

Abstract
GoxA is a glycine oxidase that possesses a cysteine tryptophylquinone (CTQ) cofactor that is formed by posttranslational modifications that are catalyzed by a modifying enzyme GoxB. It is the second known tryptophylquinone enzyme to function as an oxidase, the other being the lysine ϵ-oxidase, LodA. All other enzymes containing CTQ or tryptophan tryptophylquinone (TTQ) cofactors are dehydrogenases. Kinetic analysis of GoxA revealed allosteric cooperativity for its glycine substrate, but not O2. This is the first CTQ- or TTQ-dependent enzyme to exhibit cooperativity. Here, we show that cooperativity and homodimer stabilization are strongly dependent on the presence of Phe-237. Conversion of this residue, which is a Tyr in LodA, to Tyr or Ala eliminates the cooperativity and destabilizes the dimer. These mutations also significantly affect the kcat and Km values for the substrates. On the basis of structural and modeling studies, a mechanism by which Phe-237 exerts this influence is presented. Two active site residues, Asp-547 and His-466, were also examined and shown by site-directed mutagenesis to be critical for CTQ biogenesis. This result is compared with the results of similar studies of mutagenesis of structurally conserved residues of other tryptophylquinone enzymes. These results provide insight into the roles of specific active-site residues in catalysis and CTQ biogenesis, as well as describing an interesting mechanism by which a single residue can dictate whether or not an enzyme exhibits cooperative allosteric behavior toward a substrate.
Keywords: cooperativity, enzyme kinetics, enzyme processing, oxidase, protein self-assembly, protein structure-function, quinoprotein
Introduction
The glycine oxidase, GoxA (1), is the second enzyme to be isolated from the melanogenic marine bacterium Marinomonas mediterranea, which bears the protein-derived cysteine tryptophylquinone (CTQ)2 cofactor (2, 3), the first being the lysine-ϵ-oxidase, LodA (4). The biosynthesis of the CTQ cofactor of each enzyme requires posttranslational modifications that are catalyzed by a specific flavoenzyme, GoxB or LodB, respectively (5, 6). In each case, the gene clusters that encode these two enzymes contain only two genes, goxA and goxB, and lodA and lodB, respectively. In addition to the interesting features of the biosynthesis of CTQ in these enzymes, they exhibit atypical activities. All other known tryptophylquinone enzymes that contain either CTQ or tryptophan tryptophylquinone (TTQ) are dehydrogenases (2, 3), whereas GoxA and LodA are oxidases. The current study characterizes the steady-state kinetic properties of GoxA and variants generated by site-directed mutagenesis. Despite the similarities of GoxA and LodA, the results reveal that the kinetic mechanisms of the two enzymes and the roles of conserved active-site residues in their overall structure and function are different.
LodA catalyzes the oxidative deamination of the side chain of l-lysine to generate a semialdehyde, ammonia, and H2O2 as products (4). The enzyme is secreted, and as a consequence of the H2O2 generation, it exhibits antimicrobial properties that lead to the dispersal and differentiation of the biofilm in which the host bacterium resides (7). Steady-state kinetic analysis using its substrates, lysine and O2, showed that LodA follows a ping-pong kinetic mechanism involving a Schiff base intermediate formed between the ϵ-amino group and CTQ (8). This was verified by the crystal structure of LodA with the lysine-CTQ adduct formed (9). The roles of residues in and around the active site of the LodA in catalysis and CTQ biogenesis were identified by site-directed mutagenesis (10).
GoxA catalyzes the oxidative deamination of glycine to generate glyoxylate, ammonia, and H2O2 as products (1). Although LodA exhibits typical Michaelis-Menten behavior, the current steady-state kinetic study of GoxA reveals that it exhibits allosteric cooperativity toward the glycine substrate. It was shown previously that purified active GoxA eluted primarily as a dimer during size exclusion chromatography (11). As the crystal structure of GoxA has yet to be determined, a homology model has been constructed from the GoxA sequence and the LodA structure. Given the observed cooperativity and dimeric structure, a docking model is presented in which the GoxA monomer is docked with itself to form a putative dimer. The docking model predicts that the entrances to the active site of each of the two monomers are in the proximity of the protein-protein interface.
Phylogenetic analysis of the Integrated Microbial Genomes database of genome sequences (12) revealed that LodA-like proteins can be clustered in five different major groups that were detected in several classes of bacteria and fungi. LodA is among the proteins classified as Group IA LodA-like proteins, and GoxA is present in Group II. Comparison of the sequences of the Group IA and Group II proteins with the structure of LodA and a homology model of GoxA revealed candidates to be used as targets for site-directed mutagenesis (Fig. 1). Of particular interest is Phe-237. Although all Group II proteins have a Phe in this position of the sequences, most Group IA proteins including LodA have a Tyr in this position (Fig. 2) (12). Here we demonstrate that mutation of Phe-237 to Tyr or Ala eliminates the cooperativity exhibited by GoxA and weakens dimer formation. His-466 is of interest because all Group II proteins each have His in this position, which is in the proximity of CTQ in the active site, whereas the Group IA proteins all have a Cys at the corresponding position. Asp-547, which also resides near CTQ, was of interest as this residue is sequence-conserved in all Group IA and Group II proteins, as well as being spatially conserved in all other structurally characterized TTQ- and CTQ-dependent enzymes regardless of the different protein folds (13–15). The results presented herein provide insight into the roles of specific active-site residues in catalysis and CTQ biogenesis, as well as describing an interesting mechanism by which a single residue can dictate whether or not an enzyme exhibits cooperative allosteric behavior toward a substrate.
FIGURE 1.
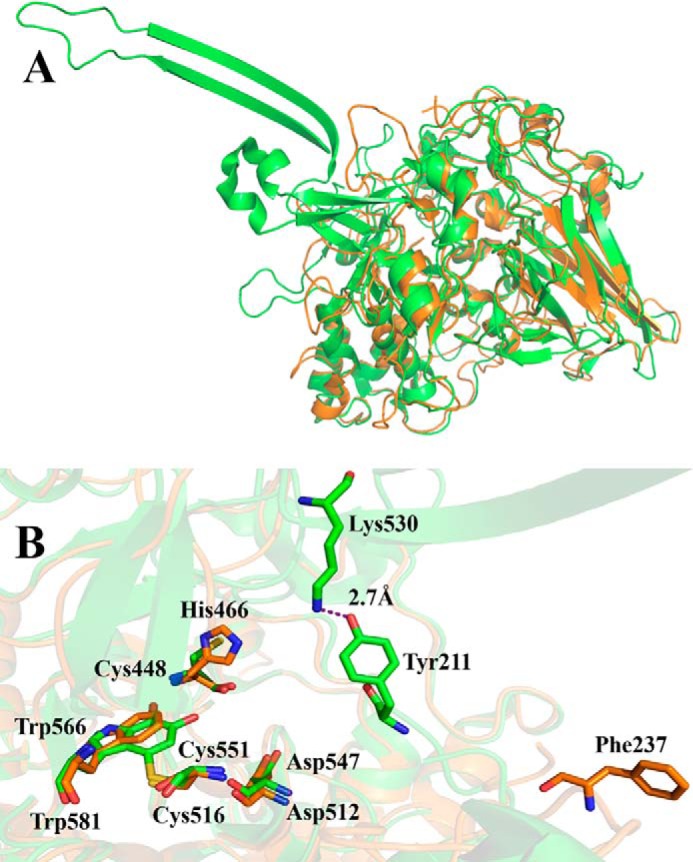
The homology model of GoxA superimposed with the crystal structure of LodA. A, the overall structures of monomers of LodA (green) and GoxA (orange) are superimposed. B, the active sites of GoxA and LodA are superimposed. The Cys and Trp residues, which form CTQ, and selected other residues are shown as sticks. Carbons on residues of LodA and GoxA are colored green and orange, respectively. Oxygen is colored red, nitrogen is blue, and sulfur is yellow. The model was generated using the sequence of the goxA gene (Marme_1655) and the crystal structure of LodA (PDB ID 3WEU) (9).
FIGURE 2.

Structure-based sequence alignment GoxA and LodA. The alignment of sequences is based on the homology model in Fig. 1. The Cys and Trp residues, which form CTQ, are in blue. The conserved residues, Cys-448 and Asp-512 of LodA and His-466 and Asp-547 of GoxA, are in purple. The loop that contains Phe-237 of GoxA and Tyr-211 of LodA is shaded yellow. The β-hairpin structural feature in LodA that contains Lys-530 and that is absent in GoxA is shaded green. The loop that contains Tyr-618 and Trp-619 in GoxA and that is absent in LodA is shaded orange. Key residues are in red.
Results
Steady-state Kinetic Analysis of WT GoxA
Initially, the concentration of the glycine was varied in the presence of 100% O2-saturated (1150 μm) buffer. Initial rates were measured, and the data were fit to the Michaelis-Menten equation (see Equation 1 under “Experimental Procedures”) (Fig. 3A). Although the fit was reasonably good (R2 = 0.97), the error ranges were high (kcat = 56 ± 5 s−1 and Km = 999 ± 172 μm) and a systematic deviation to the fit was evident (i.e. data points clustered above and below the fitted curve). The data were then fit to the Hill equation (see Equation 2 under “Experimental Procedures”) (Fig. 3B), and an improved fit was obtained (R2 = 0.99), with all data points falling on the fitted curve. The analysis yielded values of kcat = 39 ± 2 s−1, K0.5 = 514 ± 42 μm for glycine, and a Hill coefficient of 1.7 ± 0.2, which is indicative of positive cooperativity. Given the previous finding that GoxA is a homodimer (11), the h value of 1.7 (where h is the Hill coefficient) is consistent with a kinetic mechanism in which the binding of the first glycine molecule to one active site enhances the affinity of a second active site for glycine. The concentration of O2 was varied in the presence of a saturating concentration of glycine (5 mm). In this case, the data fit well to the Michaelis-Menten equation (Fig. 3C), and no improvement of the fit was obtained by using the Hill equation (Fig. 3D). Thus, GoxA does not exhibit cooperative behavior toward O2, but only toward glycine. The analysis of the data using Equation 1 (see “Experimental Procedures”) yielded values of kcat = 93 ± 18 s−1 and Km = 2590 ± 690 μm for O2. Because the apparent Km for O2 is much larger than concentration in a 100% O2-saturated solution, the perceived saturation kinetics in Fig. 3C is largely due to the single data point at 1150 μm, yielding relatively large errors in kcat/Km that are determined from the fitted parameters. As such, a second order rate constant (kcat/KmO2) was also determined from the slope of a line fit through the 0–600 μm data points (Fig. 3C, inset). Each of the values are listed in Table 1.
FIGURE 3.

Steady-state kinetic analysis of GoxA. A–D, assays of glycine oxidase activity were performed with varied concentrations of glycine in the presence of a fixed concentration of 1150 μm O2 (A and B) or with varied concentrations of O2 in the presence of a fixed concentration of 5 mm glycine (C and D). The lines are fits of each data set by either Equation 1 (A and C) or Equation 2 (B and D). The inset in C is a fit to a straight line of the data set with the last point omitted. Each data point was the average of a minimum of two replicates with error bars shown. In some cases, the error bars are not evident because the values were so similar that the bars are obscured by the data point.
TABLE 1.
Steady-state kinetic parameters for GoxA and GoxA variant proteins
NA, not applicable.
| WT GoxA | F237Y GoxA | F237A GoxA | LodAa | |
|---|---|---|---|---|
| kcat (s−1)b | 93 ± 18 | 15 ± 5 | 0.6 ± 0.1 | 0.22 ± 0.04 |
| K0.5 glycine (μm) | 514 ± 42c | NA | NA | NA |
| Km glycine (μm) | NA | 3030 ± 840 | 826 ± 123 | NA |
| Km lysine (μm) | NA | NA | NA | 3.2 ± 0.5 |
| Km O2 (μm) | 2600 ± 693 | 2270 ± 1000 | 416 ± 157 | 37.2 ± 6.1 |
| kcat/Km O2 (m−1 s−1)d | 36,000 ± 12,000 | 6600 ± 3600 | 1000 ± 400 | 5900 ± 1400 |
| kcat/Km O2 (m−1 s−1)e | 28,000 ± 870 | 5670 ± 250 | NA | NA |
a Taken from Ref. 8.
b kcat values are from experiments in which O2 was varied rather than from studies where glycine was varied because it was not possible to achieve saturating concentration of O2.
c WT GoxA fit best to Equation 2, and so the K0.5 value is used rather than a Km value.
d Determined from the fitted parameters.
Given the very large apparent Km for O2, the assays were repeated anaerobically in the presence of alternative small molecule electron acceptors to see whether GoxA could function as a dehydrogenase. However, no activity was observed when replacing O2 with NAD+, ferricyanide, phenazine ethosulfate, or dichlorophenol-indophenol. Furthermore, the kcat obtained when varying O2 was 93 s−1 as compared with 39 s−1 when varying glycine. The reason for this is that the fixed concentration of 100% O2-saturated buffer that was used, which is the highest that can be achieved, was not a saturating concentration of substrate with respect to the enzyme (i.e. it was not much greater than the Km for O2). Thus, the lower value is an apparent kcat determined at a sub-saturating concentration of the fixed substrate, and the value of 93 s−1 determined by varying O2 at a true saturating concentration of glycine is a more valid estimation of the true kcat. Because of the cooperative behavior toward glycine and the inability to achieve truly saturating concentrations of O2, it was not possible to perform the experiments in which both substrates could be varied over the appropriate range of concentrations necessary to determine whether or not GoxA obeys a ping-pong mechanism, as was done with LodA (8).
Structural Comparisons of GoxA with LodA
The crystal structure of GoxA has not yet been determined, but it was possible to construct a homology model of the structure based on its sequence and a crystal structure of LodA (Fig. 1A). Alignment of the homology model of the structure of GoxA with the crystal structure of LodA highlights strong structural conservation of the active sites (Fig. 1B) (16). It shows that the residues Cys-551 and Trp-566 are in close proximity to each other in positions similar to the residues that form CTQ in LodA. This matches the previous finding that these residues were shown by proteolysis and mass spectrometry of GoxA to form CTQ (16), thus supporting the validity of the model. For reasons discussed earlier, Asp-547, His-466, and Phe-237 of GoxA were chosen as candidates for site-directed mutagenesis to elucidate structure-function relationships. Phe-237 is of interest because it corresponds in the primary sequence to Tyr-211 of LodA (Fig. 2), which is conserved in most Group I proteins and which strongly influenced the Km values of both lysine and O2 for LodA (10). In the homology model, the position of Phe-237 is not structurally conserved with Tyr-211 in LodA, as are the positions of CTQ, Asp-547/Asp-512 and His-466/Cys-448 (GoxA/LodA numbering, respectively). Furthermore, in LodA, Lys-530 forms a hydrogen bond with Tyr-211, but no Lys is present in this position in GoxA. In fact, a β-hairpin structural feature containing this Lys in LodA is absent in GoxA (Fig. 2).
Effects of Site-directed Mutagenesis on Active-site Residues of GoxA
Asp-547
Mutation of Asp-512 in LodA affected CTQ biosynthesis, and so its effect on the activity of the mature enzyme could not be assayed (16). The analogous D547A mutation was made in GoxA. This resulted in protein with no activity. Furthermore, the inactive protein was isolated primarily in complex with the modifying enzyme GoxB. It was shown previously that a precursor of WT GoxA, which lacks CTQ, forms a tight complex with GoxB (11). As indicated in that previous study, the presence or absence of CTQ in GoxA when in complex with GoxB could be determined using a staining protocol that is specific for covalent quinoproteins (17) after transfer of proteins, which were run on SDS-PAGE by electroblot to a nitrocellulose membrane. The protein-stained gel and quinoprotein-stained blot are shown in Fig. 4. LodA, which is known to contain CTQ (9), and active WT GoxA (lanes 3 and 4) stain positive. The WT GoxA precursor lacking CTQ in complex GoxB (lane 5) shows no stain for either protein. The complex of D547A GoxA with GoxB (lane 1) does not stain, consistent with the lack of CTQ formation. It should be noted that WT GoxA typically runs as a doublet on SDS-PAGE for unknown reasons, and that D547A GoxA also exhibits some degradation.
FIGURE 4.

SDS-PAGE and quinoprotein staining of protein fractions. A, 10% gel stained for protein. B, electroblot stained for quinoproteins. A gel identical to that in A was subjected to electrophoretic transfer of the proteins and stained for quinoproteins as described under “Experimental Procedures”. The positions of migration of molecular mass markers in the stained gel are indicated. Lane 1, D547A GoxA as isolated in complex with GoxB. Lane 2, H466A GoxA as isolated in complex with GoxB. Lane 3, WT LodA. Lane 4, active WT GoxA; Lane 5, inactive WT GoxA in complex with GoxB. The molecular masses of the proteins of interest are: LodA, 82,905 kDa; GoxA, 76,285 kDa; and GoxB, 41,856 kDa.
His-466
His-466 corresponds to the Cys-448 of LodA. This LodA residue was previously mutated to Ala, Asp, and His (10). No stable C448H LodA protein was isolated, suggesting that the mutation affected the biosynthesis or stability of the protein, or both. C448A LodA and C448D LodA could be isolated. Those mutations had little effect on kcat, but each increased the Km values for both lysine and O2 with a greater impact on lysine (10). The corresponding H466C, H466A, and H466D mutations were made in GoxA. As described above for D547A GoxA, each of these three variant proteins in which His-466 was mutated could only be isolated in a complex with GoxB and exhibited no activity. As shown in Fig. 4, the H466A GoxA in complex with GoxB (lane 2) also did not stain for the presence of a quinone, indicating that this mutation also compromised CTQ formation. Thus, in GoxA, it appears that His-466 is also important for GoxB-dependent CTQ biosynthesis. As such, the roles of His-466 and Asp-547 in GoxA catalysis could not be studied further, as the mutations compromised CTQ biosynthesis.
Phe-237
Phe-237 corresponds to the Tyr-211 of LodA. That LodA residue was previously mutated to Phe and Ala. Y211A LodA was inactive, and Y211F LodA exhibited a decreased kcat and significantly increased Km for both lysine and O2 (10). Phe-237 in GoxA was converted to Tyr and Ala. The F237Y GoxA variant protein was isolated and assayed. As with WT GoxA, activity was measured at varied concentrations of the glycine in the presence of O2-saturated (1150 μm) buffer. In contrast to what was observed for WT GoxA, the data for F237Y GoxA fit well to the Michaelis-Menten model (Fig. 5A). No improvement of the fit was obtained using Equation 2 (see “Experimental Procedures”), and the fit to that equation yielded a Hill coefficient value of ∼1. This indicates that the cooperativity toward glycine exhibited by WT GoxA is lost as a consequence of the F237Y mutation. Activity was also assayed by varying the concentration of O2 in the presence of 5 mm glycine (Fig. 5B). Comparison of the fitted values of the kinetic parameters (Table 1) indicates that the F237Y mutation decreased kcat and increased Km for glycine with little effect on the Km for O2. As discussed earlier for WT GoxA, the very large apparent Km value led to large errors in the fitted parameters. As such, a second order rate constant (kcat/KmO2) was also determined from the slope of a line fit through the 0–600 mm data points (Fig. 5B, inset), and each of the values is listed in Table 1. The F237A GoxA variant protein was also isolated and analyzed (Fig. 5, C and D). This variant also obeyed Michaelis-Menten kinetics without cooperativity toward glycine. The fitted values of the kinetic parameters (Table 1) indicate that this mutation substantially reduced kcat, had little effect on the Km for glycine, and substantially decreased the Km for O2, such that saturation behavior was clearly observed (Fig. 5D). Thus, Phe-237 not only dictates cooperative behavior toward glycine but also influences the kcat and Km values for both glycine and O2.
FIGURE 5.

Steady-state kinetic analysis of GoxA variants. A and B, assays of the glycine oxidase activity of F237Y GoxA were performed with varied concentrations of glycine in the presence of a fixed concentration of 1150 μm O2 (A) or with varied concentrations of O2 in the presence of a fixed concentration of 5 mm glycine (B). C and D, assays of the glycine oxidase activity of F237A GoxA were performed with varied concentrations of glycine in the presence of a fixed concentration of 1150 μm O2 (C) or with varied concentrations of O2 in the presence of a fixed concentration of 5 mm glycine (D). The lines are fits to each data set by Equation 1. The inset in B is a fit to a straight line of the data set with the last point omitted. Each data point was the average of a minimum of two replicates with error bars shown. In some cases, the error bars are not evident because the values were so similar that the bars are obscured by the data point.
Effects of Phe-237 Mutations on the Oligomerization State of GoxA
To determine whether mutations of Phe-237, which abolished cooperativity, also affected the stabilization of the native homodimeric state of GoxA, the WT and variant proteins were analyzed by size exclusion chromatography. WT GoxA (Fig. 6A) elutes from the column primarily as a dimer, as was reported previously (11). One observes a minor peak at ∼76 kDa (16%), which corresponds to the predicted mass of a monomer, and a major peak of the dimer at ∼140 kDa (84%). In contrast, the majority of F237Y GoxA (Fig. 6B) elutes as a monomer with a major peak at 76 kDa (62%) and a minor peak at 140 kDa (38%). The majority of F237A GoxA (Fig. 6C) also elutes as a monomer with a major peak at 76 kDa (68%) and a minor peak at 140 kDa (32%). Thus, each of these mutations of Phe-237 weakens the interactions that stabilize the dimeric structure, but they do not totally abolish dimer formation.
FIGURE 6.
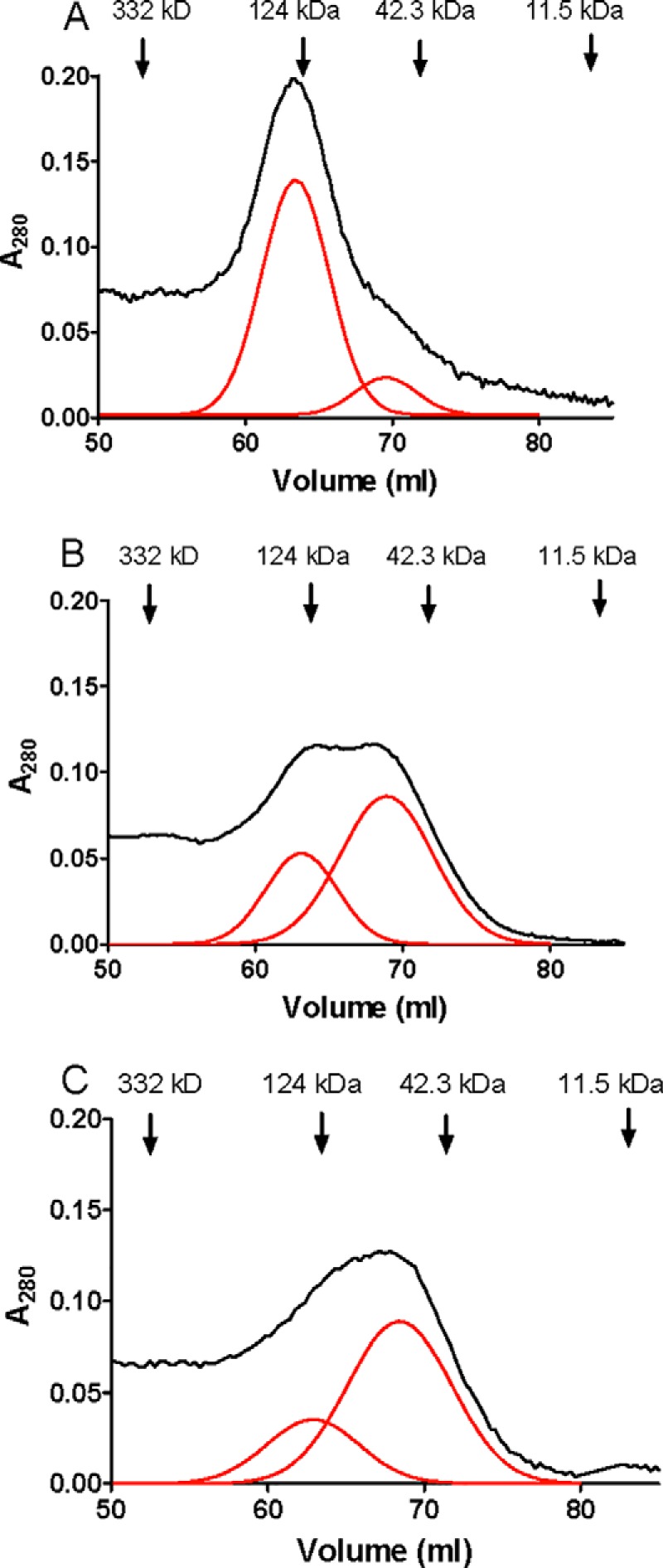
Size exclusion chromatography of WT GoxA and GoxA variants. A, WT GoxA. B, F237Y GoxA. C, F237A GoxA. The positions of the elution of the molecular mass marker proteins are indicated. The raw data are shown in black. Gaussian fits of the chromatogram that resolve overlapping peaks in the chromatogram are shown in red.
A Docking Model of the GoxA Dimer
To gain insight into how Phe-237 might be involved in the cooperativity and stabilization of dimer formation, a protein docking model was constructed using the ZDOCK program (Fig. 7). Interestingly, it shows that the Phe-237 resides at the protein-protein interface, and that the Phe-237 residues of each subunit are in close proximity to each other when present as a dimer. Phe-237 is also in a position in which it may interact with other aromatic amino acids residues that are present at the interface, Tyr-618 and Trp-619. In fact, the Tyr-618 residues of the two monomers were placed within 2 Å of each other. The distance between the Trp-691 residues of each monomer is ∼4 Å. The aromatic residues being in such close proximity suggests the possibility of π-stacking between the side chains. Because the model used to estimate the distances between these residues is a homology model, the values thus obtained are approximate. However, π-stacking interactions between residues would likely induce the other amino acids to rearrange themselves. Thus, these residues may be important in the stabilization of the dimeric structure and the communication between the two active sites that results in the observed cooperativity of WT GoxA. It is noteworthy that the loop containing these two residues of GoxA, Tyr-618 and Trp-619, is not found on LodA (Fig. 2).
FIGURE 7.
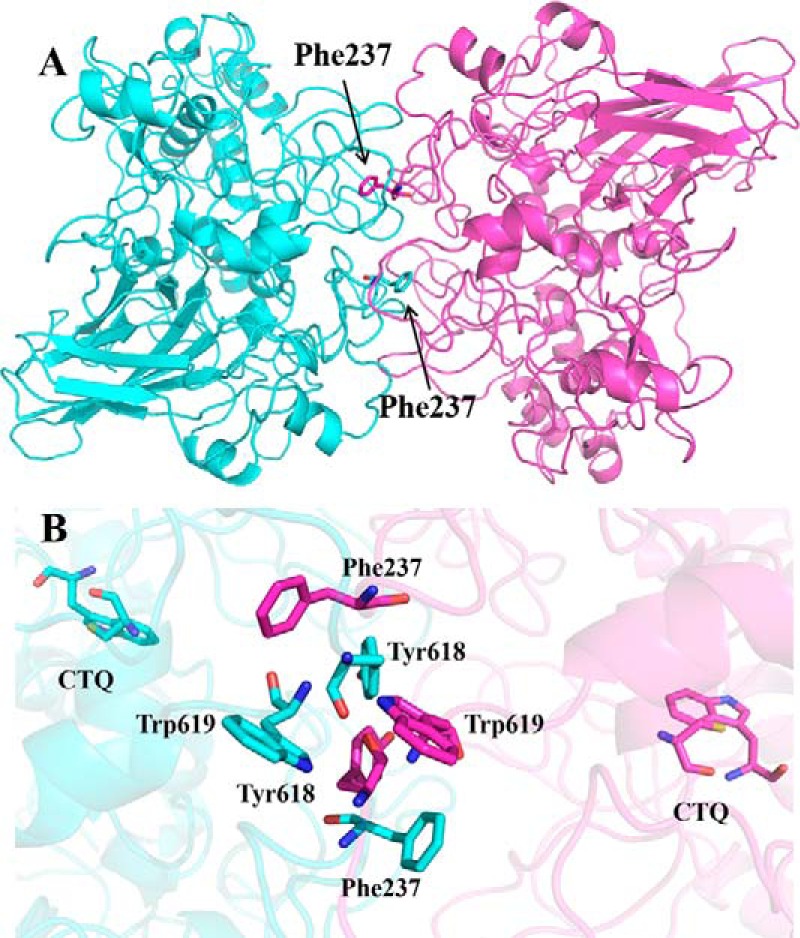
Docking model of the structure of the GoxA homodimer. The two monomers of GoxA are colored blue and pink. A, the overall structure of the dimer with residue Phe-237 shown as sticks. B, an expanded view of the protein-protein interface of the two monomers. The Cys and Trp residues, which form CTQ, and selected residues at the interface are shown as sticks. Carbons on residues of GoxA are colored blue on one subunit and pink on the other subunit. Oxygen is colored red, nitrogen is blue, and sulfur is yellow.
Discussion
GoxA and LodA are CTQ-containing oxidases discovered in the same bacteria, M. mediterranea. Each of them belongs to a newly discovered class of tryptophylquinone cofactor-containing oxidases. Analysis of the Integrated Microbial Genomes database of genome sequences identified 168 genes from 144 different species that encode LodA-like proteins (12). Prior to the characterization of LodA and GoxA, all known quinoprotein oxidases contained a tyrosylquinone cofactor (18), and in general, all amino acid oxidases contained a flavin cofactor (19). Despite the similarities in sequence and overall fold between GoxA and LodA, there are interesting differences in their kinetic mechanisms and steady-state kinetic parameters. This could be a consequence of the different physiological roles of the proteins. LodA is a secreted protein that exhibits antimicrobial activity that is important for the health of the biofilm in which the host bacterium resides (7). GoxA has also been reported to exhibit antimicrobial activity; however, the finding of cooperativity toward glycine and production of glyoxylate, which is an important metabolite (20), suggests possible metabolic and regulatory functions.
GoxA exhibits cooperative behavior toward its glycine substrate, whereas LodA exhibits no cooperativity. The K0.5 value for glycine and Km value for O2 of GoxA are each much greater than the Km values for lysine and O2 of LodA; however, the kcat value of GoxA is much greater than that of LodA. Thus, despite the very large Km value that GoxA exhibits for O2, the catalytic efficiency (kcat/KmO2) for GoxA is actually much greater than that of LodA (Table 1). Thus, LodA seems to have evolved its catalytic efficiency by increasing affinity for substrates, whereas GoxA has improved its catalytic efficiency by increasing kcat.
The protein interface of the GoxA dimer predicted from the modeling studies is completely different from what was observed in the crystal structure of LodA (9). Each LodA monomer has an unusual structure with two arms of antiparallel β-strands, one of which is a β hairpin that extends from the main body of each molecule of LodA (Fig. 1A). These extensions, which participate in subunit-subunit interactions in the proposed tetrameric structure of LodA derived from its crystal structure (9), are absent in GoxA (Figs. 1A and 2). This is consistent with GoxA exhibiting a different subunit composition and different types of subunit-subunit interactions.
The importance of Phe-237 of GoxA in directing the protein-protein interactions that stabilize the dimeric structure and dictating cooperative behavior toward glycine is remarkable. The F237Y and F237A mutations each eliminated cooperativity toward glycine. Also, the F237Y mutation decreases kcat ∼6-fold and decreases KmO2 slightly, and the F237A mutation decreases kcat 155-fold and decreases KmO2 6-fold (Table 1). Despite the differences in the effects on the kinetic parameters of substitution with Ala versus Tyr, the destabilization of the dimer by the F237A mutation is only slightly greater than for the F237Y mutation (Fig. 5). This indicates that activity is not dependent only upon the extent of dimer formation. Phe-237 of GoxA also exerts a significant influence on the kcat and affinity for substrates as does Tyr-211, the corresponding residue in LodA. In LodA, the Y211A mutation eliminated activity, whereas the Y211F mutation caused a small decrease in kcat but a large increase in Km for lysine (10). The dramatic differences in the physiological roles of the corresponding Phe-237 of GoxA and Tyr-211 of LodA may be explained by interesting differences in the sequence and thus structures of the two proteins (Fig. 2). The loop in LodA containing Lys-530, which anchors the loop containing Tyr-211 via a hydrogen bond (Fig. 1B), is absent in GoxA. The loop in GoxA containing Tyr-618 and Trp-619, which influence the position of the loop containing Phe-237 by hydrophobic interactions (Fig. 7), is absent in LodA.
At this point, in the absence of a crystal structure, one can only speculate on the details by which Phe-237 stabilizes dimer formation and facilitates communication between the active sites of each monomer that results in cooperativity. As discussed earlier, the docking model suggests the possibility that strong π-interactions and π-stacking involving Phe-237 and surrounding aromatic residues could be involved in these processes. A different structural role for Phe-237 in GoxA than for Tyr-211 in LodA is also consistent with the absence of a residue that corresponds to Lys-530 in LodA. The crystal structure of LodA suggested that Tyr-211, which resides on a flexible loop near the entry to the substrate channel, formed a hydrogen bond with Lys-530, creating a gate-like structure to control entry and exit of substrate and products from the active site. Because of the inability to hydrogen-bond with the corresponding Phe-237, the corresponding loop in GoxA (and consequently Phe-237) adopts a different position (Fig. 7), which now involves protein-protein interactions.
Asp-547 and His-466 of GoxA appear to be critical for CTQ biosynthesis. The corresponding Asp-512 in LodA was also shown to be critical for CTQ biosynthesis, whereas certain substitutions of Cys-448 of LodA, which corresponds to His-466, were tolerated (10). The tolerated mutations did, however, significantly increase the Km value for lysine and altered the efficiency of CTQ biosynthesis. These results for the CTQ-dependent enzymes are reminiscent of those from studies of TTQ biosynthesis in MADH (21). The active site of that enzyme has Asp in each of these two corresponding positions, Asp-76 and Asp-32. (Fig. 8A). Mutation of the Asp-76 in MADH that corresponds in position to Asp-547 in GoxA resulted in production of only trace amounts of protein in which the tryptophan residues that normally form TTQ were unmodified. Mutation of the Asp-32 in MADH that corresponds in position to His-466 in GoxA resulted in isolation of a mixed population in which a minor population of protein was active, albeit with significantly decreased affinity for substrate, and the majority again lacked TTQ any tryptophan modification (21). This highlights the fact that these active-site residues, which likely play roles in catalysis or substrate binding, or both, must have previously evolved to facilitate the process of both CTQ and TTQ biosynthesis.
FIGURE 8.

Residues in the active sites of MADH and QHNDH that correspond to Asp-547 and His-466 of GoxA. A, overlay of the active sites of the GoxA homology model and the MADH β-subunit (PDB ID 2BBK). The conserved active-site residues are labeled as sticks. Carbons on residues of MADH and GoxA are colored cyan and orange, respectively. B, overlay of the active sites of the GoxA homology model and the QHNDH γ-subunit (PDB ID 1JJU). Carbons on residues of QHNDH are colored yellow. Oxygen is colored red, nitrogen is blue, and sulfur is yellow.
Although GoxA and LodA have similar overall structures and protein folds, the structure and fold of MADH are quite different. That protein is an α2β2 heterotetramer with TTQ residing in each smaller 15-kDa β-subunit. The β-subunit contains 131 residues and is composed of five β-strands in two sheets, which are held together by six intrasubunit disulfide bonds, in addition to the cross-linked residues that form TTQ (13). The other know CTQ-bearing enzyme, which is not related to GoxA and LodA, is a dehydrogenase, quinohemoprotein amine dehydrogenase (QHNDH) (14). This protein is an αβγ trimer. The α-subunit contains two c-type hemes, and the γ-subunit contains CTQ. The γ-subunit is composed mostly of random coils and is held together by three unusual thioether cross-links, each involving a Cys side chain and the α-carbon of a Glu or Asp residue. The active site of CTQ also contains Asp residues in these structurally conserved positions in the active site near the quinone moiety (Fig. 8B). These observations support the notion of convergent evolution of the mechanism by which residues in the active site participate in the biogenesis of the protein-derived quinone cofactor in these diverse CTQ- and TTQ-dependent oxidases and dehydrogenases, which exhibit very different overall structures and protein folds.
Experimental Procedures
Construction, Expression, and Purification of GoxA and Its Variant Proteins
WT GoxA and GoxA variants that were generated by site-directed mutagenesis were expressed by coexpression of the goxA gene with goxB in Escherichia coli Rosetta cells as described previously (11). The GoxA proteins possess an N-terminal His6 tag, which was engineered into the gene. Preparation of cell extracts and purification using a nickel-nitrilotriacetic acid affinity column was performed as described previously (11). The following mutations were made: D547A, H466A, H466D, H466C, F237A, and F237Y. The forward and reverse primers that were used are listed in Table 2. Using these primers, the mutations were incorporated by PCR using the QuikChange Lightning kit (Agilent).
TABLE 2.
Primers used for site-directed mutagenesis
| Mutations | Primers |
|---|---|
| H466A | Forward: 5′-GCATTCGCCCCTGGCGTCG-3′ |
| Reverse: 5′-CGACGCCAGGGGCGAATGC-3′ | |
| H466D | Forward: 5′-GGCGCATTCGATCCTGGCGT-3′ |
| Reverse: 5′-ACGCCAGGATCGAATGCGCC-3′ | |
| H466C | Forward: 5′-GGCGCATTCTGTCCTGGCGTC-3′ |
| Reverse: 5′-GACGCCAGGACAGAATGCGCC-3′ | |
| F237A | Forward: 5′-GCTATTACGAGTGCCGCCGATAACGAT-3′ |
| Reverse: 5′-ATCGTTATCGGCGGCACTCGTAATAGC-3′ | |
| F237Y | Forward: 5′-GCTATTACGAGTTACGCCGATAACGAT-3′ |
| Reverse: 5′-ATCGTTATCGGCGTAACTCGTAATAGC-3′ | |
| D547A | Forward: 5′-TGGCAGTGTGCGGCCTTCAG-3′ |
| Reverse: 5′-CTGAAGGCCGCACACTGCCA-3′ |
Steady-state Kinetic Analysis
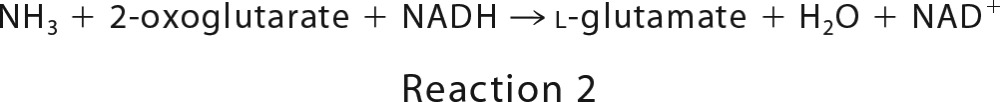
GoxA was assayed using a coupled-enzyme assay in which the formation of the NH3 product (Reaction 1) is monitored by coupling its production to the reaction of glutamate dehydrogenase (Reaction 2). The standard assay mixture contained GoxA, 5 mm 2-oxoglutarate, 0.25 mm NADH, and 20 units/ml glutamate dehydrogenase at 30 °C. The reaction was initiated by the addition of glycine. The initial velocity was determined by monitoring the rate of disappearance of NADH at 340 nm using the ϵ340 of NADH of 6220 m−1 cm−1. Initial rates were determined in the presence of varying amounts of glycine and O2. To vary [O2], a stock solution of the buffer was made anaerobic by repeated cycles of vacuum and purging with argon, and then mixed anaerobically with an appropriate amount of either air-saturated buffer ([O2] = 252 μm) or 100% O2-saturatedbuffer ([O2] = 1150 μm) buffer (22).
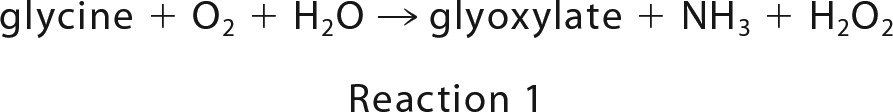 |
 |
Two alternative equations were used to fit the data from the kinetic experiments. In the Michaelis-Menten equation (Equation 1), kcat is the turnover number for the protein, Km is the Michaelis-Menten constant, [S] is the substrate concentration, [E] is the enzyme concentration, and v is the initial reaction rate. In the Hill equation (Equation 2), K0.5 is the concentration of the substrate that produces half the maximal velocity, and h is the Hill coefficient.
Quinoprotein Staining Protocol
SDS-PAGE was performed using 10% gels as described previously (11) with modifications as follows. The gel was stained for protein with EZ-Run protein staining solution (Fisher Scientific). An identical gel that had not been stained for protein was transferred by electroblot to a 0.2-μm nitrocellulose membrane. This was done at 30 V overnight at low temperature to prevent overheating. To stain for the presence of quinoproteins (17), the membrane was incubated for 45 min in the dark in a solution of 2 m potassium glycinate, pH 10, containing 0.24 mm nitro blue tetrazolium.
Size Exclusion Chromatography
Size exclusion chromatography was performed with an ÄKTA Prime FPLC system using a HiLoad 16/600 Superdex 200 (GE Healthcare). Chromatography was performed in 50 mm potassium phosphate buffer plus 150 mm NaCl at pH 7.5. The flow rate was 0.6 ml/min. The void volume was calculated using blue dextran. Proteins used as molecular mass markers were a mixture containing glutamate dehydrogenase (332 kDa), methylamine dehydrogenase (124 kDa), MauG (42 kDa), and amicyanin (11.5 kDa). A plot of the elution volume/void volume versus log molecular mass was used to estimate the masses of the WT and variant GoxA proteins. Gaussian fits of the chromatogram (using Origin and graphed in Prism GraphPad) were used to resolve overlapping peaks in the chromatogram and integrate the areas under the peaks.
Construction of Homology and Docking Models
A homology model of GoxA was constructed with the amino acid sequence of GoxA that was derived from the sequence of the goxA gene (Marme_1655) and the crystal structure of LodA (Protein Data Bank (PDB) ID 3WEU) (9) using the SWISS-MODEL server (23). A docking model of the putative GoxA dimer was constructed by using the ZDOCK Server (24). A PDB file was generated by ZDOCK of the lowest energy conformation of the homology model of a GoxA monomer docked against itself. The sequence alignment shown in Fig. 2 was derived from the homology model that was created using the SWISS-MODEL program. The program evaluates sequence identity and overlap when comparing the sequence of GoxA with the crystal structure of LodA. Comparison of the GoxA homology model with the sequence and structure of LodA yielded a sequence identity of 0.30, a sequence similarity of 0.35, and a coverage of 0.79. Alignment of the protein structures of MADH and QHNDH with the GoxA homology model to obtain overlays of the active sites was performed with PyMOL (Schrödinger, LLC).
Author Contributions
E. S. conducted most of the experiments, analyzed data, and wrote the paper. H. R. W. conducted experiments, analyzed data, and wrote the paper. V. L. D. analyzed data and wrote the paper.
Acknowledgments
We thank Drs. Carrie Wilmot at the University of Minnesota and Antonio Sanchez-Amat at the University of Murcia for helpful discussions and critically reading this manuscript.
This research was supported by internal funds from the Burnett School of Biomedical Sciences and in part by the NIGMS, National Institutes of Health Grant R37GM41574 (to V. L. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CTQ
- cysteine tryptophylquinone
- TTQ
- tryptophan tryptophylquinone
- MADH
- methylamine dehydrogenase
- QHNDH
- quinohemoprotein amine dehydrogenase.
References
- 1. Campillo-Brocal J. C., Lucas-Elio P., and Sanchez-Amat A. (2013) Identification in Marinomonas mediterranea of a novel quinoprotein with glycine oxidase activity. Microbiologyopen 2, 684–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davidson V. L. (2007) Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry 46, 5283–5292 [DOI] [PubMed] [Google Scholar]
- 3. Davidson V. L. (2011) Generation of protein-derived redox cofactors by posttranslational modification. Mol. Biosyst. 7, 29–37 [DOI] [PubMed] [Google Scholar]
- 4. Gómez D., Lucas-Elío P., Sanchez-Amat A., and Solano F. (2006) A novel type of lysine oxidase: l-lysine-ϵ-oxidase. Biochim. Biophys. Acta 1764, 1577–1585 [DOI] [PubMed] [Google Scholar]
- 5. Gómez D., Lucas-Elío P., Solano F., and Sanchez-Amat A. (2010) Both genes in the Marinomonas mediterranea lodAB operon are required for the expression of the antimicrobial protein lysine oxidase. Mol. Microbiol. 75, 462–473 [DOI] [PubMed] [Google Scholar]
- 6. Chacón-Verdú M. D., Gómez D., Solano F., Lucas-Elío P., and Sánchez-Amat A. (2014) LodB is required for the recombinant synthesis of the quinoprotein l-lysine-ϵ-oxidase from Marinomonas mediterranea. Appl. Microbiol. Biotechnol. 98, 2981–2989 [DOI] [PubMed] [Google Scholar]
- 7. Mai-Prochnow A., Lucas-Elio P., Egan S., Thomas T., Webb J. S., Sanchez-Amat A., and Kjelleberg S. (2008) Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several gram-negative bacteria. J. Bacteriol. 190, 5493–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sehanobish E., Shin S., Sanchez-Amat A., and Davidson V. L. (2014) Steady-state kinetic mechanism of LodA, a novel cysteine tryptophylquinone-dependent oxidase. FEBS Lett. 588, 752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okazaki S., Nakano S., Matsui D., Akaji S., Inagaki K., and Asano Y. (2013) X-ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in l-lysine ϵ-oxidase from Marinomonas mediterranea. J. Biochem. 154, 233–236 [DOI] [PubMed] [Google Scholar]
- 10. Sehanobish E., Chacón-Verdú M. D., Sanchez-Amat A., and Davidson V. L. (2015) Roles of active site residues in LodA, a cysteine tryptophylquinone dependent ϵ-lysine oxidase. Arch. Biochem. Biophys. 579, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sehanobish E., Campillo-Brocal J. C., Williamson H. R., Sanchez-Amat A., and Davidson V. L. (2016) Interaction of GoxA with its modifying enzyme and its subunit assembly are dependent on the extent of cysteine tryptophylquinone biosynthesis. Biochemistry 55, 2305–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campillo-Brocal J. C., Chacón-Verdú M. D., Lucas-Elío P., and Sánchez-Amat A. (2015) Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity. BMC Genomics 16, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L., Doi M., Durley R. C., Chistoserdov A. Y., Lidstrom M. E., Davidson V. L., and Mathews F. S. (1998) Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 Å resolution. J. Mol. Biol. 276, 131–149 [DOI] [PubMed] [Google Scholar]
- 14. Datta S., Mori Y., Takagi K., Kawaguchi K., Chen Z. W., Okajima T., Kuroda S., Ikeda T., Kano K., Tanizawa K., and Mathews F. S. (2001) Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc. Natl. Acad. Sci. U.S.A. 98, 14268–14273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roujeinikova A., Scrutton N. S., and Leys D. (2006) Atomic level insight into the oxidative half-reaction of aromatic amine dehydrogenase. J. Biol. Chem. 281, 40264–40272 [DOI] [PubMed] [Google Scholar]
- 16. Chacón-Verdú M. D., Campillo-Brocal J. C., Lucas-Elío P., Davidson V. L., and Sánchez-Amat A. (2015) Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-ϵ-oxidase and glycine oxidase from Marinomonas mediterranea. Biochim. Biophys. Acta 1854, 1123–1131 [DOI] [PubMed] [Google Scholar]
- 17. Paz M. A., Flückiger R., Boak A., Kagan H. M., and Gallop P. M. (1991) Specific detection of quinoproteins by redox-cycling staining. J. Biol. Chem. 266, 689–692 [PubMed] [Google Scholar]
- 18. Klinman J. P., and Bonnot F. (2014) Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev. 114, 4343–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hossain G. S., Li J., Shin H. D., Du G., Liu L., and Chen J. (2014) l-Amino acid oxidases from microbial sources: types, properties, functions, and applications. Appl. Microbiol. Biotechnol. 98, 1507–1515 [DOI] [PubMed] [Google Scholar]
- 20. Walsh K., and Koshland D. E. Jr. (1984) Determination of flux through the branch point of two metabolic cycles: the tricarboxylic acid cycle and the glyoxylate shunt. J. Biol. Chem. 259, 9646–9654 [PubMed] [Google Scholar]
- 21. Jones L. H., Pearson A. R., Tang Y., Wilmot C. M., and Davidson V. L. (2005) Active site aspartate residues are critical for tryptophan tryptophylquinone biogenesis in methylamine dehydrogenase. J. Biol. Chem. 280, 17392–17396 [DOI] [PubMed] [Google Scholar]
- 22. Grzyska P. K., Ryle M. J., Monterosso G. R., Liu J., Ballou D. P., and Hausinger R. P. (2005) Steady-state and transient kinetic analyses of taurine/α-ketoglutarate dioxygenase: effects of oxygen concentration, alternative sulfonates, and active-site variants on the FeIV-oxo intermediate. Biochemistry 44, 3845–3855 [DOI] [PubMed] [Google Scholar]
- 23. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 24. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]