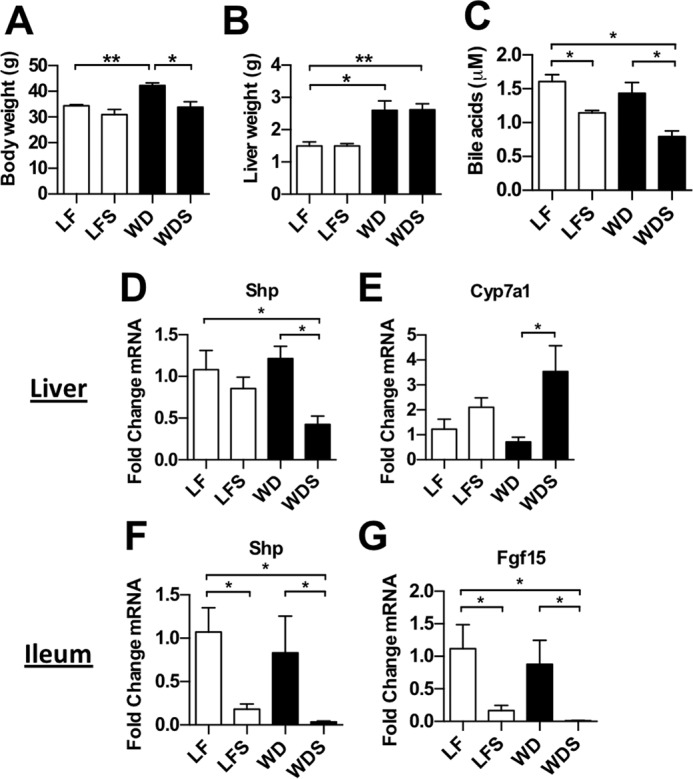
Bile acid synthesis is controlled by FXR-mediated negative feedback in the intestine and liver (29). Bile acids, the natural ligands for FXR, are reabsorbed in the terminal ileum where they activate FXR and induce FGF15 transcription (Fig. 3A). This protein is released into the portal blood, where it travels to the liver and inhibits transcription of CYP7A1 (Fig. 3A), the rate-limiting enzyme for bile acid synthesis from cholesterol (29). Reabsorbed bile acids can also travel through the portal vein and activate FXR in hepatocytes (Fig. 3A). FXR dimerizes with the nuclear receptor retinoid X receptor and induces transcription of many genes, including the inhibitory nuclear receptor small heterodimer partner (SHP). SHP, in turn, dimerizes with specific nuclear factors and prevents transcription of various genes, including CYP7A1 (29).
FIGURE 3.

Liver FXR signaling is dysregulated in NAFLD mice, and sevelamer counteracts this effect. A, enterohepatic circulation of bile acids, liver and intestinal targets of bile acids, and the role of bile acid sequestrants such as sevelamer. FXR induces Shp and Fgf15/19 transcription and inhibits Cyp7a1 transcription via SHP. B and C, liver expression of Shp and Cyp7a1 in LF, LFS, WD, and WDS mice. D and E, ileal expression of Shp and Fgf15. Columns represent mean -fold expression ± S.E. of 5–7 mice/group. *, p < 0.05.
Sevelamer acts by binding bile acids in the gut, preventing their reabsorption in the terminal ileum, and alleviating the negative feedback on bile acid synthesis (Fig. 3A). We investigated the effect of sevelamer on hepatic FXR signaling in the setting of NAFLD. Long-term WD-fed mice had markedly elevated hepatic Shp expression compared with LF controls, implying that liver FXR signaling was highly active in these mice (Fig. 3B). However, expression of liver Shp in the sevelamer-treated mice (WDS) was not significantly higher than in LF controls (Fig. 3B), suggesting that sevelamer treatment normalizes FXR signaling in NAFLD livers. The expression of Cyp7a1, which controls bile acid synthesis from cholesterol, was significantly up-regulated in both low-fat- and WD-fed mice that were given sevelamer (Fig. 3C). This increase in Cyp7a1 transcription is consistent with the Shp transcriptional data and implies that sevelamer inhibits liver FXR signaling. Supporting the liver data, Western diet feeding induced Shp overexpression in the ileum, whereas ileal Shp and Fgf15 gene expression was suppressed by sevelamer treatment (Fig. 3, D and E).
To better understand the kinetics of bile acid signaling changes induced by sevelamer and to determine whether these changes were due to an epiphenomenon, we analyzed bile acid gene expression at a time point when NAFLD pathology was still present (i.e. 12 weeks of diet followed by 2 weeks of diet with or without sevelamer). Unlike the long-term mice, these short-term mice did not demonstrate significantly higher levels of Shp in the WD diet group compared with the LF controls. This is likely due to the fact that WD mice in the long-term treatment study were fed for 10 weeks longer. Nonetheless, by week 2 of treatment, sevelamer had down-regulated hepatic Shp and increased Cyp7a1 expression (Fig. 4, D and E). Similarly, expression of Shp and Fgf15 in the terminal ileum of WDS mice was significantly lower than in WD mice (Fig. 4, F and G). No such transcriptional effect was observed in the colon (data not shown). Next we measured bile acid concentration in the portal vein and found that sevelamer-treated mice had lower levels of total bile acids than their non-treated counterparts (Fig. 4C), consistent with the mechanisms of action described above. Collectively, these data imply that chronic WD feeding induces overactivation of FXR in the liver, which is alleviated by sevelamer treatment, and that, as early as 2 weeks after initiation of sevelamer treatment, bile acid concentrations decrease in the portal vein and FXR signaling changes occur within the terminal ileum and liver.
FIGURE 4.
Effects of short-term sevelamer treatment on western diet mice. A–C, body weight, liver weight, and total bile acid concentration in the portal vein of short-term treatment mice. D and E, expression of Shp and Cyp7a1 in whole liver of short-term treatment mice. F and G, expression of Shp and Fgf15 in the terminal ileum of short-term treatment mice. Columns represent mean weight, concentration, or -fold expression ± S.E. of 4 mice/group. *, p < 0.05; **, p < 0.01.
Hepatic Innate Immune Cell Dysregulation Is Induced by Western Diet Feeding and Reversed by Sevelamer
To better characterize changes in the liver produced by Western diet feeding and sevelamer treatment, we used flow cytometry to analyze intrahepatic cells isolated from mice in each treatment group. These analyses revealed a pattern of innate immune cell dysregulation in NAFLD livers, with the most striking changes occurring in hepatic macrophages. Recent studies in macrophage ontogeny have identified two populations of macrophages in mouse livers: a tissue-resident population that is derived embryonically and an infiltrating population that arises from bone marrow-derived monocytes after birth (30, 31). These two populations are distinguished based on their level of expression of F4/80 and CD11b. Tissue-resident macrophages are defined as F4/80hiCD11blow, whereas infiltrating macrophages are defined as F4/80lowCD11bhi. In this study, we refer to the tissue-resident population as Kupffer cells (KCs) and the infiltrating population as infiltrating macrophages (IMs). We used flow cytometry to study these two populations (see gating strategy, Fig. 5A), and identified differences in KC and IM population size and phenotype between treatment and control mice in the long-term study. WD mouse livers had a significantly higher percentage of IMs than LF controls (Fig. 5, B and F); however, no significant difference in the number of KCs was observed (data not shown). These results are consistent with other studies that have reported an increase in liver IMs in the setting of NAFLD (32). Phenotype changes in both KC and IM populations were also observed in WD mice compared with LF controls. The KC population in WD mice had a significantly lower mean fluorescence intensity of the M2 marker CD206 (Fig. 5, E and J), whereas the IM population had higher levels of the pro-inflammatory markers Ly6C and CD86. The percentage of Ly6Chi-positive (Fig. 5, C and G) and CD86-positive (Fig. 5, D and I) cells in the IM population was significantly higher in WD mice compared with low-fat controls. There was also a concomitant decrease in the percentage of Ly6Clow IMs (Fig. 5, C, G, and H). No significant change in Ly6mid IMs was observed (data not shown). Ly6Chi monocytes are known to differentiate into M1-like macrophages, Ly6Clow monocytes become M2-like macrophages, and Ly6Cmid monocytes, although poorly characterized, display an intermediate phenotype (19, 33, 34). CD86 is involved in antigen presentation and a commonly used marker of M1 macrophages (35). These data demonstrate that Western diet feeding causes KCs to lose their M2-like phenotype and induces a large influx of Ly6Chi monocytes that differentiate into pro-inflammatory macrophages.
FIGURE 5.

Sevelamer reduces hepatic macrophage infiltration and alters macrophage phenotype. A, gating strategy for identifying KCs and IMs. KCs were defined as CD45+Ly6G−F4/80hiCD11blow, and IMs were defined as CD45+Ly6G−F4/80lowCD11bhi. B and F, representative plots and percent positive IMs (of CD45+ cells) in LF, WD, and WDS mouse livers. C, G, and H, representative histograms and percent positive Ly6Chi and Ly6Clow cells (of total IMs). D and I, representative histograms and percent positive CD86 cells (of total IMs) in LF, WD, and WDS mouse livers. E and J, histograms and quantification of CD206 fluorescence in the KC population of LF, WD, and WDS livers. E, black lines represent the mean fluorescence intensity (MFI) of CD206. Columns represent the mean ± S.E. of 5–7 mice/group. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Notably, Western diet feeding also induced changes in intrahepatic natural killer (NK) and natural killer T (NKT) cell numbers. WD mouse livers had significantly higher numbers of NK cells than LF controls (Fig. 6, B and D). NK cells have been reported to play dual roles in NAFLD pathogenesis, contributing directly to hepatocyte apoptosis as well as preventing fibrosis by killing early-activated hepatic stellate cells (33). In this study, hepatic NK cell accumulation occurred, but no significant hepatocellular death was detected by histological analysis (data not shown), which supports the theory that NK cells protect against NAFLD progression. In contrast, hepatic NKT cells (defined as CD1d tetramer-positive) were also depleted in WD mice (Fig. 6, A and C). This phenomenon is consistent with reports from other animal models of diet-induced NAFLD. Activated liver macrophages cause NKT cell apoptosis in an IL-12 dependent manner, and this depletion contributes to liver steatosis (34).
FIGURE 6.

Sevelamer restores NKT cell populations and reduces NK cell accumulation in NAFLD livers. A and B, quantification of NKT and NK cells in LF, LFS, WD, and WDS livers. C and D, representative plots of NK (CD45+CD3−B220−NK1.1+) and NKT (CD45+CD3intCD1d+) cell populations in LF, WD, and WDS mouse livers. Columns represent mean percent positive (of CD45+ cells) ± S.E. of 5–7 mice/group. *, p < 0.05.
Although the effect of Western diet feeding on hepatic innate immune cells was profound, the effect of sevelamer treatment on them was equally so. Treatment reversed liver macrophage infiltration and phenotype change caused by Western diet feeding. Intrahepatic cells from WDS mice had a significantly lower percentage of IMs than WD mice (Fig. 5, B and F). This reduction in IMs was accompanied by a corresponding reduction in the percentage of Ly6Chi-positive (Fig. 5, C and G) and CD86-positive IMs (Fig. 5, D and I) as well as an increase in the percentage of Ly6Clow IMs (Fig. 5, C and H). KCs in WDS mice had significantly higher CD206 fluorescence than WD mice (Fig. 5, E and J). Notably, the averages of IM percentage, CD86 percentage, Ly6Clow percentage, and CD206 fluorescence for WDS mouse samples were not significantly different from low-fat controls. Treatment also restored NK and NKT cells to LF control levels. WDS mouse livers had lower numbers of NK cells (Fig. 6, B and D) and higher numbers of NKT cells (Fig. 6, A and C) than WD mouse livers. These data demonstrate that sevelamer reverses the dysregulation of liver innate immune cells caused by Western diet feeding. Using direct ex vivo analyses of mice given short-term sevelamer treatment, we found a trend toward decreasing numbers of infiltrating macrophages and Ly6Chi monocytes as early as 2 weeks after treatment initiation despite the fact that there was no histologic improvement at that time point (data not shown).
Discussion
Bile acid sequestrants may have therapeutic benefits for NAFLD (11–14). They are commercially available drugs with few side effects and are already indicated for use in patients at risk for NAFLD (i.e. those with dyslipidemia and chronic kidney disease). However, we felt that further interrogation of the mechanistic effects of bile acid sequestrants in NAFLD was warranted. In keeping with other reports (35–39), we have found that WD feeding in a murine model induced histologic and transcriptional changes consistent with NAFLD, and sevelamer consistently reversed many of these transcriptional and phenotypic changes. In accordance with our published data and those of other laboratories (19, 40), we have shown that NAFLD is characterized by a large hepatic influx of Ly6Chi monocytes that differentiate into pro-inflammatory macrophages and a relative loss of Kupffer cells with an M2-like phenotype. For the first time, however, we provide evidence that sevelamer ameliorates the hepatic innate immune response by disrupting bile acid homeostasis.
We focused specifically on FXR signaling because of its central role in bile acid synthesis and enterohepatic circulation in the intestine and liver (41). We found that mice in the long-term study that received a Western diet 10 weeks longer than short-term mice demonstrated significant induction of Shp relative to low-fat controls. Furthermore, sevelamer inhibited FXR signaling in the liver and terminal ileum. Hepatic and ileal expression of Shp was not significantly different between Western diet and low-fat mice in the short-term study. However, after only 2 weeks, the effects of sevelamer treatment became apparent with relative down-regulation of Shp and Fgf15 despite the fact that WDS mouse livers were still enlarged, steatotic, and inflamed (Fig. 4, A and B). Thus, the short-term data show that the effect of sevelamer on FXR signaling was not simply an epiphenomenon.
Interestingly, our data suggest that, in the absence of overnutrition, sevelamer may cause mild inflammation. Mice consuming an LFS diet had elevated expression of Il1b, Il6, and Tgfb1 in the liver (Fig. 2). These data suggest that bile acid sequestrants may be used most effectively when given to NAFLD patients who have difficulty altering their diet.
We acknowledge that there are additional bile acid signaling pathways whereby bile acid sequestrants may improve obesity-caused liver disease. Recent studies have shown that bile acids bound to sequestrants improve glycemic control in obese mice by activating TGR5 in the terminal ileum (12, 42), and, because of their mechanism of action, bile acid sequestrants could potentially modulate liver X receptor signaling by altering oxysterol homeostasis (43). However, we analyzed hepatic expression of certain liver X receptor target genes (e.g. Abca1, Abcg1, ApoD) in long-term treatment mice and found that, although Western diet feeding tended to induce expression of these genes, sevelamer treatment did not significantly affect their expression levels (data not shown).
Taken as a whole, our data suggest that sevelamer also inhibits FXR signaling by binding bile acids in the intestinal lumen and causing less reabsorption in the terminal ileum. This decrease in uptake of bile acids results in lower FXR activation in ileum and liver (Figs. 3 and 4). It is worth noting, however, that inhibition of FXR in the liver may have both beneficial and pro-inflammatory effects (19, 44). Aside from hepatocytes, macrophages and sinusoidal endothelial cells are the other major cell types that express FXR in the liver. Macrophages are particularly interesting because FXR has been shown to suppress inflammation in these cells (45). Thus, inhibiting FXR in liver macrophages would be detrimental. This may explain why mice receiving an LFS diet had low-grade liver inflammation (Figs. 1 and 2) and why mice receiving a WDS diet had no significant improvement in inflammation or steatosis compared with WD mice at week 2 of treatment despite losing a significant amount of body weight (Fig. 4, A and B).
Bile acid sequestrants are known to bind a host of molecules, including sterols, phosphates, and fatty acids (8). Thus, it is also possible that the mechanism by which sevelamer improves NAFLD is not limited to the modulation of bile acid signaling alone. However, in support of other studies (12, 13), we conclude that the effects of bile acid sequestrants are at least in part due to their ability to alter bile acid signaling. We have shown that sevelamer improves NAFLD pathology and inhibits FXR signaling in the liver and terminal ileum. Although FXR activation is decidedly anti-inflammatory in immune cells like macrophages, recent reports suggest that inhibition of FXR in the intestine may be beneficial (20, 44). Furthermore, recent evidence that overexpression of FXR in the liver sensitizes it to diet-induced injury (18) implies that alleviation of chronic FXR activation in the liver may also be beneficial in the setting of NAFLD. Clearly, further study is necessary to better understand the role FXR signaling plays in NAFLD.
Experimental Procedures
Mouse Diets and Feeding Regimen
Both the Western and low-fat diets were purchased from Envigo (formerly Harlan). The Western diet (TD.88137) contained 42% kcal from fat (>60% saturated) and 34% sucrose by weight. The low-fat diet (TD.08485) contained 13% kcal from fat and 12% sucrose by weight. Sevelamer carbonate (Renvela, obtained from Genzyme) was mixed into powdered formulations of the Western and low-fat diets. The sevelamer-containing diets were 2% by weight. Male C57BL/6 mice that were 6–8 weeks old were fed a Western or low-fat diet for 12 weeks to establish NAFLD. They were then fed diet plus or minus drug for an additional 2 (short-term study) or 12 weeks (long-term study). Thus, in each study, there were four distinct treatment groups: WD, WDS, LF, and LFS. The long-term cohort contained 8–10 mice/group, whereas the short-term contained 4 mice/group.
Liver Histology
Liver tissue was formalin-fixed, paraffin-embedded, sectioned, and H&E-stained using standard procedures. Blinded liver sections were scored for NAFLD severity as described previously (21). Briefly, each section was given a score for steatosis (0–3), fibrosis (0–5), lobular inflammation (0–3), and liver cell injury (0–3). For the steatosis and inflammation metrics, the average of all scores from each treatment group (i.e. LF, LFS, WD, and WDS) was displayed in chart form (Fig. 1C).
Intrahepatic Cell Isolation
For the long-term study, fresh liver tissue was homogenized, digested with collagenase medium (0.02% collagenase IV) for 1 h at 37 °C, and washed with RPMI 1640 (Invitrogen) plus 10% FBS (HyClone). Red blood cells in the sample were lysed using 0.16 m NH4Cl and 0.17 m Tris (pH 7.65), followed by another wash and filtration through a 70-μm filter. When not being used immediately, intrahepatic cells were suspended in FBS plus 10% DMSO and stored in liquid nitrogen. For the short-term study, livers were perfused with PBS prior to cell isolation. Briefly, the inferior vena cava was severed, a 25-gauge needle was inserted into the portal vein, and sterile PBS was pumped through the liver at a flow rate of 3 ml/min. Following perfusion, liver tissue was immediately homogenized and digested as described above. Non-parenchymal cells were then isolated from homogenate using OptiprepTM density gradient medium (Sigma-Aldrich). When not being used immediately, intrahepatic cells were suspended in FBS plus 10% DMSO and stored in liquid nitrogen.
Statistical Analysis
Results are expressed as the mean ± S.E. Mann-Whitney U test was used to compare quantitative PCR data from each treatment group, and unpaired Student's t test was used to compare data from all other experiments. Prism 6.0 statistical analysis software (GraphPad Software) was used to perform the analysis, and a p value of 0.05 or less was considered significant.
Flow Cytometry
Fluor-conjugated antibodies directed against the following surface antigens were used: CD45 (30-F11, BD Biosciences), CD3 (17A2, eBioscience), NK1.1 (PK136, eBioscience), Ly6G (RB6–8C5, eBioscience), Ly6C (AL-21, BD Bioscience), CD11b (M1/70, BD Biosciences), and F4/80 (BM8, eBioscience). One antibody directed against an intracellular antigen was used: CD206 (MCA2235A647, AbD Serotec). Cells were stained for 30 min at 4 °C, washed twice (1% BSA and 0.01% sodium azide in PBS), and fixed in 200 μl of 1% paraformaldehyde (Sigma-Aldrich). When performing intracellular staining, cells were fixed and permeabilized using Fix & Perm® reagents and protocols (Invitrogen). Flow cytometry was performed using a BD FACSCanto II instrument, and data were analyzed with BD FACSDiva software (BD Biosciences).
Quantitative PCR
Fresh liver tissue was snap-frozen and stored in liquid nitrogen until use. Frozen tissue was homogenized in Buffer RLT (Qiagen) plus 1% β-mercaptoethanol (Invitrogen) using a Tissue TearorTM (Biospec, Inc.). Total RNA was then isolated from cell lysate using the RNeasy mini kit (Qiagen), and cDNA was synthesized using the QuantiTect RT kit (Qiagen) following standard protocols. The following Qiagen QuantiTect primers were used: β glucuronidase (Gusb, QT00176715), 18S ribosomal RNA (Rn18s, QT02448075), interleukin 1 β (Il1b, QT01048355), tumor necrosis factor α (Tnf, QT00104006), chemokine (C-C motif) ligand 2 (Ccl2, QT00167832), interleukin 6 (Il6, QT00098875), transforming growth factor β 1 (Tgfb1, QT00145250), α-smooth muscle actin (Acta2, QT00140119), fibroblast growth factor 15 (Fgf15, QT00102137), small heterodimer partner (Shp, QT00319333), and cytochrome P450 family 7 subfamily a polypeptide 1 (Cyp7a1, QT00319333). Samples were run in triplicate 10-μl reactions composed of 5 μl of SYBR Green PCR mixture (Qiagen), 1 μl of primer pair, 3.5 μl of H2O, and 0.5 μl of cDNA. Quantitative PCR was performed on an Applied Biosystems 7300 real-time PCR machine, and 40 amplification cycles were run as follows: 94 °C for 15 s, 50 °C for 30 s, and 72 °C for 30 s. -Fold changes in mRNA levels were calculated as described previously (22). Gusb was used as the reference gene for Tnf, Ccl2, and Acta2 calculations, whereas Rn18s was used as the reference gene for Il1b, Tgfb1, and Il6 calculations. For each gene, all samples were normalized to the average -fold change of the LF treatment group.
Author Contributions
B. M. M. performed experiments, data interpretation, and manuscript preparation and review. R. H. M. was involved in study concept and design, data acquisition and interpretation, and manuscript revision. Y. L., X. X. W., and C. P. participated in data acquisition and interpretation. D. J. O. performed semiquantitative analysis of liver histology. M. L. and H. R. R. originally conceived the study concept and design. M. L. also participated in manuscript revision. H. R. R. supervised the study and participated in manuscript preparation and revision.
This work was supported, in whole or in part, by a Veterans Affairs Merit Review grant (to H. R. R.) and National Institute of Health Grant 5K01DK096025 (to R. H. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NAFLD
- non-alcoholic fatty liver disease
- FXR
- farnesoid X receptor
- WD
- Western diet
- LF
- low-fat
- WDS
- Western diet plus sevelamer
- LFS
- low-fat plus sevelamer
- ALT
- alanine aminotransferase
- α-SMA
- α-smooth muscle actin
- SHP
- small heterodimer partner
- KC
- Kupffer cell
- IM
- infiltrating macrophage
- NK
- natural killer
- NKT
- natural killer T.
References
- 1. Vernon G., Baranova A., and Younossi Z. M. (2011) Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 34, 274–285 [DOI] [PubMed] [Google Scholar]
- 2. Levene A. P., and Goldin R. D. (2012) The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 61, 141–152 [DOI] [PubMed] [Google Scholar]
- 3. Wree A., Broderick L., Canbay A., Hoffman H. M., and Feldstein A. E. (2013) From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 10, 627–636 [DOI] [PubMed] [Google Scholar]
- 4. Hossain N., Afendy A., Stepanova M., Nader F., Srishord M., Rafiq N., Goodman Z., and Younossi Z. (2009) Independent predictors of fibrosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 7, 1224–1229, 1229.e1–2 [DOI] [PubMed] [Google Scholar]
- 5. Ratziu V., Giral P., Charlotte F., Bruckert E., Thibault V., Theodorou I., Khalil L., Turpin G., Opolon P., and Poynard T. (2000) Liver fibrosis in overweight patients. Gastroenterology 118, 1117–1123 [DOI] [PubMed] [Google Scholar]
- 6. Argo C. K., Northup P. G., Al-Osaimi A. M., and Caldwell S. H. (2009) Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 51, 371–379 [DOI] [PubMed] [Google Scholar]
- 7. Charlton M. R., Burns J. M., Pedersen R. A., Watt K. D., Heimbach J. K., and Dierkhising R. A. (2011) Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 141, 1249–1253 [DOI] [PubMed] [Google Scholar]
- 8. Braunlin W., Zhorov E., Guo A., Apruzzese W., Xu Q., Hook P., Smisek D. L., Mandeville W. H., and Holmes-Farley S. R. (2002) Bile acid binding to sevelamer HCl. Kidney Int. 62, 611–619 [DOI] [PubMed] [Google Scholar]
- 9. Rastogi A. (2013) Sevelamer revisited: pleiotropic effects on endothelial and cardiovascular risk factors in chronic kidney disease and end-stage renal disease. Ther. Adv. Cardiovasc. Dis. 7, 322–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Staels B., Handelsman Y., and Fonseca V. (2010) Bile acid sequestrants for lipid and glucose control. Curr. Diab. Rep. 10, 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi M., Ikegami H., Fujisawa T., Nojima K., Kawabata Y., Noso S., Babaya N., Itoi-Babaya M., Yamaji K., Hiromine Y., Shibata M., and Ogihara T. (2007) Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes 56, 239–247 [DOI] [PubMed] [Google Scholar]
- 12. Potthoff M. J., Potts A., He T., Duarte J. A., Taussig R., Mangelsdorf D. J., Kliewer S. A., and Burgess S. C. (2013) Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harach T., Pols T. W., Nomura M., Maida A., Watanabe M., Auwerx J., and Schoonjans K. (2012) TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci. Rep. 2, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taniai M., Hashimoto E., Tobari M., Yatsuji S., Haruta I., Tokushige K., and Shiratori K. (2009) Treatment of nonalcoholic steatohepatitis with colestimide. Hepatol. Res. 39, 685–693 [DOI] [PubMed] [Google Scholar]
- 15. Thomas C., Pellicciari R., Pruzanski M., Auwerx J., and Schoonjans K. (2008) Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 [DOI] [PubMed] [Google Scholar]
- 16. Pols T. W., Noriega L. G., Nomura M., Auwerx J., and Schoonjans K. (2011) The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 54, 1263–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kong B., Luyendyk J. P., Tawfik O., and Guo G. L. (2009) Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 328, 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng Q., Inaba Y., Lu P., Xu M., He J., Zhao Y., Guo G. L., Kuruba R., Vega R. De, Evans R. W., and Li S. (2015) Chronic activation of FXR in transgenic mice caused perinatal toxicity and sensitized mice to cholesterol toxicity. Mol. Endocrinol. 29, 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McMahan R. H., Wang X. X., Cheng L. L., Krisko T., Smith M., El Kasmi K., Pruzanski M., Adorini L., Golden-Mason L., Levi M., and Rosen H. R. (2013) Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 288, 11761–11770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang C., Xie C., Li F., Zhang L., Nichols R. G., Krausz K. W., Cai J., Qi Y., Fang Z. Z., Takahashi S., Tanaka N., Desai D., Amin S. G., Albert I., Patterson A. D., and Gonzalez F. J. (2015) Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 125, 386–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lanaspa M. A., Ishimoto T., Li N., Cicerchi C., Orlicky D. J., Ruzycki P., Ruzicky P., Rivard C., Inaba S., Roncal-Jimenez C. A., Bales E. S., Diggle C. P., Asipu A., Petrash J. M., Kosugi T., et al. (2013) Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 4, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmittgen T. D., and Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 23. Braunersreuther V., Viviani G. L., Mach F., and Montecucco F. (2012) Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 18, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lesmana C. R., Hasan I., Budihusodo U., Gani R. A., Krisnuhoni E., Akbar N., and Lesmana L. A. (2009) Diagnostic value of a group of biochemical markers of liver fibrosis in patients with non-alcoholic steatohepatitis. J. Dig. Dis. 10, 201–206 [DOI] [PubMed] [Google Scholar]
- 25. Szabo G., and Csak T. (2012) Inflammasomes in liver diseases. J. Hepatol. 57, 642–654 [DOI] [PubMed] [Google Scholar]
- 26. Kanda H., Tateya S., Tamori Y., Kotani K., Hiasa K., Kitazawa R., Kitazawa S., Miyachi H., Maeda S., Egashira K., and Kasuga M. (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance and hepatic steatosis in obesity. J. Clin. Invest. 116, 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mas E., Danjoux M., Garcia V., Carpentier S., Ségui B., and Levade T. (2009) IL-6 deficiency attenuates murine diet-induced non-alcoholic steatohepatitis. PLoS ONE 4, e7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iredale J. P. (2007) Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest. 117, 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chiang J. Y. (2009) Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S. E., Pollard J. W., Frampton J., Liu K. J., and Geissmann F. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 [DOI] [PubMed] [Google Scholar]
- 31. Gomez Perdiguero E., Klapproth K., Schulz C., Busch K., Azzoni E., Crozet L., Garner H., Trouillet C., de Bruijn M. F., Geissmann F., and Rodewald H. R. (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morinaga H., Mayoral R., Heinrichsdorff J., Osborn O., Franck N., Hah N., Walenta E., Bandyopadhyay G., Pessentheiner A. R., Chi T. J., Chung H., Bogner-Strauss J. G., Evans R. M., Olefsky J. M., and Oh D. Y. (2015) Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64, 1120–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clària J. (2012) Natural killer cell recognition and killing of activated hepatic stellate cells. Gut 61, 792–793 [DOI] [PubMed] [Google Scholar]
- 34. Kotas M. E., Lee H.-Y., Gillum M. P., Annicelli C., Guigni B. A., Shulman G. I., and Medzhitov R. (2011) Impact of CD1d deficiency on metabolism. PLoS ONE 6, e25478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaemers I. C., Stallen J. M., Kunne C., Wallner C., van Werven J., Nederveen A., and Lamers W. H. (2011) Lipotoxicity and steatohepatitis in an overfed mouse model for non-alcoholic fatty liver disease. Biochim. Biophys. Acta 1812, 447–458 [DOI] [PubMed] [Google Scholar]
- 36. Longato L. (2013) Non-alcoholic fatty liver disease (NAFLD): a tale of fat and sugar? Fibrogenesis Tissue Repair 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feldstein A. E., Werneburg N. W., Canbay A., Guicciardi M. E., Bronk S. F., Rydzewski R., Burgart L. J., and Gores G. J. (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 40, 185–194 [DOI] [PubMed] [Google Scholar]
- 38. Takahashi Y., Soejima Y., and Fukusato T. (2012) Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 18, 2300–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anstee Q. M., and Goldin R. D. (2006) Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 87, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wan J., Benkdane M., Teixeira-Clerc F., Bonnafous S., Louvet A., Lafdil F., Pecker F., Tran A., Gual P., Mallat A., Lotersztajn S., and Pavoine C. (2014) M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 59, 130–142 [DOI] [PubMed] [Google Scholar]
- 41. Matsubara T., Li F., and Gonzalez F. J. (2013) FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 368, 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watanabe M., Morimoto K., Houten S. M., Kaneko-Iwasaki N., Sugizaki T., Horai Y., Mataki C., Sato H., Murahashi K., Arita E., Schoonjans K., Suzuki T., Itoh H., and Auwerx J. (2012) Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS ONE 7, e38286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kalaany N. Y., and Mangelsdorf D. J. (2006) LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 68, 159–191 [DOI] [PubMed] [Google Scholar]
- 44. Tuominen I., and Beaven S. W. (2015) Intestinal farnesoid X receptor puts a fresh coat of wax on fatty liver. Hepatology 62, 646–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yao J., Zhou C.-S., Ma X., Fu B.-Q., Tao L.-S., Chen M., and Xu Y.-P. (2014) FXR agonist GW4064 alleviates endotoxin-induced hepatic inflammation by repressing macrophage activation. World J. Gastroenterol. 20, 14430–14441 [DOI] [PMC free article] [PubMed] [Google Scholar]