

Abstract
The Na,K-ATPase α2 subunit plays a key role in cardiac muscle contraction by regulating intracellular Ca2+, whereas α1 has a more conventional role of maintaining ion homeostasis. The β subunit differentially regulates maturation, trafficking, and activity of α-β heterodimers. It is not known whether the distinct role of α2 in the heart is related to selective assembly with a particular one of the three β isoforms. We show here by immunofluorescence and co-immunoprecipitation that α2 is preferentially expressed with β2 in T-tubules of cardiac myocytes, forming α2β2 heterodimers. We have expressed human α1β1, α2β1, α2β2, and α2β3 in Pichia pastoris, purified the complexes, and compared their functional properties. α2β2 and α2β3 differ significantly from both α2β1 and α1β1 in having a higher K0.5K+ and lower K0.5Na+ for activating Na,K-ATPase. These features are the result of a large reduction in binding affinity for extracellular K+ and shift of the E1P-E2P conformational equilibrium toward E1P. A screen of perhydro-1,4-oxazepine derivatives of digoxin identified several derivatives (e.g. cyclobutyl) with strongly increased selectivity for inhibition of α2β2 and α2β3 over α1β1 (range 22–33-fold). Molecular modeling suggests a possible basis for isoform selectivity. The preferential assembly, specific T-tubular localization, and low K+ affinity of α2β2 could allow an acute response to raised ambient K+ concentrations in physiological conditions and explain the importance of α2β2 for cardiac muscle contractility. The high sensitivity of α2β2 to digoxin derivatives explains beneficial effects of cardiac glycosides for treatment of heart failure and potential of α2β2-selective digoxin derivatives for reducing cardiotoxicity.
Keywords: cardiomyocyte, excitation-contraction coupling (E-C coupling), medicinal chemistry, molecular modeling, Na+/K+-ATPase, alpha2beta2, isoform assembly, isoform-selective inhibitors, kinetics
Introduction
The Na,K-ATPase plays a key role in cardiac muscle contractility by regulating the cytosolic Ca2+ concentration, via the Na+/Ca2+ exchanger, and hence the excitation-contraction coupling in cardiac myocytes (1). Three isoforms of the Na,K-ATPase α subunit and three isoforms of the β subunit are expressed in cardiac muscle, and their expression levels vary between species (2–4). In addition, the regulatory FXYD1 subunit and possibly other FXYD proteins are expressed in cardiac muscle (5, 6). Although α1 is the most abundant α subunit isoform in cardiomyocytes in the majority of species, the α2 isoform is functionally more important for cardiac muscle contractility (7–11). The mechanisms underlying distinct roles of α2 and α1 isoforms in the heart are unclear. In smooth and skeletal muscle, the α2 subunit is concentrated at the junctions between the tubular membrane and sarcoplasmic reticulum in close proximity to the Na+/Ca2+ exchanger and other components of the excitation-contraction complex (12), resulting in more efficient regulation of contraction by the α2 subunit in contrast to uniformly distributed α1 subunit. However, the published data on localization of the two isoforms in cardiac myocytes is inconsistent. For example, preferential T-tubular localization of α2 but uniform plasma membrane localization of the α1 have been reported by several groups (11, 13), whereas quite the opposite distribution of the isoforms was reported (3), and uniform distribution of both α1 and α2 subunits has also been reported (14). Measurements of the Na,K-ATPase function of isoforms in cardiac myocytes suggest that the α2 subunit is more concentrated in T-tubular membranes than in the external sarcolemma, whereas the α1 subunit is equally distributed in the plasma membrane (11, 15, 16).
Assembly with the β subunit is required for maturation, trafficking, membrane insertion, and transport activity of the α subunit (17), and three β isoforms differentially regulate these processes. Three β subunit isoforms differ from each other in the number of N-linked glycans, and recombinant addition or removal of N-glycans has been shown to alter the polarized sorting of the Na,K-ATPase (18, 19). The three β subunit isoforms differentially modulate voltage dependence of the Na,K-ATPase activity and the apparent affinity of the enzyme for Na+, K+ and inhibitors (20, 21). β1, but not β2 or β3, undergoes post-translational modifications, namely glutathionylation and palmitoylation, and β1 glutathionylation induced by oxidants decreases the Na,K-ATPase activity (22, 23). Thus, the specific assembly with particular β isoforms could account for distinct physiological roles of α1 and α2 isoforms. However, it is not known whether the α2 subunit has a preference for a particular β isoform in cardiac myocytes.
To examine possible functional differences between α2β(1–3) isoforms, we have expressed human α2 with all three human β subunits in Pichia pastoris, purified the complexes, and compared their functional characteristics and inhibitor sensitivity. Previous work has demonstrated some features of α2β2 and α2β3 when expressed in Xenopus oocytes and SF-9 insect cells (20, 24). Experimentally, the purified complexes are advantageous in that they allow characterization of the functional properties and inhibitor selectivity of each isoform separately (25–27) and also detailed mechanistic properties of ion binding and conformational changes (28–30).
The inhibition of the Na,K-ATPase by digitalis CGs5 has been used for years to treat heart failure. CGs increase the force of cardiac muscle contraction by reducing the inward Na+ gradient that decreases Ca2+ extrusion via the Na+/Ca2+ exchanger (NCX1), leading to increased Ca2+-induced Ca2+ release from the sarcoplasmic reticulum during excitation-contraction coupling. As a toxic side effect, excessive inhibition of the Na,K-ATPase increases bulk intracellular Na+ concentration, excessive accumulation of Ca2+ ions (i.e. “calcium overload”), and “spontaneous” Ca2+ release from sarcoplasmic reticulum that can trigger cardiac arrhythmias (1, 31). The preferential role of α2 in excitation-contraction coupling suggests that α2-specific inhibitors might be able to induce an ionotropic effect without triggering Ca2+ overload and arrhythmias. Some years ago, we demonstrated that some natural CGs, such as digoxin and digitoxin, exhibit a moderate intrinsic selectivity for α2 over α1, whereas aglycones, such as digoxigenin and digitoxigenin, show no selectivity (25). Thus, the isoform selectivity was attributed to the sugar moiety, especially the third digitoxose. It was proposed that modification of the third sugar could raise selectivity for α2. Indeed, chemical modification of the third digitoxose residue of digoxin by periodate oxidation and reductive amination by primary amines, R-NH2, produced perhydro-1,4-oxazepine derivatives with enhanced selectivity of inhibition for α2β1 over α1β1 (27). Most recently, we have described perhydro-1,4-oxazepine digoxin derivatives with various straight chain, branched, and cyclic or heterocyclic aliphatic substitutions and shown that compounds with four carbon substitutions (cyclobutyl (DcB), methyl cyclopropyl (DMcP), and isobutyl (DiB)) showed an especially high selectivity for α2β3/α1β1. α2β3 is the principal Na,K-pump isoform in non-pigmented cells of ciliary epithelium. We have shown that the digoxin derivatives with enhanced selectivity for α2β1 and especially α2β3 efficiently reduce intraocular pressure when applied topically to rabbit eyes (26, 27).
We present evidence here that the α2 and β2 isoforms preferentially assemble with each other in the heart and reside predominantly in the T-tubules. By systematic analysis of properties of purified α2β2 in comparison with α1β1, α2β1, and α2β3, we demonstrate distinctive functional properties and isoform-selective inhibition of α2β2, which explain the important role of α2 for myocardial contractility and the pharmacological potential of α2β2-selective CGs.
Results
Distribution of Na,K-ATPase α1, α2, β1, and β2 Subunits in Rat and Human Heart
Normal rat or human frozen heart sections were used to study the intracellular localization of the Na,K-ATPase subunit isoforms by immunofluorescence. In rat cardiomyocytes, the Na,K-ATPase subunits were differentially distributed (Fig. 1A). The α1 isoform was less abundant in the T-tubular membranes than in the external sarcolemma. Conversely, the α2 isoform was more abundant in the T-tubular membranes than in the sarcolemma, consistent with previously published results (11, 13, 21). Both isoforms are expressed at the intercalated discs, but α1 subunit expression is more pronounced. The immunofluorescence pattern with the Na,K-ATPase β2 antibody was similar to that of the α2 antibody with major expression observed in the T-tubules. In frozen sections of human hearts, the β1 isoform was present more abundantly in the sarcolemma than in the T-tubular membranes, whereas the β2 isoform was present exclusively in the T-tubules (Fig. 1B). These results demonstrate a differential localization of the Na,K-ATPase subunits with the α2 and β2 Na,K-ATPase subunits following the same T-tubule-specific expression pattern and the α1 and the β1 subunits ubiquitously distributed but more abundant in the sarcolemma.
FIGURE 1.
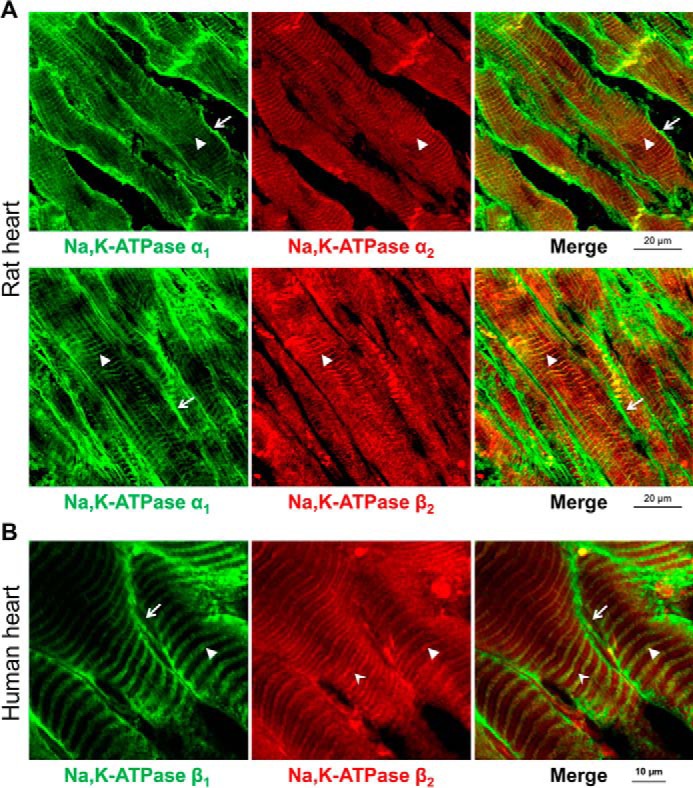
Na,K-ATPase β2 and α2 subunits are localized almost exclusively in T-tubules in cardiomyocytes, whereas the α1 and β1 subunits are localized in both sarcolemma and T-tubules. A, frozen sections of rat heart were double-stained by using mouse antibodies against α1 subunit (green) and rabbit antibodies against α2 subunit (red) (top panels) or by using mouse antibodies against α1 subunit (green) and rabbit antibodies against β2 subunit (red) (bottom panels). Anti-mouse Alexa Fluor 488-conjugated secondary antibodies were used to detect anti-α1 primary antibodies, and anti-rabbit Alexa Fluor 633-conjugated secondary antibodies were used to detect anti-β2 and anti-α2 primary antibodies. The arrows show localization of the α1 subunits, but not of α2 and β2 subunits, in the sarcolemma. The arrowheads show co-localization of α1 and β2 subunits or α1 and α2 subunits in T-tubules. B, frozen sections of human heart were double-stained by using mouse antibodies against β1 subunit (green) and rabbit antibodies against β2 subunit (red). Anti-mouse Alexa Fluor 488-conjugated secondary antibodies were used to detect anti-β1 primary antibodies, and anti-rabbit Alexa Fluor 633-conjugated secondary antibodies were used to detect anti-β2 primary antibodies. The arrows show localization of the β1 subunits, but not of β2 subunits, in the sarcolemma. The arrowheads show co-localization of β1 and β2 subunits in T-tubules. The stealth-like arrowheads show the T-tubules in which the β2 subunits, but not the β1 subunits, are present.
Because the Na,K-ATPase is a crucial component in regulating postnatal cardiac function (32), we analyzed whether the Na,K-ATPase subunits are also selectively expressed during embryogenesis. Paraffin-embedded sections of mouse embryos (embryonic day 12.5) were analyzed by immunofluorescence (Fig. 2). The Na,K-ATPase α1 subunit was expressed ubiquitously (top and bottom left panels), whereas the α2 and β2 were mostly restricted to the heart (top and bottom right panels, respectively).
FIGURE 2.

The Na,K-ATPase α2 subunit and β2 subunit are preferentially expressed in the mouse embryonic heart in contrast to the ubiquitously expressed α1 subunit. A, paraffin-embedded sections of mouse embryos (embryonic day 12.5) were double-stained by using mouse antibodies against α1 subunit (left panels) and rabbit antibodies against α2 subunit or β2 subunit (right panels). Anti-mouse Alexa Fluor 488-conjugated secondary antibodies were used to detect anti-α1 primary antibodies, and anti-rabbit Alexa Fluor 633-conjugated secondary antibodies were used to detect anti-α2 or anti-β2 primary antibodies. The rectangles show the embryonic hearts. B, a scheme demonstrating the assembly of the images shown in A. Each rectangle outlined by a dotted line represents an individual confocal microscopy image taken at ×10 magnification.
The Na,K-ATPase α2 and β2 Subunits Are Selectively Co-immunoprecipitated from Mouse Heart
To analyze the composition of the Na,K-ATPase heterodimers present in heart microsomal membranes, proteins were co-immunoprecipitated with an α2 subunit-specific antibody, and the presence of α1, β1, β2, and β3 subunits was analyzed by Western blotting. To prevent the overlap of the β subunits bands with the band corresponding to the heavy chain of the immunoprecipitating antibody, the immunoprecipitated proteins were treated with PNGase F before SDS-PAGE. Immunoprecipitation of the α2 subunit resulted in the co-immunoprecipitation of the β2 subunit and minor amounts of β1 and β3 subunits (Fig. 3A, left). No α1 subunits were detected in the immunoprecipitation with the α2-specific antibody. In contrast, when the α isoform-nonspecific antibody was used, both α1 and α2 subunits were immunoprecipitated, and approximately equal amounts of all of the three β subunits were co-immunoprecipitated (Fig. 3A, right). Conversely, immunoprecipitation with a β2-specific antibody resulted in co-immunoprecipitation of the α2 but not the α1 subunit (Fig. 3B). Taken together, these results suggest that the α2 and β2 subunit isoforms associate predominantly with each other but not with other isoforms expressed in the heart.
FIGURE 3.

The Na,K-ATPase α2 subunit and β2 subunit preferentially interact with each other in mouse heart. A, Western blotting analysis of proteins immunoprecipitated and co-immunoprecipitated from the detergent extracts of mouse heart microsome membranes by using either the α2-specific antibodies (left panels) or the α-nonspecific antibodies (right panels) shows preferential co-immunoprecipitation of the β2 subunit with the α2 subunit. B, Western blotting analysis of the immunoprecipitated β2 subunit and co-immunoprecipitated α2 subunit isoforms shows that the α2 subunit is preferentially co-precipitated with the β2 subunit. Input lanes contain 10% of the extract used for immunoprecipitation. To prevent an overlap of the β subunit bands with the heavy chain band of the antibodies used for immunoprecipitation, the immunoprecipitated proteins were treated with PNGase F before SDS-PAGE. IP, immunoprecipitation; DG, deglycosylated.
Expression and Purification of Na,K-ATPase α2β2
α2β2 and α2β3 were expressed in P. pastoris as described under “Experimental Procedures.” Under optimal expression conditions, specific ouabain binding for the α2β2 clone was 8 ± 1 pmol/mg protein and 10 ± 2 pmol/mg for the α2β3 clone, respectively. The addition of DMSO to the culture medium, which was reported to increase expression of GPCRs in P. pastoris, did not increase expression (33).
Both isoforms were purified via the N-terminal His tag of the β subunit by metal affinity chromatography on BD-Talon beads and reconstituted with purified human FXYD1 on the BD-Talon beads, as described in Experimental Procedures.6 The purity of α2β2 and α2β3 was comparable with that of purified human α1β1 and α2β1 complexes. As depicted in Fig. 4, at least five bands of β2 were observed, in comparison with two bands for β3 and β1. The two bands of β1 were shown previously to represent two glycosylated versions of the β1 subunit of the ManX-GlcNAc2 type, typical for P. pastoris (34). An increase in heterogeneity of glycoforms of β2 as compared with β3 and β1 is consistent with the presence of eight N-glycosylation sites in β2 and only two and three in β3 and β1, respectively. When deglycosylated by PNGase treatment, all three β isoforms migrated at their expected molecular mass of approximately 35, 31, and 33 kDa for β1, β3, and β2, respectively.
FIGURE 4.

Expression of purified Na,K-ATPase isoforms. Coomassie-stained SDS-PAGE of purified isoforms (5 μg/lane). For deglycosylation, samples were denatured and treated with PNGase F for 60 min at 37 °C.
Recent studies have demonstrated that α2β1 is less stable to thermal and detergent-mediated inactivation than α1β1, due to suboptimal interaction with phosphatidylserine (35). We investigated the relative effect of the β subunit on Na,K-ATPase isoform stability by thermal inactivation of [3H]ouabain binding to the membranes and Na,K-ATPase activity of the purified proteins, as described in previous publications (see for example Refs. 29 and 65). By both criteria, the α2 complexes were significantly less thermally stable than α1, and α2β2 was somewhat less stable than α2β1 and α2β3. Thus, the relative instability of the α2β(1–3) complexes is a feature attributable primarily to the α2 subunit. The differences between β isoforms, β2 < β3 ≈ β1, are rather small.
Functional Properties of Purified α1β1, α2β1, α2β2, and α2β3 Complexes
The specific Na,K-ATPase activity of the purified isoforms was highest for α1β1 (16.4 ± 0.7 μmol/mg/min), followed by similar values for α2β1 (10.9 ± 0.6), α2β3 (10.7 ± 1.9), and α2β2 (8.4 ± 1.4; Table 1, column 2). Note that FXYD1 itself inhibits the Na,K-ATPase activity of the purified human α1β1 by about 25% and α2β1 by about 15% compared with the αβ complexes alone (30). One kinetic property showing large differences between the isoform complexes was the K0.5K+ for activation of Na,K-ATPase activity, with values of 1.5 ± 0.1 mm for α1β1 and 2.7 ± 0.1 mm for α2β1, whereas the K0.5K+ for both α2β2 and α2β3 was much higher, with apparent K0.5 values of 7.4 ± 0.2 and 6.4 ± 0.5 mm, respectively (Table 1, column 4). Sodium titrations for activation of Na,K-ATPase activity revealed that K0.5Na+ was not different between α1β1 and α2β1, whereas K0.5Na+ for α2β2 and α2β3 was significantly lower (Table 1, column 3). We have also looked at the affinity for inhibition of Na,K-ATPase activity by vanadate, which is a phosphate analogue that binds to the E2(2K) conformation, mimicking the transition state E2P2K during dephosphorylation (36). All three α2 isoforms have a higher Ki vanadate compared with α1β1 (Ki = 0.48 μm), and the effects are greatest with β2 and β3 in the order α2β2 (Ki = 34 μm) > α2β3 (Ki = 19 μm) > α2β1 (Ki = 3.5 μm) (Table 1, column 5).
TABLE 1.
Functional properties of purified Na,K-ATPase complexes
Specific activity, K0.5Na+, K0.5K+, and Ki vanadate of purified Na,K-ATPase complexes are shown. Values represent averages of at least three different experiments ± S.E. The reaction medium contained sodium plus potassium as indicated, 1 mm ATP, 3 mm MgCl2, 25 mm histidine, pH 7.4, 1 mm EGTA, 0.01 mg/ml SOPS, 0.001 mg/ml cholesterol, and 0.005 mg/ml C12E8. Maximal Na,K-ATPase activity was measured in the presence of 120 mm NaCl, 20 mm KCl. For ion titrations, ATPase activity was measured in medium containing 80 mm KCl and 0–120 mm NaCl or 120 mm NaCl and 0–40 mm KCl. Ionic strength was maintained constant with choline chloride. K0.5 values were obtained from least square fits of the data points to the Hill equation. Ki vanadate was determined in medium containing 120 mm NaCl, 20 mm KCl, and 1 mm ATP, and data points were fitted to a one-side inhibition model.
| Isoform complexes | Specific Na,K-ATPase activity | K0.5Na+ (nH) | K0.5K+ (nH) | Vanadate Ki |
|---|---|---|---|---|
| μmol/mg/min | mm | mm | μm | |
| α1β1 | 16.4 ± 0.7 | 16 ± 0.4 (1.7 ± 0.1) | 1.5 ± 0.1 (1.8 ± 0.1) | 0.5 ± 0.1 |
| α2β1 | 10.9 ± 0.6 | 17.7 ± 0.5 (1.9 ± 0.2) | 2.7 ± 0.1 (2.0 ± 0.2) | 3.5 ± 0.3 |
| α2β2 | 8.4 ± 1.4 | 9.8 ± 0.7 (1.8 ± 0.2) | 7.4 ± 0.2 (1.7 ± 0.1) | 34.0 ± 2.0 |
| α2β3 | 10.7 ± 1.9 | 13.0 ± 0.2 (1.9 ± 0.2) | 6.4 ± 0.5 (1.8 ± 0.2) | 19.0 ± 1.5 |
A simple explanation of the raised K0.5K+ in Na,K-ATPase activity assays could be that the β2 and β3 subunit reduce the binding affinity of 2K ions for their extracellular sites. K+ and Na+ binding was determined by using the electrochromic shift dye RH421 in a medium of fixed ionic strength (containing also 5 mm magnesium ions) (Fig. 5 and Table 2) (29, 30, 37). The inset of Fig. 5A shows the typical RH421 responses upon the addition of Na+ ions (E1-E1(3Na)) and then ATP (to E2P) and K+ ions (E2(2K)) for the α2β2 complex, as explained in recent papers (29). By varying Na+ or K+ concentrations, equilibrium titrations of either 3Na+ binding to cytoplasmic sites or 2K+ binding to extracellular sites are readily obtained, leading to curves such as those in Fig. 5, A and B. The binding parameters derived from best fits of the curves to the Hill equation are collected in Table 2, where K0.5Na+ and K0.5K+ represent the intrinsic binding affinity for 3Na+ and 2K+ ions, respectively. Evidently, the intrinsic binding affinity for 3Na+ ions is the same for all of the isoform complexes. By contrast, in these conditions, the binding affinity of 2K+ ions to α2β1 is significantly lower than to α1β1, and the binding affinity for both α2β2 and α2β3 is further strongly reduced compared with α2β1. Note that, compared with K0.5K+ for α1β1, the K0.5K+ for α2β2 or α2β3 is almost an order of magnitude higher. Because vanadate binds primarily to the E2(2K) conformation, the differences in Ki for vanadate inhibition of Na,K-ATPase activity could be secondary to the differences in K0.5K+ values, with the same order for the Ki as for K0.5K+ values (α1β1 < α2β1 < α2β3 < α2β2; Table 1).
FIGURE 5.

Sodium and potassium binding and conformational changes of the isoforms measured with RH421. A, equilibrium titration of sodium binding to the E1 conformation. The inset shows a standard experiment and fluorescence changes associated with ion binding and release to α2β2 (sodium binding to E1, sodium release and conformational transition to E2P, and potassium binding). B, equilibrium titration of potassium binding to E2P. C, stopped-flow trace of the E1Na3 → E2P transition of α2β2 fitted to a double exponential function. The average of 15 traces is shown. D, stopped-flow trace of the α2β2 E2(Rb2)ATP → E1Na3ATP transition fitted to a single exponential function.
TABLE 2.
Binding affinities of sodium and potassium detected with RH421
Sodium or potassium titration curves in Fig. 5 were fitted to the Hill equation. The K0.5 and nH values represent averages from three experiments ±S.E.
| Isoform | K0.5Na+ (nH) | K0.5K+ (nH) |
|---|---|---|
| mm | mm | |
| α1β1 | 7.7 ± 0.5 (1.7 ± 0.13) | 0.6 ± 0.04 (1.8 ± 0.1) |
| α2β1 | 8.0 ± 0.3 (1.9 ± 0.2) | 1.5 ± 0.1 (1.5 ± 0.1) |
| α2β2 | 8.1 ± 0.3 (1.7 ± 0.2) | 5.1 ± 0.1 (1.6 ± 0.1) |
| α2β3 | 7.3 ± 0.6 (1.8 ± 0.1) | 4.8 ± 0.6 (1.5 ± 0.2) |
An alternative or additional explanation to that just given is that raised K0.5K+ of the α2β2 and α2β3 complexes is caused by an α2β2- or α2β3-induced shift in poise of the E1-E2 conformational equilibrium toward E1 or E1-P. This explanation would also be consistent with the parallel reduction in apparent K0.5Na+ and increase in Ki for vanadate (Table 1, column 3). A direct test of the relative effects of β1, β2, and β3 on the conformational changes was made by measuring the rates of the E1P(3Na) → E2P and E2(2Rb)ATP → E13Na·ATP transitions using RH421 in stopped-flow experiments, as described in our recent publications (29, 30). Traces for the α2β2 complex are shown in Fig. 5, C and D, and the rate constants for all of the isoform complexes are collected in Table 3. These data show directly that for α2β1, the rates of E1P(3Na) → E2P are indeed slower than for α1β1, and for both α2β2 and α2β3, the rate is still slower than for α2β1.
TABLE 3.
Rates of conformational changes for the different isoform complexes determined by stopped-flow measurements
All rates were measured at 23 °C as described under “Experimental Procedures.” The turnover rate was calculated from the function (a × b)/(a + b).
The rates of E2(2Rb)ATP → E13Na·ATP for α2β1, α2β2, and α2β3 are all significantly slower than for α1β1, but they are indistinguishable from each other. The turnover rate in s−1 for each isoform was calculated from the expression (a × b)/(a + b), where a is the rate of E1P(3Na) → E2P and b is the rate of E2(2Rb)ATP → E13Na·ATP (Table 3), assuming that the rates of phosphorylation E1Na + ATP → E1P(3Na) and dephosphorylation E2P2Rb → E2(2Rb) are fast and do not significantly limit the turnover rate. The calculated value for α1β1 (14 s−1) is greater than for α2β1, α2β2, and α2β3, which are not significantly different from each other (9.17, 8.76, and 8.75 s−1, respectively; average of 8.89). The ratios of the calculated turnover rates for α1β1/α2β1, α1β1/α2β2, and α1β1/α2β3 are essentially the same, with an average of 1.6. These ratios are close to those of Na,K-ATPase activities in Table 1, validating the assumptions that underlie the calculation of the turnover rates.
Inhibition of Na,K-ATPase Isoforms by Perhydro-1,4-oxazepine Derivatives of Digoxin
As reported recently, chemical modification of the third digitoxose residue of digoxin (by periodate oxidation and reductive amination with R-NH2) to produce perhydro-1,4-oxazepine derivatives increases selectivity of inhibition for α2β1 and α2β3 over α1β1 (26, 27). Here we have compared inhibition of α1β1 and all three complexes α2β1, α2β2, and α2β3 by several derivatives described previously (26) and six new ones (DAz, DTh, DcB2,2dM, DMSM, DEMS, and DESA). The structures of the substituents, abbreviated names, and masses of all of the derivatives are given in Fig. 6. Fitted Ki values and selectivity ratios for all of these compounds are summarized in Table 4. Compounds that have the highest selectivity, compared with digoxin itself, are indicated by double asterisks, and compounds with significant but lower selectivity are shown with a single asterisk. There is a clear peak for the cyclobutyl derivative with four carbon atoms, DcB, with 16.9-, 22.2-, and 33.6-fold selectivity for α2β1, α2β2, and α2β3 over α1β1, respectively. Overall, Table 4 shows that aliphatic substituents with four carbon are most selective (DcB > DMcP > DiB), whereas the substituents with three carbons (DiP ≈ DcP > DP) are also quite selective, and the selectivity ratios decline for substituents with 5, 6, or 7 carbon atoms. The sulfonyl derivatives DMSM, DEMS, and DESA also showed strongly increased selectivity over α1β1 in the order α2β3 > α2β2 > α2β1.
FIGURE 6.

Substituent structures, abbreviated names, and predicted and found masses of perhydro-1,4-oxazepine derivatives of digoxin.
TABLE 4.
Ki values and selectivity ratios for the inhibition of purified Na,K-ATPase isoforms by perhydro-1,4-oxazepine derivatives of digoxin
The reaction medium contained 130 mm NaCl, 5 mm KCl, 3 mm MgCl2, 25 mm histidine, pH 7.4, 1 mm EGTA, 0.01 mg/ml SOPS, 0.001 mg/ml cholesterol, and 0.005 mg/ml C12E8. Each experiment was carried out at least three times. The calculated Ki values represent averages ± S.E. *, compounds with significantly higher selectivity (Ki α1β1/α2β1–3) than digoxin (>4- and <10-fold). **, compounds with the highest selectivity (Ki α1β1/α2β1–3) compared with digoxin (>12-fold).
| CG |
Ki ± S.E. |
Selectivity |
n | |||||
|---|---|---|---|---|---|---|---|---|
| α1β1 | α2β1 | α2β2 | α2β3 | α1β1/α2β1 | α1β1/α2β2 | α1β1/α2β3 | ||
| nm | ||||||||
| Digoxin | 268.0 ± 13.8 | 58.7 ± 5.4 | 58.0 ± 1.9 | 42.8 ± 3.0 | 4.5 | 4.6 | 6.2 | 7 |
| DMe | 103.0 ± 5.6 | 15.3 ± 1.2 | 20.4 ± 1.8 | 10.8 ± 0.6 | 6.7* | 5.1 | 9.5* | 7 |
| DEt | 137.9 ± 12.6 | 23.2 ± 0.9 | 16.4 ± 1.6 | 14.4 ± 1.3 | 5.9 | 8.3* | 9.5* | 4 |
| DP | 87.7 ± 7.9 | 18.3 ± 1.7 | 10.5 ± 1.8 | 9.8 ± 1.1 | 4.8 | 8.3* | 8.8* | 5 |
| DiP | 149.0 ± 20.7 | 28.9 ± 1.7 | 16.7 ± 1.9 | 10.3 ± 1.8 | 5.1 | 8.9* | 14.4** | 4 |
| DcP | 109.0 ± 6.2 | 14.6 ± 11.6 | 13.0 ± 1.3 | 8.1 ± 1.36 | 7.5* | 8.5* | 13.4** | 3 |
| DiB | 92.0 ± 8.9 | 20.6 ± 1.4 | 10.0 ± 0.8 | 5.8 ± 0.6 | 4.4 | 9.0* | 16.0** | 5 |
| DMcP | 95.8 ± 13.7 | 18.3 ± 1.6 | 8.0 ± 0.8 | 4.3 ± 0.6 | 5.2 | 12.0** | 22.2** | 4 |
| DcB | 135.0 ± 11.0 | 8.0 ± 1.3 | 6.0 ± 1.0 | 4.0 ± 0.15 | 16.9** | 22.2** | 33.6** | 3 |
| DAz | 222.0 ± 11.6 | 52.0 ± 1.7 | 28.0 ± 3.8 | 26.5 ± 2.3 | 4.2 | 7.7* | 8.4* | 3 |
| DTh | 260.0 ± 41.0 | 108.0 ± 22.6 | 170.0 ± 10.4 | 196.0 ± 34.8 | 2.4 | 1.5 | 1.3 | 3 |
| DcPe | 138.0 ± 21.0 | 33.4 ± 7.5 | 33.5 ± 11.9 | 27.6 ± 9.5 | 4.1 | 4.1 | 5.0 | 3 |
| DcHe | 70.4 ± 4.1 | 15.2 ± 3.7 | 15.3 ± 2.9 | 11.7±.5 | 4.6 | 4.6 | 10.1* | 3 |
| DcB2,2dM | 102.0 ± 4.0 | 49.0 ± 18.0 | 39.0 ± 6.0 | 45.0 ± 14.0 | 2.1 | 2.6 | 2.3 | 3 |
| DMcB3,3dM | 31.6 ± 0.5 | 8.6 ± 1.4 | 5.1 ± 0.5 | 3.9 ± 0.7 | 3.7 | 6.2 | 8.2* | 5 |
| DMSM | 944.0 ± 123.0 | 137.0 ± 9.8 | 123.0 ± 7.3 | 89.0 ± 8.7 | 6.9* | 7.7* | 10.6* | 3 |
| DEMS | 464.0 ± 14.0 | 49.2 ± 1.9 | 31.7 ± 3.2 | 24.7 ± 2.1 | 9.4* | 14.6** | 18.8** | 3 |
| DESA | 301.0 ± 23.0 | 38.9 ± 2.2 | 31.5 ± 4.4 | 20.1 ± 0.9 | 7.7* | 9.5* | 15** | 4 |
Potassium-Cardiac Glycoside Antagonism
CGs bind with high affinity to E2P, and potassium binding causes rapid dephosphorylation to E2(2K), which has a much lower affinity for CGs than E2P (38). Because K0.5K+ of α2β2, α2β3 and also α2β1 for activating Na,K-ATPase are higher than for α1β1 in the order α2β2 > α2β3 > α2β1 > α1β1 (Table 1), a higher selectivity for α2β2 and α2β3 and also α2β1 might be due, at least partially, to a weaker K+-CG antagonism. To assess the effect of K+-CG antagonism more systematically, we have measured Ki values for digoxin, the digoxin derivatives and ouabain at increasing K+ concentrations from 2.5, 5, 10, and 20 mm K+. Fig. 7 presents the data for digoxin, but very similar data were obtained for the isobutyl derivative of digoxin (DiB) and also ouabain (not shown). For α1β1, the Ki values increased by 3–4-fold in the range of 2.5–20 mm K+, respectively. By contrast, the Ki values for α2β1 were much less affected by the increased potassium concentration, and inhibition of α2β2 and α2β3 was essentially unaffected by potassium ions in the range 2.5–20 mm. Consequently, the selectivity for inhibition of α2β2 over α1β1 by digoxin (or other CGs) significantly increased over the range 2.5, 5, 10, and 20 mm potassium from 3.67, 4.77, 11.6, to 15.0, respectively. The differences in K+-digoxin antagonism were also investigated more directly by K+-[3H]digoxin displacement assays (Fig. 8). Yeast membranes harboring either α1β1, α2β1, α2β2, or α2β3 were equilibrated with [3H]digoxin and subsequently incubated with increasing amounts of KCl (up to 150 mm). In the conditions of digoxin binding (with vanadate/magnesium but sodium-free), the apparent K0.5K+ for digoxin displacement was 0.62 mm for α1β1 (see also Ref. 25), 1.8 ± 0.1 mm for α2β1, 2.8 ± 0.1 mm for α2β2, and 3.2 ± 0.5 mm for α2β3. Furthermore, at high K+ concentrations, displacement of digoxin binding was incomplete for α2β1, α2β2, and α2β3, the remaining fraction at saturating potassium being 20.3 ± 1.5, 26 ± 1.8, and 30.3 ± 1.5% of control, respectively. Thus, K+-CG antagonism reflects both the affinity for potassium ions and the maximal degree of displacement by potassium ions.
FIGURE 7.

Potassium-digoxin antagonism depicted for α1β1, α2β1, α2β2, and α2β3. Inhibition of purified Na,K-ATPase isoforms by digoxin was measured, and Ki values were plotted against the potassium concentration in the assay medium. Error bars, S.E.
FIGURE 8.
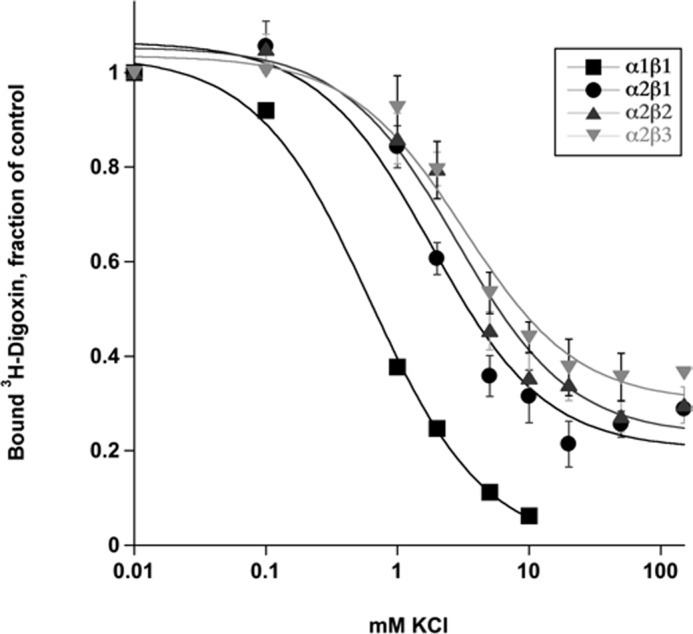
Potassium-[3H]digoxin antagonism. Yeast membranes harboring α2β1, α2β2, or α2β3 Na,K-ATPase were incubated with varying concentrations of KCl, and residual [3H]digoxin binding was measured. The lines represent Hill fits. Data points for α1β1 were taken from Ref. 25. Error bars, S.E. from 3 experiments.
Overall, in conditions similar to the extracellular physiological medium (140 mm sodium, 5 mm potassium or with potassium elevated to 20 mm), K-CG antagonism is prominent for α1β1, weak for α2β1 and negligible for α2β2 and α2β3. Note, however, that, in similar conditions, the selectivity ratios for all six derivatives DiB, DMcP, DcB, DMSM, DEMS, and DESA for α2β3/α1β1 > α2β2/α1β1 > α2β1/α1β1 are significantly greater than for digoxin itself (Table 4). This shows that reduced K-CG antagonism can indeed account only partially for the increased selectivity for α2β3 and α2β2, whereas the structure of the derivatives is a crucial element determining the isoform selectivity.
Discussion
We discuss here the data showing selective assembly of α2β2 in cardiac myocytes and distinct functional properties and isoform-selective inhibition of human α2β2 and α2β3, together with possible molecular explanations and physiological or pharmacological implications. Comparisons of functional properties and inhibition of α2β1 with α1β1 reveal the differences between α2 and α1, whereas comparisons of α2β2 or α2β3 with α2β1 reveal the influence of β2 or β3 compared with β1. As one general conclusion, it is the combined effects of α2 and β2 (or β3) that give rise to the distinctive functional properties and isoform-selective inhibition of α2β2 (or α2β3).
Specific Assembly of α2β2 in Heart
Three of the four Na,K-ATPase α subunit isoforms and all three β subunit isoforms are expressed in the heart. Although α1 is the most abundant α subunit isoform, the α2 isoform rather than the α1 isoform plays a key role in cardiac muscle contractility by regulating the cytosolic Ca2+ concentration in cardiac myocytes. The transient rise in Ca2+ concentration associated with electrical excitability then triggers cardiac muscle contraction (1). Despite extensive studies of isoform-specific properties of the Na,K-ATPase, the reasons for differential roles of α2 and α1 isoforms in cardiac muscle contractility are not fully understood.
The data presented here indicate that the α2 and β2 isoforms preferentially assemble with each other in the heart as detected by co-immunoprecipitation (Fig. 3). These data are consistent with previous reports on selective assembly (17) and co-purification of α2 and β2 isoforms in the brain (39). Immunofluorescence data in rat and human heart sections indicate that both α2 and β2 isoforms specifically localize to the T-tubular membranes, whereas α1 and β1 isoforms are distributed in both T-tubular and external sarcolemma membranes (Fig. 1). The data on preferential T-tubular localization of the α2 and ubiquitous distribution of the α1 are in agreement with previous reports on measurements of the isoform-specific Na,K-ATPase activity in cardiac myocytes (11, 15, 16, 40) and several immunofluorescence reports (11, 13). On the other hand, our data contrast with several other reports on immunodetection of the Na,K-ATPase α isoforms in the heart (3, 14). The uniform distribution of the β1 isoform has been reported previously (3), whereas this is the first report describing the specific localization of the β2 isoform in the heart.
It seems paradoxical that there are two isoform complexes, α2β2 and α2β3, with similar functional properties, as shown in this paper, but in the heart, α2 assembles with β2 rather than with β3. Assembly with a particular β isoform is known to affect trafficking and polarized sorting of the α subunit (19, 41, 42), suggesting that association with the β2 can be important for the specific location of the α2 isoform in the T-tubules. Particularly, recombinant addition of N-glycosylation sites to the β1 subunit has been shown to alter localization of the Na,K-ATPase from the basolateral to the apical domain of the plasma membrane in gastric epithelial cells (19). It is possible that eight N-glycans versus two or three in the β3 or β1 isoform play a role in this specific targeting of the α2 to the microdomains of T-tubular membranes that contain Na+/Ca2+ exchanger and are proximal to other components of Ca2+-regulating complex, which can explain the importance of the α2 isoform in cardiac muscle contraction.
Another difference between β3 and β2 is a role of β2 in cell adhesion (39, 43). Whether this adhesive role is required for specific location of α2β2 in T-tubules is not clear, but α2β2 differs from α2β3 in this crucial aspect.
Functional Properties of the α2β1, α2β2, and α2β3 Isoform Complexes
The major functional differences between α2β2 and α2β3 when compared with α2β1, and especially α1β1, is the raised K0.5K+ for activating Na,K-ATPase, reduced K0.5Na+, and turnover rate (Table 1). These results confirm previous evidence that α2 raises K0.5K+ compared with α1 when it is complexed with β1 (20, 24, 44) and now show that both β2 and β3 lower K0.5Na+ as well as raising K0.5K+ when complexed with α2, in comparison with complexes with β1. The RH421 experiments show that the principal mechanism of the effects of α2 versus α1 and β2 and β3 versus β1 to raise K0.5K+ is reduction of the intrinsic binding affinity for potassium ions at the extracellular surface, with K0.5K+ values in the order α1β1 < α2β1 < α2β3 ≤ α2β2, respectively (Fig. 5 and Table 2). In RH421 experiments, the potassium affinity is determined in the presence of 50 mm sodium, and, in principle, the reduced affinity for Kexc ions could reflect an increased affinity for Naexc and competition with Kexc. Nevertheless, displacement of digoxin by potassium ions in the absence of sodium ions (K0.5K+ 0.6 ± 0.07 mm for α1β1, 1.8 ± 0.1 mm for α2β1, 2.8 ± 0.1 mm for α2β2, and 3.2 ± 0.5 mm for α2β3) shows that there is a true and large reduction in intrinsic Kexc affinity (Fig. 8). The vanadate titrations with Ki values in the order α1β1 < α2β1 < α2β3 < α2β2 (Table 1) are consistent with the order of decreasing affinities for potassium ions.
Compared with α1β1, α2β1, α2β2, and α2β3 significantly reduced the rate of the conformational transition E1P(3Na) → E2P, with the order α1β1 > α2β1 > α2β2 ≈ α2β3 (Fig. 5 and Table 3). In steady-state Na,K-ATPase conditions, a shift of the E1P(3Na) → E2P conformational equilibrium toward E1P would contribute to the higher K0.5K+ as well as lower K0.5Na+ (and higher Ki vanadate) (Table 1). Note that the reduced K0.5Na+ of α2β2 and α2β3 compared with α1β1 and α2β1 is not explained by a change in intrinsic binding affinity for sodium ions (see Fig. 5A).
α2β1 also displayed a reduced rate of E2(2Rb)ATP→E1(3Na)ATP when compared with α1β1, but there was no further decrease in α2β2 and α2β3. This finding explains the reduced turnover rate for all α2 complexes compared with α1. The different β isoforms appear to have no isoform-selective influence on the turnover rate, which is governed by the slow rate-determining step E2(2Rb)ATP → E1(3Na)ATP, reduced by α2, and not by the faster E1P(3Na) → E2P transition.
In the absence of high resolution structures of the α2 complexes, one can only speculate on possible explanations of the kinetic effects of α2 and β2 or β3, such as the reduced Kexc binding affinity. Using chimeras of Na,K-ATPase and H,K-ATPase, the extracellular domain was identified as the main modifier of the apparent potassium affinity (for displacing bound ouabain) (45), and the main interaction site was shown to be an SYGQ motif in the M7-8 loop of α (46). In an older study (47), the extracellular domain of β1 was suggested to cover the extracellular domain of the α subunit and control access to the Rb(K) binding sites. In general, this concept appears compatible with the molecular structures of Na,K-ATPase (α1β1) (48–50). Thus, compared with α1β1, the Kexc entry and exit pathway may be more accessible in α2β1 itself and even more so in α2β2 and α2β3, leading to the large decrease in Kexc binding affinity.
Digoxin Derivatives with Strong Selectivity for α2β2 and α2β3 Complexes
The conclusion from all of the experiments in Table 4 is that digoxin perhydro-1,4-oxazepine derivatives with four carbon substitutions (cyclobutyl (DcB), methyl cyclopropyl (DMcP), and isobutyl (DiB)) have the highest selectivity for α2β3/α1β1 and α2β2/α1β1 compared with compounds with 1–3 or 5 or a greater number of carbon atom substitutions. This confirms the data in Ref. 26 for α2β3/α1β1 and extends them to α2β2/α1β. The optimal size of the aliphatic substituents (four carbon atoms), cyclic and non-cyclic, may indicate a size restriction of the space between the α and β subunits. Replacement of one methylene group in the cyclobutyl DcB with a single NH (DAz) or sulfur (DTh) atom strongly reduces selectivity, whereas the three new sulfonyl derivatives, DMSM, DEMS, and DESA, showed enhanced selectivity for α2β2 and α2β3 and to some extent also α2β1, relative to digoxin itself (Table 4). Thus, the highest selectivity for the α2β2 and α2β3 isoform complexes depends crucially on the structure of the derivatives, providing the strongest evidence for selective interactions of the substituent groups with β2 and β3, respectively.
We have attempted to explain the selectivity of DcB for α2β3 and α2β2 with molecular docking models, starting with a structure of renal Na,K-ATPase (α1β1) with bound digoxin (51) (Protein Data Bank code 4RET) (Fig. 9). The models display the optimal positions found for DcB relative to digoxin and emphasize the major attractive interactions, particularly with the β subunit. The modeling supports the experimental result whereby DcB binds to α2β3 with the highest potency and selectivity, showing a hydrophobic interaction between the DcB-cyclobutyl moiety and β3Val-88. There is also an electrostatic interaction of the protonated nitrogen of perhydro-1,4-oxazepine ring with Asp-889 of α2. In the case of α2β2, to which DcB also binds with good potency and selectivity, the modeling shows a hydrogen bond interaction between the protonated perhydro-1,4-oxazepine ring nitrogen and β2Glu-89 and also an additional hydrogen bond between the lactone ring and the structural water molecule located at the bottom of the binding site. It is noticeable that the steroid and lactone moieties are somewhat rotated compared with the digoxin itself. By contrast with the α2β3 and α2β2 models, in the α1β1 model, the steroid-lactone moiety of DcB almost exactly overlaps that of digoxin and displays only a weak interaction with β1Gln-84. This result also appears to be consistent with the experimental result for α1β1 which displays a much lower potency for DcB and only a small difference from digoxin itself.
FIGURE 9.

Models for docking of DcB to α1β1, α2β2, and α2β3. DcB was docked into homology models of human Na,K-ATPase isoforms derived from the porcine E2P·Mg·digoxin structure (Protein Data Bank code 4RET) as described under “Experimental Procedures.” α and β subunits are shown in green and blue ribbon representations. Transmembrane helices 3 and 5 of the α-subunit are removed for clarity. Digoxin (yellow) and DcB (purple) are shown in a stick representation, and water and a magnesium ion are shown as spheres. The residues of the β subunit closest to the cyclobutyl moiety, β1Gln-84, β2Glu-89, and β3Val-88, are shown as sticks, and distances to DcB are indicated.
Taken together, it is evident that both the α and the β subunits determine the selectivity of the digoxin derivatives, and the β subunit, in particular, has favorable interactions with the substituted third sugar residue.
Physiological Role of α2β2
In relation to the physiological function, major differences of α2β2 compared with α1β1 include the high K0.5K+ values (low affinity) for extracellular potassium ions, a somewhat lower turnover rate, and a significantly lower K0.5Na+. Although the current experiments have been done with detergent-soluble purified proteins, a very similar effect of α2β2 to strongly raise K0.5K+ values for extracellular potassium ions compared with α1β1 and also α2β1 has been described with the proteins expressed in Xenopus oocytes (20, 21, 52). A property that we cannot assess in experiments with detergent-soluble proteins is the dependence of activity on membrane potential. As described previously (20), α2β1 shows a steeper dependence on voltage than α1β1 or α3β1, and recent work shows that α2β2 shows a particularly steep dependence (21). The voltage dependence of the pump current in physiological conditions is mainly a reflection of Na+exc-mediated competition with K+exc leading to inhibition at negative potentials. The conditions of our experiments are equivalent to those in cells at 0 mV membrane potential, and compared with physiological conditions, the raised K0.5K+ of α2β2 represents, if anything, an underestimated value compared with α1β1. At physiological K+exc of 4.5 mm and resting membrane potentials in the heart or skeletal muscle or brain glial cells (−70 to 90 mV), α2β2 is largely inactive. Thus, α2β2 acts as a “reserve pump,” which responds to acutely increased K+exc or positive membrane potentials by increasing its rate and moderating the changes in K+exc and then restoring the ion gradients after the change (see Refs. 52 and 53). For example, increased activity of skeletal muscles leads to loss of potassium ions and a large increase in K+exc, which can reach as high as 8.3 mm in serum, 10–12 mm in muscle interstitial fluid, and locally as high as 25 mm in T-tubules (54). In heart muscle, the α2β2 is also almost inactive at resting potentials but becomes acutely activated during raised cardiac activity due to the fact that the membrane potential is positive for a significant fraction of the time during the extended action potentials (21). As in skeletal muscle, the potassium concentration in T-tubules can be presumed to rise significantly. The lower K0.5Na+ of α2β2 should also contribute significantly to rapid restoration of the sodium gradient associated with increased activity and raised cytoplasmic sodium. Due to coupling of the sodium fluxes mediated by α2β2 with sodium and calcium fluxes mediated by NCX1, the kinetic features of α2β2 described are expected to play an important role in regulation of the calcium dynamics of active versus resting cardiac muscle. Indeed, an important role of α2 overexpression in calcium dynamics in myocytes, associated with a decreased K0.5Na+, has been proposed recently (55), although it was not known which β isoform is coupled with α2. The α1β1 complex maintains ion gradients in resting conditions but is not suited for the regulatory role of α2β2 just discussed because the potassium sites (K0.5K+ = 1.5 ± 0.1 mm) are almost saturated even in resting conditions, and the voltage dependence of α1β1 is shallow.
Pharmacological Implications
As discussed in the Introduction, an α2-selective CG could be an efficient inotropic agent. The present findings have the additional interesting implication that an α2β2-selective CG, such as the digoxin derivatives described here, could also have reduced cardiotoxicity compared with digoxin.
For many years, digoxin was used routinely to treat heart failure, due to its inotropic and chronotropic effects, but it is now used much less on account of the narrow therapeutic range and cardiotoxicity resulting from the well known phenomena of calcium overload and cardiac arrhythmias (31). The incidence of digitalis toxicity has decreased in parallel with its decreased use (56). The main cause of digitalis toxicity is accumulation of digoxin, secondary to decreased renal function, and a major exacerbating factor is hypokalemia, which is prevalent in subjects treated with diuretics in addition to digitalis (56) (for a recent study, see Ref. 57). Indeed, due to the potentiation of digitalis toxicity by hypokalemia, it was even recommended, in the past, to treat severe cases by raising serum potassium (58).
The salient present finding is that inhibition of α1β1 by digoxin and all other CGs is strongly antagonized by raising potassium in the range of 2.5–20 mm, whereas α2β2 is unaffected (Fig. 7). Assuming that digoxin toxicity in vivo, exacerbated by hypokalemia, is associated with excessive Na,K-pump inhibition, this implies that toxicity is indeed mediated by α1β1. Conversely, the insensitivity of inhibition of α2β2 to potassium (2.5–20 mm) would imply that α2β2 does not play a major role in digitalis toxicity. This conclusion fits well with the rationale that α2β2-selective derivatives could be effective positive inotropic agents and also have reduced toxic effects.
Considerable evidence exists for the presence of endogenous CG-like compounds in mammalian tissues, such as ouabain or marinobufagenin, that may serve to regulate Na,K-ATPase activity (59–61). Our unpublished experiments show a higher sensitivity of α2β2 over α1β1 for ouabain (Ki = 63.7 ± 9.8 versus 153.0 ± 11.6 nm, respectively),7 suggesting that endogenous ouabain-like compounds may bind more efficiently to α2β2 isoform and thus specifically regulate the α2β2 ion pumping activity or α2-dependent signaling pathways. In particular, α2-mediated signaling may explain a specific role of the α2 subunit in the modulation of blood pressure under stress conditions (60–63).
Conclusions
In summary, our data demonstrate the specific association of the Na,K-ATPase α2 isoform with the β2 and the specific intracellular location of the α2β2 heterodimer. The distinct functional properties of human α2β2 are consistent with an important regulatory role in cardiac muscle contraction. Furthermore, isoform-selective inhibition by digoxin derivatives, such as DcB, suggests that they could be safer cardiac inotropic agents compared with digoxin itself.
Experimental Procedures
Materials
n-Dodecyl-β-d-maltopyranoside (catalogue no. D310) and C12E8 (25% (w/w), catalogue no. 0330) were purchased from Anatrace, and BD-Talon metal affinity resin was from Clontech (catalogue no. 635503). 1-Stearoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (SOPS) was purchased from Avanti Polar Lipids. PiColorLockTM was purchased from Innova Bioscience, and RH421 was from MoBiTec. All other reagents were purchased from Merck or Sigma-Aldrich at the highest quality level available.
Primary Antibodies
For immunofluorescent staining, the following monoclonal antibodies were used: Na,K-ATPase α1 subunit (mouse, clone C464.6, 1:20; Millipore) and Na,K-ATPase β1 subunit (mouse, clone M17 P5 F11, 1:100; Affinity Bioreagents). The polyclonal antibodies used were Na,K-ATPase α2 subunit (rabbit, 1:200; Millipore) and Na,K-ATPase β2 subunit (rabbit, 1:200; Millipore).
For Western blotting analysis, the following monoclonal antibodies were used: against the Na,K-ATPase α1 subunit (mouse, clone C464.6, 1:1000; Millipore), against the Na,K-ATPase β2 subunit (mouse, clone 35; BD Transduction Laboratories), and Na,K-ATPase β3 subunit (goat, 1:500; Santa Cruz Biotechnology, Inc.). Na,K-ATPase β1 subunit polyclonal antibody (rabbit; 1:5000) was a generous gift of Dr. W. James Ball, Jr. (University of Cincinnati).
Methods
Confocal Microscopy
Confocal microscopy images were acquired using the Zeiss LSM 510 laser scanning confocal microscope and ZEN 2009 software.
Isolation of Membrane Fractions from Mouse Heart
Mouse heart was homogenized with a tight Dounce homogenizer (Wheaton, Millwille, NY). Cell debris was removed by centrifugation (2000 × g, 10 min). The cleared homogenate was layered onto a 42% sucrose solution in 10 mm PIPES, 2 mm EGTA, 2 mm EDTA, pH 7.0, and spun in a Beckman SW28 swinging bucket rotor at 25,000 rpm for 1 h at 4 °C. The fraction at the interface of buffer/sucrose was collected and diluted to a total volume of 15 ml of 10 mm PIPES, 2 mm EGTA, 2 mm EDTA, pH 7.0. Membranes were spun down by centrifugation in a Beckman 75Ti rotor (35,000 rpm, 4 °C, 1 h). The pellet was resuspended in 10 mm PIPES/Tris buffer containing 2 mm EGTA and 2 mm EDTA, pH 7.0, by homogenization with a 2-ml Teflon homogenizer (Wheaton). The membranes were aliquoted, flash-frozen, and stored at −80 °C. Proteins were extracted by incubating membranes with 50 mm Tris, pH 7.5, containing 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and complete protease inhibitor mixture (1 tablet/50 ml at 4 °C for 30 min). Membrane extracts were clarified by centrifugation (100,000 × g, 1 h) at 4 °C. Where indicated, protein extracts were treated by PNGase F from Flavobacterium meningosepticum (New England Biolabs) according to the manufacturer's instructions before loading on SDS-PAGE.
Immunoprecipitation
Protein extracts from mouse heart membrane fractions (100–300 μg of protein) were incubated with 30 μl of the protein A-agarose suspension (Roche Diagnostics) in a total volume 1 ml of the extraction buffer at 4 °C with continuous rotation for at least 3 h (or overnight) to remove the components that non-specifically bind to protein A. The precleared cell extract was mixed with 10 μl of polyclonal antibodies against the Na,K-ATPase α2 subunit (Millipore) or 10 μl of polyclonal antibodies against the Na,K-ATPase α subunit (64) or 10 μl of polyclonal antibodies against the Na,K-ATPase β2 subunit (Millipore) and incubated with continuous rotation at 4 °C for 60 min. After the addition of 30 μl of the protein A-agarose suspension, the mixture was incubated at 4 °C with continuous rotation overnight. The bead-adherent complexes were washed three times on the beads and then eluted as described previously (65).
Where indicated, the bead-adherent proteins were treated with PNGase F. Deglycosylation by PNGase F was performed by incubation of the bead-adherent proteins with 1 μl of PNGase F in 30 μl of 50 mm sodium phosphate, pH 7.5, containing 1% Nonidet P-40 at 37 °C for 1 h. After incubation with glycosidases, the reaction mixture was separated from the beads. The adherent proteins were eluted from the beads by incubation in 30 μl of 2× SDS-PAGE sample buffer for 5 min at 80 °C. To account for possible dissociation of immunoprecipitated proteins from the beads during deglycosylation, the eluted proteins were combined with the reaction mixture. After separation by SDS-PAGE, the immunoprecipitated and co-immunoprecipitated proteins were analyzed by Western blotting by using appropriate antibodies.
Western Blotting Analysis
1–10 μg of proteins extracted from mouse heart membranes or 5–20 μl of proteins eluted from the protein A-conjugated agarose beads were loaded onto 4–12% gradient SDS-polyacrylamide gels (Invitrogen). Proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane (Bio-Rad), and detected by Western blotting analysis as described previously (65).
Immunofluorescent Staining
Mouse embryo sections and frozen tissue sections on FDA standard frozen tissue rat or human arrays (BioChain) were incubated with Dako Protein Block serum-free solution (Dako Corp.) for 30 min. Immunofluorescent staining was performed by a 1-h incubation with the primary antibodies followed by a 1-h incubation with Alexa Fluor 633- or Alexa Fluor 488-conjugated anti-mouse or anti-rabbit antibodies (Invitrogen).
Plasmid Construction for the Expression of α2β2 α2β3 Na,K-ATPase
Generation of pHil-D2 expression vector containing cDNA of human α1 and His10-tagged porcine (p) or human (h) β1 was described previously (35). cDNAs of human β2 and β3 in pSD5 were a gift from K. Geering (University Lausanne, Switzerland). Open reading frames and flanking regions of human β2 and β3 were amplified by PCR using primers containing BglII and SalI cleavage sites. The resulting fragments were subcloned into pHil-D2-hα2/His10-pβ1 to create pHil-D2-hα2/His10-hβ2 and pHil-D2-hα2/His10-hβ3, respectively. Correct integration and sequence was confirmed by sequencing.
Yeast transformation and clone selection have been described in detail (35). P. pastoris SMD1165 was grown in BMG (100 mm potassium phosphate, pH 6, 1.34% yeast nitrogen base, 4 × 10−5% biotin, 0.3% glycerol) to OD 6–8, and expression was induced in BMM (100 mm potassium phosphate, pH 6, 1.34% yeast nitrogen base, 4 × 10−5% biotin, 0.5% methanol added daily). Yeasts were transformed with linearized pHil-D2-human α2-human His10-β2 or human His10-β3, and His+/MutS clones were selected and grown at 20 °C for 3 days in baffled Erlenmeyer flasks, as described previously for α1β1 and α2β1 (35, 66). Under these conditions, only weak expression was observed for α2β2 and α2β3. It was then determined that expression of both α2β3 and α2β2 was transient and peaked between 15 and 19 h for α2β3 and between 16 and 28 h for α2β2. Further screening of expression temperature revealed 23 °C to be optimal. This transient expression was not observed for α1β1, α2β1, and α3β1 that were stably expressed for 3–5 days, which also resulted in a higher cell density and protein yield per liter of culture. To overcome this limitation for α2β3 and α2β2, the following three-phase growth/induction protocol was used. In phase I, glycerol batch cultivation, yeasts were grown in BMG in an aerated 10-liter vessel until OD reached 6–8. In phase II, the glycerol-fed batch phase, 0.05% glycerol/h was added to the culture. Glycerol feeding was continued until OD reached 13–15. Extending the fed batch to higher cell densities often led to excessive foaming and a loss of cells. In phase III, the induction phase, expression was induced by adding 0.5% methanol/day. No increase of OD was observed during this phase. All three phases were carried out at 23 °C. In addition to the increase in cell density, fed batch cultivation increased expression levels of Na,K-ATPase. Membranes prepared from cells grown in baffled spinner flasks showed >30% higher specific ouabain binding when compared with membranes from cells that did not undergo fed batch cultivation. The same increase was observed in Western blots of these membranes, indicating that most of the expressed protein was properly folded and functional.
Expression and Purification of α1β1FXYD1, α2β1FXYD1, α2β2FXYD1, and α2β3FXYD1 Complexes
The experiments have utilized the purified detergent-soluble αβFXYD1 complexes rather than the αβ complexes alone, because FXYD1 strongly stabilizes the proteins against thermal inactivation (67) and does not affect inhibition by ouabain or digoxin (25, 68). Membrane preparation and His tag purification on BD-Talon beads of recombinant human α1β1FXYD1 and α2β1FXYD1 Na,K-ATPase were done essentially as described previously (69–71). Yeast membrane expression and purification of recombinant α2β2 and α2β3 Na,K-ATPase were similar except where indicated (for details, see “Results”). Human FXYD1 was expressed in Escherichia coli and purified and reconstituted with αβ complexes on the beads to yield αβFXYD1 complexes (35, 67, 71). Elution buffers consisted of 200 mm imidazole, 100 mm NaCl, 20 mm MOPS/Tris, pH 7.4, 0.1 mg/ml C12E8, 0.01 mg/ml cholesterol, and 0.07 mg/ml SOPS. 25% glycerol was added, and the soluble protein complexes were stored at −80 °C.
Biochemical Assays
ATPase activity assays as well as titrations with NaCl, KCl, and vanadate were performed as described previously (29, 35) using PiColorLockTM malachite green assay (Innova Bioscience). For ion titrations, activity was measured at varying concentrations of sodium plus choline chloride and KCl (80 mm), with constant total ionic strength (170 mm total). K0.5Na+ and K0.5K+ values were obtained by fitting the data to the Hill equation using KaleidaGraph (Synergy Software) (30).
Inhibition of Na,K-ATPase activity and [3H]ouabain binding and K-[3H]digoxin displacement assays were performed as reported (25). For derivation of Ki values, percentage inhibition of Na,K-ATPase activity, VCG/V0, was calculated, and Ki values were obtained by fitting the data to the function, VCG/V0 = Ki/([CG] + Ki) + c. Inhibition was estimated in 3–7 separate experiments, and average Ki values ± S.E. were calculated (27).
Equilibrium and Stopped-flow Fluorescence Measurements Using RH421
Equilibrium fluorescence experiments were carried out in a Varian fluorimeter at room temperature. 10 μg of purified Na,K-ATPase was added to 1 ml of 20 mm MOPS/Tris, pH 7.2, 5 mm MgCl2, and 200 nm RH421. NaCl, ATP, and KCl where then added successively. NaCl and KCl titrations were performed as described previously (37) with emission and excitation wavelength set to 580 and 680 nm with a 5-nm slit width.
Stopped-flow measurements were made using an Applied Photophysics SX20 system (see Ref. 29). Using a combined xenon/mercury lamp, excitation wavelength was 577 nm, and fluorescence was measured at ≥665 nm using cut-off filters. Solutions were mixed 1:1 using ∼120 μl/syringe, and the temperature of the measurements was set to 23 °C. All solutions were buffered to pH 7.2 using MOPS/Tris, and ionic strength was kept constant at 120 mm for all measurements using choline chloride.
For measurement of E2(2Rb)ATP → E1·3NaATP, syringe 1 contained 20 μg/ml enzyme non-covalently labeled with 200 nm RH421 in 20 mm RbCl, 1 mm EDTA and was mixed with 80 mm NaCl, 2 mm ATP, 1 mm EDTA in syringe 2. For measurement of E1·3Na → E2P, 10 μg/ml Na,K-ATPase non-covalently labeled with 200 nm RH421 in 100 mm NaCl and 4 mm MgCl2 in syringe 1 was mixed with 1 mm ATP in the same solution in syringe 2.
Data were fitted using KaleidaGraph (Synergy Software). Ion titrations were fitted using the Hill equation,
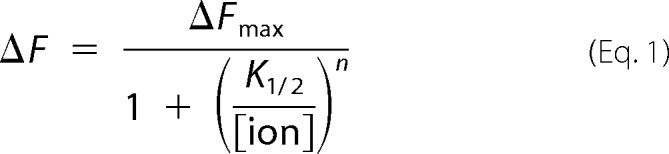 |
where n is the Hill coefficient, [ion] is the free concentration of the respective ion, and K½ is the concentration required to obtain the half-maximal fluorescence signal.
Stopped-flow traces were fitted to a monoexponential function,
or double exponential function,
where A is the amplitude of the fluorescence signal, k is the rate of the reaction, and c is the equilibrium fluorescence level after the reaction is complete. All values are expressed as averages of 2–4 experiments.
Molecular Modeling
Homology modeling of human Na,K-ATPase α2β1, α2β2, and α2β3 isoform complexes was carried out using the template α1β1 with bound digoxin (Protein Data Bank code 4RET) as described previously (26). The final model exhibits the highest Profiles 3D score (72) and the lowest number of Ramachandran violations (73). Rather consistent profiles were observed for each model (α2β1, α2β2, and α2β3), demonstrating that the human Na,K-ATPase models were reasonable and could be employed for the further docking study. The magnesium ion and three structural waters were positioned in each final model. Models were eventually refined by energy minimization using the CHARMm force field (74).
Molecular Docking
Molecular docking of DcB to the different human Na,K-ATPase isoform models was carried out in Discovery Studio version 4.0 (Biovia, Dassault Systemes, San Diego, CA) with CDOCKER, which is an implementation of a CHARMm-based docking tool using a rigid receptor (75). The DcB ligand was prepared before docking using the Prepare Ligand module to evaluate ionization states for a given pH, isomers, and tautomers, correct bad valences, and generate three-dimensional conformations. The digoxin bioactive binding conformation was copied from the 4RET crystal structure and positioned into each model. The model binding site was defined as a sphere with radius that stays within 15 Å from the geometric centroid of the digoxin ligand using the Define and Edit Binding Site tool.
DcB was docked into the active site of each Na,K-ATPase human model. Different ligand orientations were generated, and for each orientation, the CHARMm energy (interaction energy plus ligand strain) and the interaction energy alone were calculated. The ligand orientations were sorted by CHARMM energy, and the top scoring (most negative, thus favorable to binding) orientations were retained. The final orientations selected were chosen based on their docking scores, which favor interactions with amino acids from the β subunit.
Synthesis of Digoxin Derivatives
Detailed protocols for synthesis, purification, and analysis of perhydro-1–4-oxazepine derivatives of digoxin have been described previously (26, 27, 76).
Author Contributions
M. H. designed and performed cloning, expression, and biochemical experiments; analyzed data; and wrote and edited the manuscript. E. T. designed and performed experiments, analyzed data, and edited the manuscript. Y. N. performed expression and biochemical experiments and analyzed data. S. P. F. designed and performed experiments, analyzed data, and edited the manuscript. R. J. K. provided mouse embryos. E. B. Z. performed the molecular modeling. E. B.-D. performed molecular cloning and expression experiments. L. A. D. aided in conceptual experimental design and wrote sections of the manuscript. Z. F. wrote the section on digitalis toxicity. J. H. K. provided the isoform-nonspecific antibody against α subunit and edited the manuscript. G. S. edited the manuscript. D. M. T. synthesized digoxin derivatives. A. K. performed experiments and analyzed data. O. V. designed the study, performed experiments, analyzed data, and wrote the manuscript. S. J. D. K. designed and coordinated the study, planned experiments, and wrote the manuscript.
Acknowledgments
We thank Dr. W. James Ball, Jr. (University of Cincinnati) for providing an antibody against the Na,K-ATPase β1 subunit.
This work was supported by Israel Science Foundation (789/12) and US-Israel Binational Science Foundation (711993, to S. J. D. K.) and, in part, by National Institutes of Health Grants R37-HL48129 (to L. A. D.), RO1HL113350 (to L. A. D. and O. V.), USVA 2I01BX001006 (to G. S.), and 1RO1DK105156-01 (to G. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
All purified proteins described here are the αβFXYD1 complexes, although, for simplicity, when referring to or naming the different isoform complexes, the FXYD1 has been omitted.
A. Katz, O. Vagin, and S. J. D. Karlish, unpublished results.
- CG
- cardiac glycoside
- NCX1
- Na/Ca exchanger isoform 1
- C12E8
- octaethylene glycol monodecyl ether
- PNGase F
- peptide:N-glycosidase F
- RH421
- N-(4-sulfobutyl)-4-(4-(4-(dipentylamino)phenyl) butadienyl)pyridinium
- SOPS
- 1-stearoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- DMe
- methylamine
- DEt
- ethylamine
- DP
- propylamine
- DiP
- isopropylamine
- DcP
- cyclopropylamine
- DiB
- isobutylamine
- DMcP
- methylcyclo-propylamine
- DcB
- cyclobutylamine
- DAz
- azetidine-3-amine
- DTh
- thietane-3-amine
- DcPe
- cyclopentylamine
- DcHe
- cyclohexylamine
- DcB2,2dM
- 2,2-dimethylcyclobutylamine
- DMcB3,3dM
- methylcyclobutylamine-3,3-dimethyl
- DMSM
- (2-methylsulfonyl)methylamine
- DEMS
- (2-ethylsulfonyl)methylamine
- DESA
- (2-sulfonamide)ethylamine.
References
- 1. Bers D. M. (2008) Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 [DOI] [PubMed] [Google Scholar]
- 2. Harada K., Lin H., Endo Y., Fujishiro N., Sakamoto Y., and Inoue M. (2006) Subunit composition and role of Na+,K+-ATPases in ventricular myocytes. J. Physiol. Sci. 56, 113–121 [DOI] [PubMed] [Google Scholar]
- 3. McDonough A. A., Zhang Y., Shin V., and Frank J. S. (1996) Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am. J. Physiol. 270, C1221–1227 [DOI] [PubMed] [Google Scholar]
- 4. Sweadner K. J., Herrera V. L., Amato S., Moellmann A., Gibbons D. K., and Repke K. R. (1994) Immunologic identification of Na+,K+-ATPase isoforms in myocardium: isoform change in deoxycorticosterone acetate-salt hypertension. Circ. Res. 74, 669–678 [DOI] [PubMed] [Google Scholar]
- 5. Geering K. (2006) FXYD proteins: new regulators of Na-K-ATPase. Am. J. Physiol. Renal Physiol. 290, F241–F250 [DOI] [PubMed] [Google Scholar]
- 6. Pavlovic D., Fuller W., and Shattock M. J. (2013) Novel regulation of cardiac Na pump via phospholemman. J. Mol. Cell. Cardiol. 61, 83–93 [DOI] [PubMed] [Google Scholar]
- 7. Despa S., Lingrel J. B., and Bers D. M. (2012) Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc. Res. 95, 480–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dostanic I., Paul R. J., Lorenz J. N., Theriault S., Van Huysse J. W., and Lingrel J. B. (2005) The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am. J. Physiol. Heart Circ. Physiol. 288, H477–H485 [DOI] [PubMed] [Google Scholar]
- 9. James P. F., Grupp I. L., Grupp G., Woo A. L., Askew G. R., Croyle M. L., Walsh R. A., and Lingrel J. B. (1999) Identification of a specific role for the Na,K-ATPase α2 isoform as a regulator of calcium in the heart. Mol. Cell 3, 555–563 [DOI] [PubMed] [Google Scholar]
- 10. Shattock M. J., Ottolia M., Bers D. M., Blaustein M. P., Boguslavskyi A., Bossuyt J., Bridge J. H., Chen-Izu Y., Clancy C. E., Edwards A., Goldhaber J., Kaplan J., Lingrel J. B., Pavlovic D., Philipson K., Sipido K. R., and Xie Z. J. (2015) Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 593, 1361–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swift F., Tovsrud N., Enger U. H., Sjaastad I., and Sejersted O. M. (2007) The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc. Res. 75, 109–117 [DOI] [PubMed] [Google Scholar]
- 12. Juhaszova M., and Blaustein M. P. (1997) Distinct distribution of different Na+ pump α subunit isoforms in plasmalemma: physiological implications. Ann. N.Y. Acad. Sci. 834, 524–536 [DOI] [PubMed] [Google Scholar]
- 13. Silverman B. Z., Fuller W., Eaton P., Deng J., Moorman J. R., Cheung J. Y., James A. F., and Shattock M. J. (2005) Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc. Res. 65, 93–103 [DOI] [PubMed] [Google Scholar]
- 14. Mohler P. J., Davis J. Q., and Bennett V. (2005) Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3, e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berry R. G., Despa S., Fuller W., Bers D. M., and Shattock M. J. (2007) Differential distribution and regulation of mouse cardiac Na+/K+-ATPase α1 and α2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc. Res. 73, 92–100 [DOI] [PubMed] [Google Scholar]
- 16. Despa S., and Bers D. M. (2007) Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 293, C321–C327 [DOI] [PubMed] [Google Scholar]
- 17. Tokhtaeva E., Clifford R. J., Kaplan J. H., Sachs G., and Vagin O. (2012) Subunit isoform selectivity in assembly of Na,K-ATPase α-β heterodimers. J. Biol. Chem. 287, 26115–26125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tokhtaeva E., Sachs G., and Vagin O. (2009) Assembly with the Na,K-ATPase α1 subunit is required for export of β1 and β2 subunits from the endoplasmic reticulum. Biochemistry 48, 11421–11431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vagin O., Turdikulova S., and Sachs G. (2005) Recombinant addition of N-glycosylation sites to the basolateral Na,K-ATPase beta1 subunit results in its clustering in caveolae and apical sorting in HGT-1 cells. J. Biol. Chem. 280, 43159–43167 [DOI] [PubMed] [Google Scholar]
- 20. Crambert G., Hasler U., Beggah A. T., Yu C., Modyanov N. N., Horisberger J. D., Lelièvre L., and Geering K. (2000) Transport and pharmacological properties of nine different human Na,K-ATPase isozymes. J. Biol. Chem. 275, 1976–1986 [DOI] [PubMed] [Google Scholar]
- 21. Stanley C. M., Gagnon D. G., Bernal A., Meyer D. J., Rosenthal J. J., and Artigas P. (2015) Importance of the voltage dependence of cardiac Na/K ATPase isozymes. Biophys. J. 109, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Figtree G. A., Liu C. C., Bibert S., Hamilton E. J., Garcia A., White C. N., Chia K. K., Cornelius F., Geering K., and Rasmussen H. H. (2009) Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ. Res. 105, 185–193 [DOI] [PubMed] [Google Scholar]
- 23. Howie J., Tulloch L. B., Shattock M. J., and Fuller W. (2013) Regulation of the cardiac Na+ pump by palmitoylation of its catalytic and regulatory subunits. Biochem. Soc. Trans. 41, 95–100 [DOI] [PubMed] [Google Scholar]
- 24. Blanco G., Koster J. C., Sánchez G., and Mercer R. W. (1995) Kinetic properties of the α2β1 and α2β2 isozymes of the Na,K-ATPase. Biochemistry 34, 319–325 [DOI] [PubMed] [Google Scholar]
- 25. Katz A., Lifshitz Y., Bab-Dinitz E., Kapri-Pardes E., Goldshleger R., Tal D. M., and Karlish S. J. (2010) Selectivity of digitalis glycosides for isoforms of human Na,K-ATPase. J. Biol. Chem. 285, 19582–19592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katz A., Tal D. M., Heller D., Habeck M., Ben Zeev E., Rabah B., Bar Kana Y., Marcovich A. L., and Karlish S. J. (2015) Digoxin derivatives with selectivity for the α2β3 isoform of Na,K-ATPase potently reduce intraocular pressure. Proc. Natl. Acad. Sci. U.S.A. 112, 13723–13728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katz A., Tal D. M., Heller D., Haviv H., Rabah B., Barkana Y., Marcovich A. L., and Karlish S. J. (2014) Digoxin derivatives with enhanced selectivity for the α2 isoform of Na,K-ATPase: effects on intraocular pressure in rabbits. J. Biol. Chem. 289, 21153–21162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cirri E., Katz A., Mishra N. K., Belogus T., Lifshitz Y., Garty H., Karlish S. J., and Apell H. J. (2011) Phospholemman (FXYD1) raises the affinity of the human α1β1 isoform of Na,K-ATPase for Na ions. Biochemistry 50, 3736–3748 [DOI] [PubMed] [Google Scholar]
- 29. Habeck M., Haviv H., Katz A., Kapri-Pardes E., Ayciriex S., Shevchenko A., Ogawa H., Toyoshima C., and Karlish S. J. (2015) Stimulation, inhibition, or stabilization of Na,K-ATPase caused by specific lipid interactions at distinct sites. J. Biol. Chem. 290, 4829–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mishra N. K., Habeck M., Kirchner C., Haviv H., Peleg Y., Eisenstein M., Apell H. J., and Karlish S. J. (2015) Molecular mechanisms and kinetic effects of FXYD1 and phosphomimetic mutants on purified human Na,K-ATPase. J. Biol. Chem. 290, 28746–28759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wasserstrom J. A., and Aistrup G. L. (2005) Digitalis: new actions for an old drug. Am. J. Physiol. Heart Circ. Physiol. 289, H1781–H1793 [DOI] [PubMed] [Google Scholar]
- 32. Schwinger R. H., Bundgaard H., Müller-Ehmsen J., and Kjeldsen K. (2003) The Na,K-ATPase in the failing human heart. Cardiovasc. Res. 57, 913–920 [DOI] [PubMed] [Google Scholar]
- 33. André N., Cherouati N., Prual C., Steffan T., Zeder-Lutz G., Magnin T., Pattus F., Michel H., Wagner R., and Reinhart C. (2006) Enhancing functional production of G protein-coupled receptors in Pichia pastoris to levels required for structural studies via a single expression screen. Protein Sci. 15, 1115–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohen E., Goldshleger R., Shainskaya A., Tal D. M., Ebel C., le Maire M., and Karlish S. J. (2005) Purification of Na+,K+-ATPase expressed in Pichia pastoris reveals an essential role of phospholipid-protein interactions. J. Biol. Chem. 280, 16610–16618 [DOI] [PubMed] [Google Scholar]
- 35. Lifshitz Y., Petrovich E., Haviv H., Goldshleger R., Tal D. M., Garty H., and Karlish S. J. (2007) Purification of the human α2 Isoform of Na,K-ATPase expressed in Pichia pastoris: stabilization by lipids and FXYD1. Biochemistry 46, 14937–14950 [DOI] [PubMed] [Google Scholar]
- 36. Cantley L. C. Jr., Cantley L. G., and Josephson L. (1978) A characterization of vanadate interactions with the (Na,K)-ATPase: mechanistic and regulatory implications. J. Biol. Chem. 253, 7361–7368 [PubMed] [Google Scholar]
- 37. Habeck M., Cirri E., Katz A., Karlish S. J., and Apell H. J. (2009) Investigation of electrogenic partial reactions in detergent-solubilized Na,K-ATPase. Biochemistry 48, 9147–9155 [DOI] [PubMed] [Google Scholar]
- 38. Forbush B. (1983) Cardiotonic steroid binding to Na,K-ATPase. Curr. Top. Membr. Transp. 19, 167–201 [Google Scholar]
- 39. Gloor S., Antonicek H., Sweadner K. J., Pagliusi S., Frank R., Moos M., and Schachner M. (1990) The adhesion molecule on glia (AMOG) is a homologue of the β subunit of the Na,K-ATPase. J. Cell Biol. 110, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bossuyt J., Despa S., Han F., Hou Z., Robia S. L., Lingrel J. B., and Bers D. M. (2009) Isoform specificity of the Na/K-ATPase association and regulation by phospholemman. J. Biol. Chem. 284, 26749–26757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vagin O., Turdikulova S., and Tokhtaeva E. (2007) Polarized membrane distribution of potassium-dependent ion pumps in epithelial cells: different roles of the N-glycans of their β subunits. Cell Biochem. Biophys. 47, 376–391 [DOI] [PubMed] [Google Scholar]
- 42. Vagin O., Sachs G., and Tokhtaeva E. (2007) The roles of the Na,K-ATPase β1 subunit in pump sorting and epithelial integrity. J. Bioenerg. Biomembr. 39, 367–372 [DOI] [PubMed] [Google Scholar]
- 43. Antonicek H., Persohn E., and Schachner M. (1987) Biochemical and functional characterization of a novel neuron-glia adhesion molecule that is involved in neuronal migration. J. Cell Biol. 104, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmalzing G., Kröner S., Schachner M., and Gloor S. (1992) The adhesion molecule on glia (AMOG/β2) and α1 subunits assemble to functional sodium pumps in Xenopus oocytes. J. Biol. Chem. 267, 20212–20216 [PubMed] [Google Scholar]
- 45. Eakle K. A., Kabalin M. A., Wang S. G., and Farley R. A. (1994) The influence of beta subunit structure on the stability of Na+/K+-ATPase complexes and interaction with K+. J. Biol. Chem. 269, 6550–6557 [PubMed] [Google Scholar]
- 46. Colonna T. E., Huynh L., and Fambrough D. M. (1997) Subunit interactions in the Na,K-ATPase explored with the yeast two-hybrid system. J. Biol. Chem. 272, 12366–12372 [DOI] [PubMed] [Google Scholar]
- 47. Lutsenko S., and Kaplan J. H. (1993) An essential role for the extracellular domain of the Na,K-ATPase β-subunit in cation occlusion. Biochemistry 32, 6737–6743 [DOI] [PubMed] [Google Scholar]
- 48. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., and Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 49. Shinoda T., Ogawa H., Cornelius F., and Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 50. Kanai R., Ogawa H., Vilsen B., Cornelius F., and Toyoshima C. (2013) Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 502, 201–206 [DOI] [PubMed] [Google Scholar]
- 51. Laursen M., Gregersen J. L., Yatime L., Nissen P., and Fedosova N. U. (2015) Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. U.S.A. 112, 1755–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Larsen B. R., Assentoft M., Cotrina M. L., Hua S. Z., Nedergaard M., Kaila K., Voipio J., and MacAulay N. (2014) Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62, 608–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. DiFranco M., Hakimjavadi H., Lingrel J. B., and Heiny J. A. (2015) Na,K-ATPase α2 activity in mammalian skeletal muscle T-tubules is acutely stimulated by extracellular K+. J. Gen. Physiol. 146, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clausen T. (2008) Regulatory role of translocation of Na+-K+ pumps in skeletal muscle: hypothesis or reality? Am. J. Physiol. Endocrinol. Metab. 295, E727–E728; author reply 729 [DOI] [PubMed] [Google Scholar]
- 55. Correll R. N., Eder P., Burr A. R., Despa S., Davis J., Bers D. M., and Molkentin J. D. (2014) Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 114, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang E. H., Shah S., and Criley J. M. (2012) Digitalis toxicity: a fading but crucial complication to recognize. Am. J. Med. 125, 337–343 [DOI] [PubMed] [Google Scholar]
- 57. Chan K. E., Lazarus J. M., and Hakim R. M. (2010) Digoxin associates with mortality in ESRD. J. Am. Soc. Nephrol. 21, 1550–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoffman B. K., and Bigger J. T. (1985) Digitalis and allied cardiac glycosides. in The Pharmacological Basis of Therapeutics, 7th Ed (Goodman A., Gilman L. S., Rall T. W., and Murad F., eds) pp. 716–747, Macmillan, New York [Google Scholar]
- 59. Bagrov A. Y., Shapiro J. I., and Fedorova O. V. (2009) Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 61, 9–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Blaustein M. P., Zhang J., Chen L., Song H., Raina H., Kinsey S. P., Izuka M., Iwamoto T., Kotlikoff M. I., Lingrel J. B., Philipson K. D., Wier W. G., and Hamlyn J. M. (2009) The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension 53, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lingrel J. B. (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 72, 395–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rindler T. N., Dostanic I., Lasko V. M., Nieman M. L., Neumann J. C., Lorenz J. N., and Lingrel J. B. (2011) Knockout of the Na,K-ATPase α2-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 301, H1396–H1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Loreaux E. L., Kaul B., Lorenz J. N., and Lingrel J. B. (2008) Ouabain-Sensitive α1 Na,K-ATPase enhances natriuretic response to saline load. J. Am. Soc. Nephrol. 19, 1947–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gatto C., Wang A. X., and Kaplan J. H. (1998) The M4M5 cytoplasmic loop of the Na,K-ATPase, overexpressed in Escherichia coli, binds nucleoside triphosphates with the same selectivity as the intact native protein. J. Biol. Chem. 273, 10578–10585 [DOI] [PubMed] [Google Scholar]
- 65. Tokhtaeva E., Sachs G., and Vagin O. (2010) Diverse pathways for maturation of the Na,K-ATPase β1 and β2 subunits in the endoplasmic reticulum of Madin-Darby canine kidney cells. J. Biol. Chem. 285, 39289–39302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Strugatsky D., Gottschalk K. E., Goldshleger R., Bibi E., and Karlish S. J. (2003) Expression of Na+,K+-ATPase in Pichia pastoris: analysis of wild type and D369N mutant proteins by Fe2+-catalyzed oxidative cleavage and molecular modeling. J. Biol. Chem. 278, 46064–46073 [DOI] [PubMed] [Google Scholar]
- 67. Mishra N. K., Peleg Y., Cirri E., Belogus T., Lifshitz Y., Voelker D. R., Apell H. J., Garty H., and Karlish S. J. (2011) FXYD proteins stabilize Na,K-ATPase: amplification of specific phosphatidylserine-protein interactions. J. Biol. Chem. 286, 9699–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Crambert G., Fuzesi M., Garty H., Karlish S., and Geering K. (2002) Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc. Natl. Acad. Sci. U.S.A. 99, 11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haviv H., Cohen E., Lifshitz Y., Tal D. M., Goldshleger R., and Karlish S. J. (2007) Stabilization of Na+,K+-ATPase purified from Pichia pastoris membranes by specific interactions with lipids. Biochemistry 46, 12855–12867 [DOI] [PubMed] [Google Scholar]
- 70. Haviv H., Habeck M., Kanai R., Toyoshima C., and Karlish S. J. (2013) Neutral phospholipids stimulate Na,K-ATPase activity: a specific lipid-protein interaction. J. Biol. Chem. 288, 10073–10081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kapri-Pardes E., Katz A., Haviv H., Mahmmoud Y., Ilan M., Khalfin-Penigel I., Carmeli S., Yarden O., and Karlish S. J. (2011) Stabilization of the α2 isoform of Na,K-ATPase by mutations in a phospholipid binding pocket. J. Biol. Chem. 286, 42888–42899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bowie J. U., Lüthy R., and Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 [DOI] [PubMed] [Google Scholar]
- 73. Lovell S. C., Davis I. W., Arendall W. B. 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Calpha geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 74. Brooks B. R., Brooks C. L. 3rd, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu G., Robertson D. H., Brooks C. L. 3rd, and Vieth M. (2003) Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 24, 1549–1562 [DOI] [PubMed] [Google Scholar]
- 76. Tal D. M., Karlish S. J., and Gottlieb H. E. (2016) An NMR study of new cardiac glycoside derivatives. Magn. Reson. Chem. 54, 260–262 [DOI] [PubMed] [Google Scholar]