

Abstract
Patients with chronic pancreatitis (CP) frequently have genetic risk factors for disease. Many of the identified genes have been connected to trypsinogen activation or trypsin inactivation. The description of CP in patients with mutations in the variable number of tandem repeat (VNTR) domain of carboxyl ester lipase (CEL) presents an opportunity to study the pathogenesis of CP independently of trypsin pathways. We tested the hypothesis that a deletion and frameshift mutation (C563fsX673) in the CEL VNTR causes CP through proteotoxic gain-of-function activation of maladaptive cell signaling pathways including cell death pathways. HEK293 or AR42J cells were transfected with constructs expressing CEL with 14 repeats in the VNTR (CEL14R) or C563fsX673 CEL (CEL maturity onset diabetes of youth with a deletion mutation in the VNTR (MODY)). In both cell types, CEL MODY formed intracellular aggregates. Secretion of CEL MODY was decreased compared with that of CEL14R. Expression of CEL MODY increased endoplasmic reticulum stress, activated the unfolded protein response, and caused cell death by apoptosis. Our results demonstrate that disorders of protein homeostasis can lead to CP and suggest that novel therapies to decrease the intracellular accumulation of misfolded protein may be successful in some patients with CP.
Keywords: apoptosis, disulfide, endoplasmic reticulum stress (ER stress), lipase, unfolded protein response (UPR), carboxyl ester lipase, chronic pancreatitis, disulfide bonds
Introduction
Pancreatitis is an inflammatory disease with significant health and economic burdens (1, 2). There are no therapies to prevent recurrent episodes or progression to chronic disease. The absence of effective therapies stems in part from our limited understanding about the pathophysiology of pancreatitis. The prevailing model holds that intracellular trypsinogen activation in the acinar cell and failure of protective mechanisms responsible for trypsin inactivation are central to pathogenesis (3). Accumulating evidence suggests that the acinar cell responds to injury through multiple pathways and that trypsin activation is not the only potential player in this process (4, 5). The recent clinical reports of patients who develop chronic pancreatitis in association with mutations in a pancreatic digestive lipase, carboxyl ester lipase (CEL),2 offers a unique opportunity to study the pathogenesis of chronic pancreatitis independently of trypsin (6–9). CEL has no known role in trypsinogen activation or trypsin degradation, nor does CEL require activation by trypsin for function. Thus, CEL variants likely cause disease through a trypsin-independent mechanism. Understanding the molecular details of the pathophysiology underlying CEL variant-associated disease can provide insight into novel therapies for chronic pancreatitis.
CEL is expressed in the pancreas and lactating mammary glands (10). The protein consists of two domains, an N-terminal α/β-hydrolase fold and a C-terminal domain containing a variable number of proline-rich tandem repeats (VNTR) (11). The number of tandem repeats in humans ranges from three to 28 (12, 13). About 40% of humans are homozygous for 16 repeats (12). The proline-rich repeats are heavily O-glycosylated and are important for protein stability (14). Furthermore, the number of repeats influences intracellular processing of CEL and may alter stability in an aqueous milieu after secretion (15, 16). The number of tandem repeats does not associate with increased risk for chronic pancreatitis (17).
Conversely, alterations in the VNTR sequence can cause pancreatic disease. A hybrid allele formed by recombination between CEL and the adjacent pseudogene, CELP, encodes a fusion protein of CEL and the abnormal VNTR of CELP (9). The presence of the hybrid allele increased the risk for chronic pancreatitis in a northern European population about 5-fold (9). Interestingly, the hybrid allele did not associate with chronic pancreatitis in an Asian population (18).
In another study, mutations in CEL were identified in two Norwegian families with pedigrees of nonketotic, autosomal dominant diabetes (7). Each disease-associated mutation causes a frameshift in the VNTR, resulting in an altered amino acid sequence and premature truncation of CEL. All of the carriers have pancreatic exocrine insufficiency evidenced by low fecal elastase or steatorrhea and irreversible anatomical changes in the pancreas (such as fibrosis and fat infiltration) by computed tomography and histopathology. Abdominal pain was not a prominent feature of the carriers. Still, mild, recurrent abdominal pain was present in almost 60% of the carriers (7). Hence, all mutation carriers meet criteria for chronic pancreatitis. The diabetes is likely a consequence of chronic pancreatitis rather than an isolated effect of the CEL variant. Two recent publications reported the cellular fate of c.1686delT (p.C563fsX673) CEL also called CEL MODY, the most common variant associated with chronic pancreatitis (19, 20). The first publication demonstrated insoluble intracellular and extracellular aggregates of CEL MODY in HEK293 cells (19). Expression of CEL MODY variant modestly activated the protein kinase RNA-like endoplasmic reticulum kinase/eukaryotic translation initiation factor 2A arm of the unfolded protein response (UPR) pathway but did not inhibit growth or cause cell death in HEK293 cells (19). The authors speculated that the UPR offset the toxic consequences arising from intracellular accumulation of CEL MODY and was not the cause of cell injury. The authors also concluded that the formation of insoluble aggregates causes disease through an undefined mechanism. In a later study, aggregates of CEL MODY were observed on the cell surface and inside cytoplasmic vacuoles (20). The authors speculated that many of the vacuoles resulted from endocytosis of secreted CEL MODY. Furthermore, exposure of pancreatic exocrine and endocrine cell lines to conditioned medium from stably transfected HEK293 cells expressing CEL MODY showed a significant decrease in viability. The authors speculated that extracellular aggregates of CEL MODY or their degradation products caused pancreatic cell injury. In this alternate model, CEL MODY has a direct cytotoxic effect on beta cells.
In this study, we sought to rectify this controversy and tested an alternative hypothesis: CEL MODY causes pancreatitis through proteotoxic gain-of-function activation of maladaptive cell signaling pathways including cell death pathways. To identify the CEL MODY-dependent adaptive cell signaling pathways, we expressed CEL and CEL MODY in HEK293T cells and in a pancreatic acinar cell line, AR42J cells. We then determined the fate of the expressed proteins and measured activation of the UPR and downstream effects that activate inflammatory and cell death pathways. Our findings indicate that CEL MODY forms insoluble intracellular aggregates that activate the UPR and trigger apoptosis.
Results
Expression and Activity of CEL VNTR Variants
To characterize the CEL MODY mutation associated with pancreatic disease, we transfected Pichia pastoris GS115 yeast with vectors containing wild-type CEL14R or with a frameshift mutation in the VNTR, CEL MODY. Immunoblotting and lipase activity assays both suggested impaired secretion of CEL mutant variants by transformed yeasts with the most substantial secretion defect observed for CEL MODY (data not shown). Even so, we were able to purify each of the recombinant CEL variants from culture medium. The proteins were purified as determined by SDS-PAGE and protein staining (Fig. 1B). We assayed each CEL variant for activity against medium and long chain triglyceride substrates trioctanoin and triolein. The activity of the mutant variants was indistinguishable from the activity of CEL14R, indicating that the structure of the VNTR did not influence CEL activity (Fig. 1C).
FIGURE 1.

VNTR amino acid sequence, expression, and assay of VNTR variants. A, the VNTR amino acid sequence for CEL with 16, 14, and three repeats and the sequence of the VNTR from patients with pancreatic disease are presented with the single-letter amino acid code (7). B, a GelCode Blue-stained SDS-polyacrylamide gel of CEL variants expressed in P. pastoris and purified to near homogeneity. Sections of the same gel are presented after the removal of a lane containing a sample from cells expressing CEL with the VNTR deleted. C, activity against trioctanoin and triolein of the VNTR variants is presented. The assays were done as described under “Experimental Procedures.” Error bars represent S.D.
Intracellular Accumulation of Aggregated CEL MODY Expressed in HEK293T Cells
We further investigated the impact of mutations on CEL secretion in a mammalian cell system by transfecting HEK293T cells to transiently express CEL protein variants. We focused only on CEL MODY because there is more clinical information on patients with this particular mutation compared with the other variants (6, 7, 21). Seventy-two hours after transfection, no lipase activity against trioctanoin was detected in the medium from cells transfected with a control plasmid. In contrast, lipase activity was easily detectable in the medium from cells expressing the CEL variants. The medium from cells expressing CEL14R had significantly more activity than did the medium from cells expressing CEL MODY (8.7 ± 0.4 versus 0.7 ± 0.2 units/ml) (p < 0.01). Western blotting analysis confirmed the presence of both CEL variants in the medium. The amount of CEL14R was 10-fold higher than the amount of CEL MODY (Fig. 2, A and B) (p < 0.001). In contrast, the amount of intracellular CEL MODY was nearly 2-fold higher than the amount of CEL14R (p < 0.001). Furthermore, unlike CEL14R, the majority of intracellular CEL MODY was present in a detergent-insoluble fraction, and only a small fraction of CEL MODY was soluble. Altogether, these results suggest that the CEL MODY mutation causes CEL protein to accumulate as insoluble aggregates inside cells, which leads to a secretion defect.
FIGURE 2.
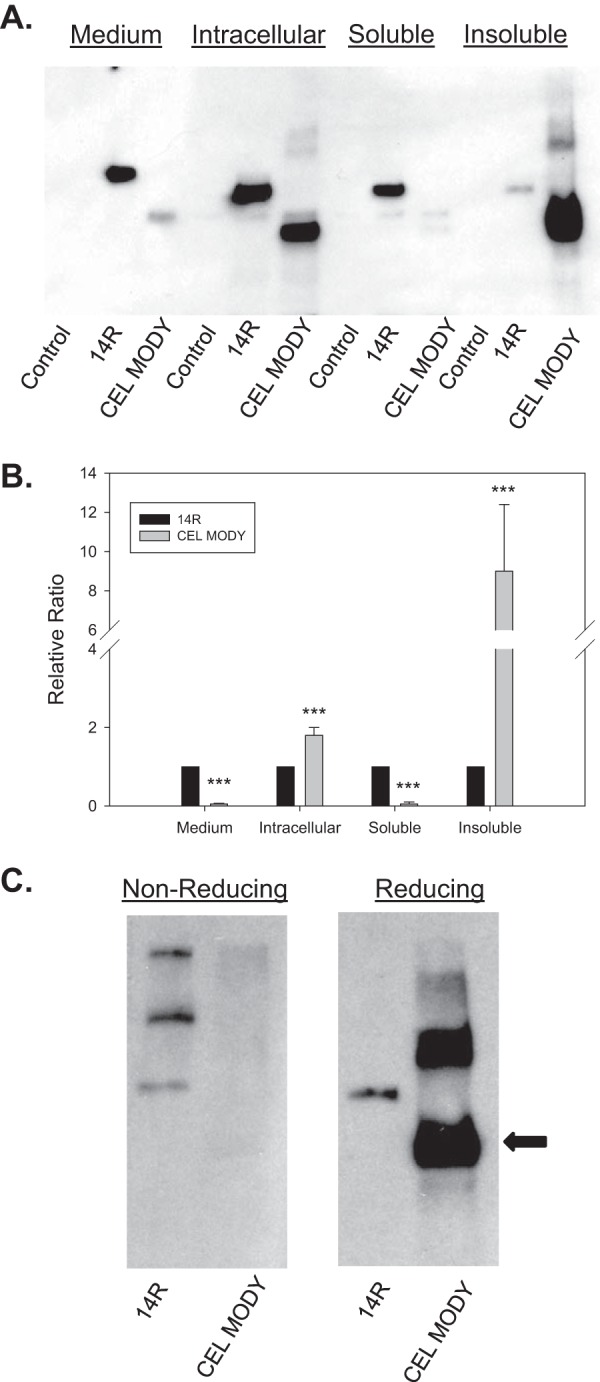
Expression of CEL14R and CEL MODY in HEK293T cells. A, the cells were transfected with the indicated plasmid as described under “Experimental Procedures.” Samples were obtained from the medium and from the intact cells. In separate experiments, the intracellular CEL was separated into soluble and insoluble fractions as described under “Experimental Procedures.” The proteins in each sample were separated by SDS-PAGE, and immunoblotting was performed with CEL antiserum. Sections of the same gel are presented after the removal of a lane containing a sample from cells expressing CEL with the VNTR deleted. B, the amount of immunoreactive protein was quantified by densitometry and presented in bar graphs. The medium and soluble fractions contained significantly more CEL14R (p = 0.001). The total intracellular sample and the insoluble fraction contained significantly more CEL MODY (p = 0.001) (n = 6). C, cells were transfected with the indicated plasmids, and the insoluble fraction was isolated. The samples were separated by SDS-PAGE with and without β-mercaptoethanol, and CEL was detected by immunoblotting. A representative example of three separate experiments is shown. The arrow indicates the location of CEL MODY. Error bars represent S.D. *** indicates p = 0.001.
Because the frameshift introduces 10 additional cysteine residues in the VNTR, we investigated whether the formation of inappropriate disulfide bonds contributed to CEL MODY aggregation. To test this hypothesis, we separated the detergent-soluble and -insoluble fractions of CEL MODY and CEL14R by SDS-PAGE under non-reducing and reducing conditions and looked at the pattern of bands by protein immunoblotting (Fig. 2C). Under both conditions, a band corresponding to the expected mass of CEL14R was present in both the detergent-soluble and -insoluble fractions. In the samples containing CEL MODY, a faint band of the expected mass was detected in both fractions on non-reducing gels, whereas a much stronger band was detected in both fractions when separated by reducing gels. The results are consistent with the hypothesis that the mechanism of insoluble aggregation of CEL MODY is the formation of inappropriate disulfide bonds.
Expression of CEL MODY Activates the UPR
Because most of the intracellular CEL MODY is present as an insoluble aggregate, we next determined whether CEL MODY expression caused ER stress and activated the UPR. First, we measured the expression of the major ER chaperones BiP and GRP94 by protein immunoblotting (Fig. 3A). Only BiP was significantly increased in cells expressing CEL14R and CEL MODY compared with control-transfected cells (p < 0.001). However, there was a distinctive distribution of BiP in extracts from cells expressing the two CEL variants (Fig. 3B). In cells expressing CEL14R, BiP was mostly present in the soluble fraction with trace amounts in the insoluble fraction. Conversely, in cells expressing CEL MODY, BiP was mostly in the insoluble fraction with only a small amount in the soluble fraction. These results suggest that BiP may attempt to aid folding or degradation of the aggregation-prone CEL MODY protein but then becomes trapped upon substrate aggregation. Trapping of protein quality control factors by an aggregating substrate is also evident in cytoplasmic Lewy bodies (22).
FIGURE 3.

Markers of ER stress in HEK293T cells transfected with CEL14R or CEL MODY expression plasmids. A, total cell extracts from cells transfected with control plasmid (mock), CEL14 R, or CEL MODY were separated by SDS-PAGE, and immunoblotting was performed with antiserum against BiP or GRP94. The amount of protein was determined by densitometry. The amount of BiP in the cells expressing CEL14R or CEL MODY was significantly greater than in cells transfected with a plasmid control (p < 0.001) (n = 8). B, total cell extracts of cells transfected with a control plasmid or the CEL14R or CEL MODY expression plasmid were separated into the soluble and insoluble fractions, and BiP was detected by immunoblotting. Sections of the same gel are presented after the removal of a lane containing a sample from cells expressing CEL with the VNTR deleted. C, total RNA was isolated from cells transfected with the control plasmid or the CEL14R or CEL MODY expression plasmid. The amount of mRNA encoding the spliced and unspliced forms of XBP1 mRNA was determined by RT-PCR. GAPDH was examined as a loading control. The samples were separated on 2% agarose gels and stained with ethidium bromide. D, the amounts of XBP1U and XBP1S were determined by densitometry, and the results were plotted as the ratio of XBP1S/XBP1U. The ratio is significantly higher in the CEL MODY-expressing cells compared with the CEL14R-expressing cells (p = 0.01) (n = 6). Error bars represent S.D.
We next investigated whether CEL MODY expression triggered the UPR in HEK293T cells by measuring X-box-binding protein-1 (XBP1) mRNA splicing. Like BiP, the overexpression of both CEL variants increased XBP1 mRNA splicing as opposed to the control (Fig. 3, C and D). The ratio of XBP1 mRNA splicing was significantly higher in the cells expressing CEL MODY than in cells expressing CEL14R (p = 0.002). Collectively, these findings support the hypothesis that CEL MODY expression causes ER stress and triggers the UPR.
CEL MODY Expression Induces Cell Death
The UPR can either be adaptive to restore normal ER function and to promote cell survival, or it can be maladaptive and activate cell death pathways (23). First, we studied whether the expression of CEL MODY triggered cell death by measuring lactate dehydrogenase (LDH) activity in the medium at 48, 72, or 96 h after transfection (Fig. 4A). At each time point, the activity of LDH in the medium from cells transfected with CEL MODY was significantly higher than that from cells transfected with CEL14R or the mock control (p < 0.001). The difference increased with time. The LDH activity for the latter two groups did not differ significantly throughout the experiment period. The LDH activity was consistently higher in the medium from cells transfected with CEL MODY, independent of the initial seeding density (ranging from 0.2 to 1.0 × 106 cell/ml) (Fig. 4B). This finding ruled out cell overpopulation as a potential contributing factor for elevated LDH activity in the medium from cells transfected with CEL MODY. Interestingly, applying additional environmental stress to the transfected cells including cold stress, glucose deprivation, oxidative stress with menadione, and hypoxia mimicked by treatment with cobalt chloride did not increase the rate or amount of LDH release (data not shown). The results suggest that the UPR in cells expressing CEL MODY is overwhelmed, and cell death pathways are activated.
FIGURE 4.

Activation of apoptosis and cell death in HEK293T cells transfected with the CEL14R or CEL MODY expression plasmid. A, cells were transfected with the control plasmid or the CEL14R or CEL MODY expression plasmid. Aliquots of the medium were removed at the indicated times, and the cumulative LDH concentration was determined. At each time point, the amount of LDH in the CEL MODY medium was significantly higher than the amount in the CEL14R medium (p < 0.001) (n = 8). There was no difference in the LDH content in the medium from control and CEL14R cells. B, cells were plated at the indicated densities and transfected with the control, CEL14R, or CEL MODY vectors 12 h after plating. Aliquots of each medium were removed at 72 h and assayed for LDH activity. The results are the average of three determinations at each cell density. C, total cellular extracts were separated by SDS-PAGE, and PARP was detected by immunoblotting. A representative blot is shown. Sections of the same gel are presented after the removal of a lane containing a sample from cells expressing CEL with the VNTR deleted. D, the amount of cleaved PARP relative to the empty vector sample is presented as a bar graph. The amount of cleaved PARP in the cells transfected with CEL MODY is significantly greater than the amount in the CEL14R-transfected cells. (p < 0.001) (n = 6). Error bars represent S.D.
Others have reported that prolonged or profuse UPR can cause cell death through apoptosis (24). We therefore next examined whether apoptosis was triggered by expression of CEL MODY by Western blotting analysis of cleaved PARP, a marker of apoptosis; staurosporine-treated cells were used as a positive control (Fig. 4C). Only a faint band representing the cleaved PARP was detected in cells transfected with vector control and CEL14R. In contrast, the abundance of cleaved PARP was 10-fold higher in the cells expressing CEL MODY compared with CEL14R (p < 0.001) with the level at roughly 60% of that in the cells treated with staurosporine (Fig. 4D). Furthermore, propidium iodide staining at 24, 72, and 96 h showed that cell death through necrosis did not occur in cells expressing CEL MODY (data not shown). The results demonstrate that the expression of CEL MODY caused apoptotic cell death.
Insoluble Aggregates of CEL MODY Accumulate in AR42J Cells
We next characterized CEL MODY in AR42J cells, a model system for pancreatic acinar cells (25). These cells synthesize and store a number of pancreatic exocrine proteins in secretory granules that are secreted through both a basal and a regulated secretory pathway. The acinar phenotype of AR42J cells is enhanced by treatment with dexamethasone. We found that AR42J cells are difficult to transfect with plasmids but are readily transduced with adenovirus. Therefore, we transduced dexamethasone-treated AR42J cells with control, CEL14R, or CEL MODY adenovirus. The samples were harvested 72 h after transduction. First, real time quantitative PCR analysis showed that the abundance of mRNA encoding CEL was not statistically higher in the CEL MODY-transduced cells compared with CEL14R (p > 0.05) (Fig. 5A). This insured comparable levels of transduction with each CEL variant. We then measured the levels of CEL protein in the medium, in whole cell lysates, and in the soluble and insoluble intracellular fractions (Fig. 5, B and C). The amount of CEL14R in the medium was 40% higher than the amount of CEL MODY (p < 0.001). The total intracellular levels of the two variants were similar, but CEL14R was mostly present in the soluble fraction, whereas CEL MODY was more abundant in the insoluble fraction (p < 0.001). The results from AR42J cells and HEK293T cells are comparable except the AR42J cells exhibit slightly improved secretion and slightly more soluble CEL MODY protein compared with HEK293T cells.
FIGURE 5.

Expression of CEL14R and CEL MODY in AR42J cells transduced with adenovirus constructs. A, total RNA was isolated from cells transduced with a control or a CEL14R- or CEL MODY-containing adenovirus. The amount of mRNA encoding human CEL and GAPDH was determined by qRT-PCR as described under “Experimental Procedures.” There is no significant difference in the amount of mRNA encoding CEL14R and CEL MODY (n = 6). B, the cells were transfected with the indicated adenovirus as described under “Experimental Procedures.” Samples were obtained from the medium and from the intact cells. In separate experiments, the intracellular CEL was separated into soluble and insoluble fractions as described under “Experimental Procedures.” The proteins in each sample were separated by SDS-PAGE, and immunoblotting was performed with CEL antiserum. C, the amount of immunoreactive protein was quantified by densitometry and presented in bar graphs. The medium and soluble fractions contained significantly more CEL14R (p = 0.001). The total intracellular sample and the insoluble fraction contained significantly more CEL MODY (p = 0.001) (n = 8). D, cells were transduced with the indicated adenoviral constructs, and the insoluble fraction was isolated. The samples were separated by SDS-PAGE in the presence or absence of β-mercaptoethanol, and CEL was detected by immunoblotting. Representative gels from three separate experiments are shown. Error bars represent S.D.
We next investigated whether CEL MODY aggregates are disulfide-bonded as found in HEK293T cells. As aforementioned, we separated the insoluble fraction of CEL MODY and CEL14R by SDS-PAGE under non-reducing and reducing conditions and performed immunoblotting analysis (Fig. 5D). Under both conditions, a band corresponding to the expected size of CEL14R was present. In the samples containing CEL MODY, a faint band of the expected size was detected among the smear under non-reducing condition, whereas a much stronger band was detected under reducing condition. The results are consistent with what we found in HEK293T cells. Importantly, our findings support the hypothesis that aggregation of CEL MODY arises from the formation of inappropriate disulfide bonds.
Expression of CEL MODY in AR42J Cells Increases ER Stress and Triggers the UPR
To determine whether CEL MODY expression also triggered ER stress and UPR activation in AR42J cells, we first analyzed the ultrastructure of the ER in cells expressing a green fluorescent protein (GFP) control, CEL14R, or CEL MODY by transmission electron microscopy. A well documented marker of ER stress is pronounced dilation of the ER (20). In cells expressing GFP or CEL14R, the ER was not dilated (Fig. 6A). In contrast, cells expressing CEL MODY showed marked dilation of the ER cisterna with granular material. The findings suggest the increased insoluble aggregates of CEL MODY reside in the ER and consequently lead to ER stress. Second, we measured the levels of several major chaperones in cells expressing CEL14R or CEL MODY (Fig. 6, B and C). Total intracellular BiP protein and mRNA were both significantly increased in cells expressing CEL MODY compared with CEL14R (p < 0.01 for protein and p < 0.001 for mRNA). Furthermore, levels of GRP94 and calreticulin were also increased in cells expressing CEL MODY in comparison with CEL14R (p < 0.01). As in HEK293T cells, BiP was predominantly present in the insoluble fraction from AR42J cells expressing CEL MODY (Fig. 6B). Third, we investigated whether the increased ER stress in cells expressing CEL MODY up-regulated the UPR by measuring XBP1 mRNA splicing. Although the overexpression of CEL14R increased XBP1 mRNA splicing compared with control, XBP1 mRNA splicing was increased significantly further in cells expressing CEL MODY compared with cells expressing CEL14R (p < 0.01) (Fig. 6D). Altogether, these findings clearly indicate that the expression of CEL MODY results in ER stress and triggers the UPR.
FIGURE 6.

ER stress response in AR42J cells transduced with control, CEL14R, or CEL MODY adenovirus. A, transmission electron micrographs of cells transduced with control, CEL14R, or CEL MODY adenovirus. Two separate experiments with CEL MODY are shown. The arrows mark ER. B, the ER stress markers BiP, calreticulin, and GRP94 were detected by immunoblotting from lysates from cells transduced with control, CEL14R, or CEL MODY adenovirus. We determined the amount of BiP in the total cell extract as well as in the soluble and insoluble intracellular fractions. GAPDH was used as the loading control. C, left panel, the relative BiP protein abundance in the total cellular extract and the soluble fraction is presented as a ratio to the BiP content in the control cells. The insoluble fraction was not quantitated because no detectable BiP was present in the CEL14R samples. BiP was significantly higher in the total cellular extract of cells transduced with CEL MODY compared with cells transduced with CEL14R (p < 0.01) (n = 6). The insoluble fraction is not plotted because BiP was never detected in the cells expressing CEL14R. Middle panel, the amount of BiP mRNA was determined by qRT-PCR and plotted relative to the control sample. There was significantly more BiP mRNA in cells expressing CEL MODY than in cells expressing CEL14 R (p < 0.001) (n = 4). Right panel, the relative abundance of GRP94 and calreticulin determined by immunoblotting. There was significantly more GRP94 and calreticulin in the cells expressing CEL MODY than in cells expressing CEL14R (p = 0.01) (n = 6). D, total RNA was isolated from cells transduced with the control adenovirus or the CEL14R or CEL MODY adenovirus. The amount of mRNA encoding the spliced and unspliced forms of XBP1 mRNA was determined by RT-PCR. GAPDH was used as a loading control. The samples were separated on 2% agarose gels and stained with ethidium bromide. In AR42J cells, the hybrid form of XBP1 mRNA (XBP1H) was detected (44). The amounts of XBP1U and XBP1S were determined by densitometry, and the results plotted as a ratio of XBP1S/XBP1U. The ratio is significantly higher in the CEL MODY cells compared with the CEL14R cells (p = 0.01) (n = 6). Error bars represent S.D. NS, not significant. **, p = 0.01; ***, p = 0.001.
CEL MODY Also Leads to the Death of AR42J Pancreatic Acinar Cells
As observed in HEK293T cells, the expression of CEL MODY caused increased cell death in AR42J cells (Fig. 7A). Cell death was minimal in cells transduced with GFP, whereas about 5% of cells expressing CEL14R died over 72 h. About 30% of the cells expressing CEL MODY underwent cell death, which was significantly higher compared with CEL14R (p < 0.001). Next, we evaluated several biomarkers associated with the activation of apoptosis to determine whether CEL MODY-induced death occurred through apoptosis. First, we evaluated the mRNA levels of ATF4 and CHOP in transduced cells. ATF4, a basic leucine zipper domain transcription factor, regulates UPR target genes including CHOP that mediate ER stress-mediated apoptosis. The levels were similar between the cells transduced with the control or CEL14R adenovirus (Fig. 7, B and C). The levels of ATF4 and CHOP mRNA were significantly increased in the cells expressing CEL MODY (p = 0.007 and p < 0.01, respectively). Second, we evaluated PARP cleavage. Cells expressing CEL MODY had about 2-fold more cleaved PARP compared with cells expressing CEL14R (p < 0.001) and about 70% of the amount in the cells treated with staurosporine (Fig. 7D). Cleaved PARP was not detected in control cells. Collectively, these results provide strong evidence that AR42J cells expressing CEL MODY undergo apoptosis.
FIGURE 7.

Cell death and apoptosis in AR42J cells transduced with control, CEL14R, or CEL MODY adenovirus. A, cell death was determined as described under “Experimental Procedures.” The level of cell death was significantly higher in cells expressing CEL MODY than in cells expressing CEL14R (p < 0.001) (n = 3). B, the amount of mRNA encoding ATF4 in each cell line was determined by qRT-PCR 48 h after transduction. The amount of ATF4 was significantly higher in cells expressing CEL MODY than in cells expressing CEL14R (p = 0.007) (n = 4). C, the amount of mRNA encoding CHOP in each cell line was determined by qRT-PCR 48 h after transduction. The amount of CHOP was significantly higher in cells expressing CEL MODY than in cells expressing CEL14R (p = 0.01) (n = 3). D, total cellular extracts from cells transduced with control, CEL14R, or CEL MODY adenovirus or treated with staurosporine were separated by SDS-PAGE, and cleaved PARP was detected by immunoblotting. A representative blot is shown. The amount of cleaved PARP relative to the mock sample is presented as a bar graph. The amount of cleaved PARP in the cells transfected with CEL MODY is significantly greater than the amount in the CEL14R-transfected cells (p < 0.001) (n = 5). E, activation of the NF-κB promoter was determined in a luciferase assay as described under “Experimental Procedures.” There was significantly more activation of the NF-κB promoter in cells transduced with adenovirus expressing CEL MODY than in cells transduced with CEL14R (p < 0.001) (n = 3). There was no difference among the mock, GFP, and CEL14R expressed proteins. Error bars represent S.D. **, p = 0.01; ***, p = 0.001.
Induction of NF-κB in AR42J Cells Expressing CEL MODY
One of the pathological consequences of ER stress is the activation of NF-κB (23). Additionally, NF-κB is activated in experimental models of both acute and chronic pancreatitis (26, 27). NF-κB regulates genes that control the inflammatory response and cell survival. To determine whether expression of CEL MODY induced NF-κB expression, we co-transduced cells with an adenovirus expressing luciferase under the control of the NF-κB promoter and with adenovirus expressing GFP, CEL14R, or CEL MODY (Fig. 7E). Only the cells expressing CEL MODY showed an increase in luciferase activity, indicating that NF-κB expression was turned on in the CEL MODY cells.
Discussion
In this study, we investigated the trafficking and function of CEL MODY in HEK293T cells, a human epithelial cell line, and in AR42J cells, a rat pancreatic acinar cell line. The behavior and consequences of the disease-causing mutant CEL protein were similar in both cell lines. The findings provide additional insight into the molecular details of the pathophysiology underlying chronic pancreatitis present in patients carrying this mutation. The biochemical characterization of purified recombinant CEL variant proteins suggested that their enzymatic activity against triglycerides was similar. However, CEL MODY was poorly secreted from the transfected cell lines and largely retained inside the cells, and the majority of the intracellular mutant protein was present in detergent-insoluble aggregates, which is in line with previous reports (19). In addition, the retention of mutant CEL inside cells caused ER stress and activated the UPR, which resulted in NF-κB activation and apoptosis in both cell lines. Therefore, our results support the hypothesis that CEL MODY causes chronic pancreatitis through proteotoxic gain of function.
In both transfected cell lines, CEL MODY formed detergent-insoluble aggregates. The mechanism underlying the aggregation of CEL MODY was partially investigated in this study. Unlike CEL14R, CEL MODY was primarily detected at the expected size on SDS-PAGE only when a reducing agent was included, indicating that disulfide bonds likely contribute to the formation of aggregates. Of note, the frameshift caused by the single base deletion introduces 10 additional cysteine residues in the VNTR region of CEL MODY as opposed to no cysteine residues in the CEL14R VNTR (Fig. 1A). Nevertheless, it remains possible that other structural or biophysical features of the acquired VNTR sequence in CEL MODY contribute to protein misfolding and aggregation.
Overexpression of either CEL14R or CEL MODY increased ER stress likely in response to the increased requirements for protein synthesis. Multiple criteria suggested important differences in the response of cells transfected with CEL14R compared with cells transfected with CEL MODY with the latter inducing cell death. First, the ER in AR42J cells expressing CEL MODY was markedly dilated, a hallmark of ER stress. Second, the expression of CEL MODY in AR42J cells up-regulated the expression of several major ER chaperones beyond the levels in cells expressing CEL14R. Third, the intracellular distribution of BiP was quite different between cells expressing CEL14R and CEL MODY. The majority of BiP was in the insoluble fraction in CEL MODY cells, whereas virtually all of the BiP was in the soluble fraction in CEL14R cells. Our studies do not distinguish between BiP binding to the aggregates to effect disaggregation or BiP sequestration in the aggregates. Presumably, the binding of BiP to insoluble CEL MODY depletes the pool of soluble BiP, which provides a feasible explanation for the differential distribution of BiP in cells expressing CEL MODY compared with cells expressing CEL14R. Fourth, cells expressing CEL MODY showed evidence of a maladaptive UPR as evidenced by increased levels of spliced XBP1 mRNA and activation of inflammatory and apoptosis.
Expression of CEL MODY alone was sufficient stress to activate NF-κB and apoptosis. In fact, treatment of the transfected cells with various metabolic stressors did not increase cell death over that induced by CEL MODY. The observation that a defect in protein homeostasis alone can trigger cell death opens new windows in understanding the pathogenesis of pancreatitis. The concept that perturbations of protein homeostasis cause disease in the exocrine pancreas fits well with the function of the acinar cell. Pancreatic acinar cells are specialized for protein synthesis, storage, and secretion. They synthesize more protein than other cells in the body. In particular, acinar cells synthesize large amounts of a small number of proteins, the 20 or so digestive enzymes (28). Because protein synthesis is inherently error-prone, there is a constant flux of destabilized proteins, and any increase in destabilized proteins is potentially devastating for acinar cells. However, pancreatic acinar cells have adapted to handle a large flux of newly synthesized proteins in the ER. In fact, five transcription factors known to regulate genes encoding key players in protein homeostasis are among the 22 most highly expressed transcription factors in acinar cells (29).
Because they make large quantities of select digestive enzymes, pancreatic acinar cells are susceptible to injury from small perturbations in the homeostasis of these proteins. Any change that leads to the excessive accumulation of misfolded proteins can trigger maladaptive cell signaling pathways (30). Recent studies of transfected cells expressing proteases or pancreatic lipases containing polymorphisms associated with higher risk for chronic pancreatitis (p.R116C in PRSS1, p.A73T in chymotrypsin C, p.N256K in CPA1, and p.T221M in pancreatic lipase) have demonstrated that the polymorphisms increased protein misfolding and decreased secretion, leading to ER stress and UPR activation (31–35). It was suggested that the ER stress response played a role in the pathogenesis of chronic pancreatitis in patients with these polymorphisms. Of these examples, only the p.A73T chymotrypsin C mutant was shown to activate apoptosis (36). It remains unclear whether the other mutations elicit a similar cascade of events as occurs in cells expressing CEL MODY as shown in this study.
Our findings differ in important ways from previous studies of CEL MODY (19, 20). Most importantly, previous studies showed that expression of CEL MODY in stably transfected HEK293 cells activated an ER stress response without activating cell death pathways. The activation of NF-κB was not examined in these studies. The most likely explanation for the discrepancy between previous and our current work is the relatively lower expression levels of CEL variant proteins in past work. It is possible that the lower expression levels were a result of the selection process, which may have resulted in enrichment for cells with lower expression levels because cells with higher expression levels would trigger apoptosis and would not survive the selection process. A similar explanation could also account for the results obtained in CEL MODY transgenic mice in which the level of mRNA encoding CEL MODY was less than one-tenth of the endogenous mRNA that encodes mouse CEL (37). As a result, the CEL MODY transgenic mice failed to mimic the pancreatic pathophysiology seen in the patients. These results suggest that the physiological expression levels of mutant CEL MODY protein are critical to study the cellular responses induced by this mutant protein.
In another study, the same investigators found evidence that CEL MODY is toxic to cells. They treated various cell lines including pancreatic acinar and beta cell model cell lines with conditioned medium from HEK293 cells expressing either CEL16R or CEL MODY. Interestingly, CEL MODY was taken up by all of the cell lines to a much greater extent than CEL16R. Prolonged exposure of the pancreatic exocrine and endocrine cell lines to conditioned medium containing CEL MODY caused increased cell death via apoptosis relative to medium containing CEL16R (20). The authors proposed that CEL MODY endocytosis was an essential feature of the events leading to cell death and that endocytosis plays a role in the development of pancreatic insufficiency and diabetes in patients carrying the CEL variant. Our results suggest that ER stress triggered by aggregated CEL MODY is more important in inducing cell death pathways in pancreatic acinar cells.
Consistent with our data, ER stress is an early pathogenic feature present in a variety of models of experimentally induced acute pancreatitis (27, 30, 38–40). For instance, the reduction of ER stress by tauroursodeoxycholic acid, a chemical chaperone, lessened acinar cell damage and systemic inflammation in rat models of acute pancreatitis, suggesting ER stress as a potential contributing factor for pancreatitis (41). Ethanol-induced pancreatitis in mice triggers an ER stress response that is thought to be a protective mechanism that improves cell survival (5).
In sum, our results provide direct evidence that a mutation in CEL is sufficient to cause ER stress, induction of the UPR, NF-κB activation, and acinar cell death. In our model, the accumulation of mutant protein CEL MODY overwhelms the first line adaptive and protective mechanism offered by the ER stress response. Apoptosis may serve as a protective response to decrease the severity of cell injury by eliminating damaged cells without eliciting significant inflammation. As the purified CEL14R and CEL MODY proteins have similar functional properties, future studies should address the clearance of the toxic mutant protein inside cells to alleviate its proteotoxicity. This knowledge has the potential to provide the basis for novel therapeutic approaches to treat chronic pancreatitis.
Experimental Procedures
Plasmid DNA Constructions
All DNA manipulations were carried out by standard methods unless stated otherwise. The full-length human CEL cDNA clone was purchased from OpenBiosystems (clone ID 5187959; GenBankTM accession number BC042510.1). The cDNA encodes for a 14-repeat VNTR, the variant present in the CEL MODY family. The EcoRI/XhoI fragment containing the entire CEL cDNA coding region was subcloned into yeast (pPICZA) and mammalian (pcDNA3) protein expression vectors. The desired mutations were introduced by using customized primers and a QuikChange II XL site-directed mutagenesis kit (Stratagene). If stated, a FLAG tag was inserted in between human CEL secretory signal peptide and its mature polypeptide by overlap PCR to facilitate protein detection, and the insertion had no detectable effect on the properties of the CEL variant proteins (data not shown). Dideoxynucleotide sequencing validated all plasmid DNA constructs.
Expression, Purification, and Characterization of Recombinant Proteins
Recombinant human CEL protein variants were produced as secretory proteins in P. pastoris GS115 and purified as described in detail previously (34). Six micrograms of each purified protein was resolved by 4–15% SDS-PAGE and stained with GelCode Blue stain reagent (Pierce) to evaluate their homogeneity and integrity. The lipase activity was carried out with 10 μg of purified protein and trioctanoin or triolein as substrate in assay buffer containing 12 mm sodium cholate using the pH-stat method (34). The lipolytic activities are expressed in international lipase units/mg of enzyme. One unit corresponds to 1 μmol of fatty acid released/min.
Recombinant Adenovirus
The cDNAs for human CEL14R, which is a normal variant of CEL, and CEL MODY were subcloned into the pVQAd CMV K-NpA shuttle vector at KpnI/XbaI sites, and the recombinant adenoviruses were then custom-made by Viraquest. Adenovirus expressing enhanced GFP was purchased from Viraquest.
Cell Culture of HEK293T and AR42J Cells
Both cell lines were purchased from ATCC and maintained at 37 °C in a 5% CO2-humidified incubator following the manufacturer's instructions with HEK293T cells in DMEM + 10% FBS and AR42J cells in F-12K + 20% FBS. The transfection of HEK293T cells was carried out using FuGENE 6 (Roche Diagnostics) following the protocol described previously (34). Unless otherwise stated, culture medium and cells were harvested following routine procedures 48 or 72 h after transfection. Prior to transduction, AR42J cells were seeded in 10-cm dishes at a density of 107 cells/dish in 10 ml of medium containing 100 nm dexamethasone (Sigma) and maintained for 24 h. The cells were transduced with 1 × 109 particles of recombinant adenovirus (multiplicity of infection, 100) to express GFP, CEL14R, and CEL MODY proteins. Samples were harvested for analysis 72 h after transduction unless stated otherwise.
Collection of Culture Media and Cells
Samples were harvested as described previously (33). Briefly, 72 h after transfection or transduction, conditioned medium were withdrawn and subjected to 200 × g centrifugation for 5 min to obtain cell-free conditioned medium. Collected cells were washed twice with ice-cold PBS followed by 200 × g × 5-min centrifugation. Whole cell lysates were extracted using 1× Laemmli buffer followed by 3 × 15-s sonication. As for soluble and insoluble fractions, the soluble fraction was defined as the supernatant of radioimmune precipitation assay buffer extracts of cells after 16,000 × g centrifugation for 20 min. The final pellets were resuspended in 1× Laemmli buffer and sonicated until no particle was visible. This part was designated as the insoluble lysate fraction. The protein concentration of cell lysate samples was determined by BCA Protein Assay (Thermo Scientific).
Western Blotting Analysis
Aliquots of conditioned medium (20 μl) or cell lysates (10–50 μg for soluble fractions; 0.1–5 μg for insoluble fractions) were prepared as described previously with the exception of the non-reducing SDS-PAGE in which 2-mercaptoethanol was omitted (34). The prepared samples were resolved on 4–15% Mini-PROTEAN TGX pre-cast gel (Bio-Rad) and transferred onto Immobilon PVDF membranes (Millipore). The blots were incubated sequentially in 5% nonfat milk in TBS-Tween 20, with a primary antibody for 1 h at room temperature or overnight at 4 °C, and then with a corresponding secondary antibody conjugated with HRP at room temperature for 1 h. The HRP signal was detected using the SuperSignal West Femto chemiluminescent substrate (Pierce), and the band density was quantified by densitometry. For CEL analysis, medium (40 μl), insoluble (1.5 μg), and soluble (5 μg) samples were loaded. For other proteins, insoluble (1.2 μg) and soluble (20 μg) samples were loaded.
Antibodies used in this study included rabbit polyclonal antibody against human CEL (1:20,000). To create the antigen, we introduced a premature stop codon into the cDNA encoding CEL by site-directed mutagenesis. The mutation introduced a premature stop codon prior to the VNTR at p.V562X, thereby preventing transcription of the entire VNTR. The cDNA was cloned into pPICZA and expressed in yeast as above. The protein was purified from the medium. Rabbit antiserum against the protein was produced by Cocalico Biologicals (Stevens, PA). Rabbit anti-BiP (catalog number 3183; 1:1000), GRP94 (catalog number 2104; 1:1000), PARP (catalog number 9545, 1:4000), and GAPDH (catalog number 5174, 1:4000) were purchased from Cell Signaling Technology.
Quantitative and Semiquantitative PCR
For transduced AR42J cells, total RNA was purified using an RNeasy Plus Mini kit (Qiagen) and reverse transcribed into cDNA using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). XBP1 mRNA splicing was analyzed by RT-PCR and visualized by ethidium bromide staining after gel electrophoresis on 2% agarose gels. The mRNA expression of human CEL, BiP, CHOP, ATF4, and GAPDH (endogenous control) was evaluated by qRT-PCR using the TaqMan Gene Expression Assay (Applied Biosystems) on a 7300 Real Time PCR System (Applied Biosystems) and primer/probes from Life Technology. The measurement of XBP1 mRNA splicing in transfected HEK293T cells expressing the CEL variant proteins or control was carried out as described (34).
Assays for Cell Death
To determine cell death in HEK293T cells, we measured LDH release into the medium following the manufacturer's instructions (Sigma-Aldrich, catalog number TOX7). For determination of the kinetics of LDH release from HEK293T cells, we plated the cells in a 24-well culture dish. Cells were transfected with control plasmid, CEL14R, and CEL MODY as described above. Another set of wells was not transfected. Medium (100 μl) was removed from the transfected cells at the indicated times for the LDH assay. The medium was removed from a well of the untreated cells, and the cells were lysed with 1 ml of 1% Triton X-100 in PBS. A separate well was harvested for each time point. The insoluble material was pelleted in a microcentrifuge. Total LDH activity in the cells was determined by assaying 100 μl. The amount of LDH in the medium of the transfected cells divided by the total LDH was plotted. To determine the effect of cell density on cell toxicity, we plated HEK293T cells in 6-well plates at the indicated densities. Transfections were done as above. A 100-μl aliquot of medium was assayed for LDH.
We determined cell death in AR42J cells by measuring the number of living cells by the dehydrogenase activity against a water-soluble tetrazolium with Cell Counting Kit-8 (Dojindo Molecular Technologies) (36). Seventy hours after viral transduction in a 24-well plate, 40 μl of the tetrazolium substrate was added to each well. After the incubation at 37 °C for 2 h, the absorbance at 450 nm was measured using a Synergy H1 Hybrid plate reader (BioTek). Absorbance values were normalized to cells transduced to express GFP.
Measurement of NF-κB Activity
NF-κB-luciferase activity was assayed using a luciferase assay kit (E397A, Promega). Fifty-six hours after viral transduction to express GFP, CEL14R, or CEL MODY, AR42J cells were then transduced with Ad-NF-κB-luciferase (42). Sixteen hours later, cells were harvested and lysed using 5× reporter lysis buffer. The treatment of cells with 500 μm taurolithocholic acid 3-sulfate for 2 h prior to sample harvest served as a positive control. Samples were then rigorously vortexed and subjected to a 2-min 12,000 × g centrifugation. The supernatants were plated in a 96-well plate, and the luciferase activities were measured using a Synergy H1 plate reader.
Transmission Electron Microscopy (TEM)
Cell monolayers of AR42J cells were prepared for TEM as described previously (43). Briefly, the cells were washed with PBS and fixed with 2.5% glutaraldehyde in PBS for 1 h at room temperature. After washing with PBS three times, the samples were postfixed in 1% osmium tetroxide and 1% potassium ferricyanide for 1 h at room temperature. After washing with PBS and dehydration in a graded series of ethanol, the samples were infiltrated with Poly/Bed 812. Beam capsules filled with resin were placed on the monolayer and allowed to polymerize. The beam capsules were removed from the culture dishes and sectioned en face for TEM. Sections were stained with 1% uranyl acetate and 1% lead citrate. Images were acquired with a JEM-1400 transmission electron microscope.
Author Contributions
X. X. helped conceive and coordinate the study. He performed or directed construction of all plasmids, prepared recombinant proteins, helped with all cell studies, and directed others involved in the work. He helped write the first draft of the manuscript. G. J. performed studies on AR42J cells including cell culture, transfection, immunoblotting, and PCR analysis. W. A. S. performed studies on HEK293T cells including cell culture, immunoblotting, and cell death assays. D. B. S. was responsible for the electron microscopy. K. E. M. and M. H. helped with studies in HEK293T cells including transfection and immunoblots. A. M. helped conceive studies evaluating cell death pathways and helped direct those studies. Y. W. also helped conceive studies evaluating cell death pathways, helped direct those studies, and contributed to the preparation of the figures. M. E. L. helped conceive the project and directed all aspects of the project. He analyzed data, prepared figures, and along with X. X. wrote the initial draft of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Jeffrey Brodsky for comments on the manuscript.
This work was supported by National Institutes of Health Grant DK808820. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CEL
- carboxyl ester lipase
- ATF4
- activating transcription factor 4
- BiP
- binding immunoglobulin protein
- CEL14R
- wild-type CEL with 14 repeats in the VNTR
- MODY
- maturity onset diabetes of youth with a deletion mutation in the VNTR
- ER
- endoplasmic reticulum
- CHOP
- C/EBP homologous protein
- GRP94
- glucose-regulated protein 94
- LDH
- lactate dehydrogenase
- PARP
- poly(ADP-ribose) polymerase
- NF-κB
- nuclear factor κB
- UPR
- unfolded protein response
- VNTR
- variable number of tandem repeat(s)
- XBP1
- X-box-binding protein-1
- qRT-PCR
- quantitative RT-PCR
- TEM
- transmission electron microscopy
- XBP1U
- unspliced XBP1
- XBP1S
- spliced XBP1.
References
- 1. Fagenholz P. J., Fernández-del Castillo C., Harris N. S., Pelletier A. J., and Camargo C. A. Jr. (2007) Direct medical costs of acute pancreatitis hospitalizations in the United States. Pancreas 35, 302–307 [DOI] [PubMed] [Google Scholar]
- 2. Pandol S. J., Saluja A. K., Imrie C. W., and Banks P. A. (2007) Acute pancreatitis: bench to the bedside. Gastroenterology 132, 1127–1151 [DOI] [PubMed] [Google Scholar]
- 3. Sahin-Tóth M. (2006) Biochemical models of hereditary pancreatitis. Endocrinol. Metab. Clin. North Am. 35, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ji B., and Logsdon C. D. (2011) Digesting new information about the role of trypsin in pancreatitis. Gastroenterology 141, 1972–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pandol S. J., Gorelick F. S., and Lugea A. (2011) Environmental and genetic stressors and the unfolded protein response in exocrine pancreatic function—a hypothesis. Front. Physiol. 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raeder H., Haldorsen I. S., Ersland L., Grüner R., Taxt T., Søvik O., Molven A., and Njølstad P. R. (2007) Pancreatic lipomatosis is a structural marker in nondiabetic children with mutations in carboxyl-ester lipase. Diabetes 56, 444–449 [DOI] [PubMed] [Google Scholar]
- 7. Raeder H., Johansson S., Holm P. I., Haldorsen I. S., Mas E., Sbarra V., Nermoen I., Eide S. A., Grevle L., Bjørkhaug L., Sagen J. V., Aksnes L., Søvik O., Lombardo D., Molven A., and Njølstad P. R. (2006) Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat. Genet. 38, 54–62 [DOI] [PubMed] [Google Scholar]
- 8. Torsvik J., Johansson S., Johansen A., Ek J., Minton J., Raeder H., Ellard S., Hattersley A., Pedersen O., Hansen T., Molven A., and Njølstad P. R. (2010) Mutations in the VNTR of the carboxyl-ester lipase gene (CEL) are a rare cause of monogenic diabetes. Hum. Genet. 127, 55–64 [DOI] [PubMed] [Google Scholar]
- 9. Fjeld K., Weiss F. U., Lasher D., Rosendahl J., Chen J. M., Johansson B. B., Kirsten H., Ruffert C., Masson E., Steine S. J., Bugert P., Cnop M., Grützmann R., Mayerle J., Mössner J., et al. (2015) A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 47, 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hui D. Y., and Howles P. N. (2002) Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J. Lipid Res. 43, 2017–2030 [DOI] [PubMed] [Google Scholar]
- 11. Moore S. A., Kingston R. L., Loomes K. M., Hernell O., Bläckberg L., Baker H. M., and Baker E. N. (2001) The structure of truncated recombinant human bile salt-stimulated lipase reveals bile salt-independent conformational flexibility at the active-site loop and provides insights into heparin binding. J. Mol. Biol. 312, 511–523 [DOI] [PubMed] [Google Scholar]
- 12. Lindquist S., Bläckberg L., and Hernell O. (2002) Human bile salt-stimulated lipase has a high frequency of size variation due to a hypervariable region in exon 11. Eur. J. Biochem. 269, 759–767 [DOI] [PubMed] [Google Scholar]
- 13. Strömqvist M., Hernell O., Hansson L., Lindgren K., Skytt A., Lundberg L., Lidmer A. S., and Bläckberg L. (1997) Naturally occurring variants of human milk bile salt-stimulated lipase. Arch. Biochem. Biophys. 347, 30–36 [DOI] [PubMed] [Google Scholar]
- 14. Bruneau N., Nganga A., Fisher E. A., and Lombardo D. (1997) O-Glycosylation of C-terminal tandem-repeated sequences regulates the secretion of rat pancreatic bile salt-dependent lipase. J. Biol. Chem. 272, 27353–27361 [DOI] [PubMed] [Google Scholar]
- 15. Bruneau N., Lombardo D., and Bendayan M. (1998) Participation of GRP94-related protein in secretion of pancreatic bile salt-dependent lipase and in its internalization by the intestinal epithelium. J. Cell Sci. 111, 2665–2679 [DOI] [PubMed] [Google Scholar]
- 16. Bruneau N., Lombardo D., Levy E., and Bendayan M. (2000) Roles of molecular chaperones in pancreatic secretion and their involvement in intestinal absorption. Microsc. Res. Tech. 49, 329–345 [DOI] [PubMed] [Google Scholar]
- 17. Ragvin A., Fjeld K., Weiss F. U., Torsvik J., Aghdassi A., Mayerle J., Simon P., Njølstad P. R., Lerch M. M., Johansson S., and Molven A. (2013) The number of tandem repeats in the carboxyl-ester lipase (CEL) gene as a risk factor in alcoholic and idiopathic chronic pancreatitis. Pancreatology 13, 29–32 [DOI] [PubMed] [Google Scholar]
- 18. Zou W. B., Boulling A., Masamune A., Issarapu P., Masson E., Wu H., Sun X. T., Hu L. H., Zhou D. Z., He L., Fichou Y., Nakano E., Hamada S., Kakuta Y., Kume K., et al. (2016) No association between CEL-HYB hybrid allele and chronic pancreatitis in Asian populations. Gastroenterology 150, 1558.e5–1560.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johansson B. B., Torsvik J., Bjørkhaug L., Vesterhus M., Ragvin A., Tjora E., Fjeld K., Hoem D., Johansson S., Ræder H., Lindquist S., Hernell O., Cnop M., Saraste J., Flatmark T., et al. (2011) Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): a protein misfolding disease. J. Biol. Chem. 286, 34593–34605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torsvik J., Johansson B. B., Dalva M., Marie M., Fjeld K., Johansson S., Bjørkøy G., Saraste J., Njølstad P. R., and Molven A. (2014) Endocytosis of secreted carboxyl ester lipase in a syndrome of diabetes and pancreatic exocrine dysfunction. J. Biol. Chem. 289, 29097–29111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vesterhus M., Raeder H., Aurlien H., Gjesdal C. G., Bredrup C., Holm P. I., Molven A., Bindoff L., Berstad A., and Njølstad P. R. (2008) Neurological features and enzyme therapy in patients with endocrine and exocrine pancreas dysfunction due to CEL mutations. Diabetes Care 31, 1738–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baek J. H., Whitfield D., Howlett D., Francis P., Bereczki E., Ballard C., Hortobágyi T., Attems J., and Aarsland D. (2016) Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol. 42, 352–365 [DOI] [PubMed] [Google Scholar]
- 23. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 24. Urra H., Dufey E., Lisbona F., Rojas-Rivera D., and Hetz C. (2013) When ER stress reaches a dead end. Biochim. Biophys. Acta 1833, 3507–3517 [DOI] [PubMed] [Google Scholar]
- 25. Christophe J. (1994) Pancreatic tumoral cell line AR42J: an amphicrine model. Am. J. Physiol. Gastrointest. Liver Physiol. 266, G963–G971 [DOI] [PubMed] [Google Scholar]
- 26. Gukovsky I., Gukovskaya A. S., Blinman T. A., Zaninovic V., and Pandol S. J. (1998) Early NF-κB activation is associated with hormone-induced pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 275, G1402–G1414 [DOI] [PubMed] [Google Scholar]
- 27. Sah R. P., Garg S. K., Dixit A. K., Dudeja V., Dawra R. K., and Saluja A. K. (2014) Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J. Biol. Chem. 289, 27551–27561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whitcomb D. C., and Lowe M. E. (2007) Human pancreatic digestive enzymes. Dig. Dis. Sci. 52, 1–17 [DOI] [PubMed] [Google Scholar]
- 29. MacDonald R. J., Swift G. H., and Real F. X. (2010) Transcriptional control of acinar development and homeostasis. Prog. Mol. Biol. Transl. Sci. 97, 1–40 [DOI] [PubMed] [Google Scholar]
- 30. Logsdon C. D., and Ji B. (2013) The role of protein synthesis and digestive enzymes in acinar cell injury. Nat. Rev. Gastroenterol. Hepatol. 10, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kereszturi E., Szmola R., Kukor Z., Simon P., Weiss F. U., Lerch M. M., and Sahin-Tóth M. (2009) Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum. Mutat. 30, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Witt H., Beer S., Rosendahl J., Chen J. M., Chandak G. R., Masamune A., Bence M., Szmola R., Oracz G., Macek M. Jr., Bhatia E., Steigenberger S., Lasher D., Bühler F., Delaporte C., et al. (2013) Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 45, 1216–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szabó A., Xiao X., Haughney M., Spector A., Sahin-Tóth M., and Lowe M. E. (2015) A novel mutation in PNLIP causes pancreatic triglyceride lipase deficiency through protein misfolding. Biochim. Biophys. Acta 1852, 1372–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiao X., Mukherjee A., Ross L. E., and Lowe M. E. (2011) Pancreatic lipase-related protein-2 (PLRP2) can contribute to dietary fat digestion in human newborns. J. Biol. Chem. 286, 26353–26363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beer S., Zhou J., Szabó A., Keiles S., Chandak G. R., Witt H., and Sahin-Tóth M. (2013) Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 62, 1616–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szmola R., and Sahin-Tóth M. (2010) Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut 59, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ræder H., Vesterhus M., El Ouaamari A., Paulo J. A., McAllister F. E., Liew C. W., Hu J., Kawamori D., Molven A., Gygi S. P., Njølstad P. R., Kahn C. R., and Kulkarni R. N. (2013) Absence of diabetes and pancreatic exocrine dysfunction in a transgenic model of carboxyl-ester lipase-MODY (maturity-onset diabetes of the young). PLoS One 8, e60229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kubisch C. H., and Logsdon C. D. (2007) Secretagogues differentially activate endoplasmic reticulum stress responses in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1804–G1812 [DOI] [PubMed] [Google Scholar]
- 39. Kubisch C. H., and Logsdon C. D. (2008) Endoplasmic reticulum stress and the pancreatic acinar cell. Expert. Rev. Gastroenterol. Hepatol. 2, 249–260 [DOI] [PubMed] [Google Scholar]
- 40. Kubisch C. H., Sans M. D., Arumugam T., Ernst S. A., Williams J. A., and Logsdon C. D. (2006) Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G238–G245 [DOI] [PubMed] [Google Scholar]
- 41. Seyhun E., Malo A., Schäfer C., Moskaluk C. A., Hoffmann R. T., Göke B., and Kubisch C. H. (2011) Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G773–782 [DOI] [PubMed] [Google Scholar]
- 42. Jin S., Orabi A. I., Le T., Javed T. A., Sah S., Eisses J. F., Bottino R., Molkentin J. D., and Husain S. Z. (2015) Exposure to radiocontrast agents induces pancreatic inflammation by activation of nuclear factor-κB, calcium signaling, and calcineurin. Gastroenterology 149, 753.e11–764.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tafaleng E. N., Chakraborty S., Han B., Hale P., Wu W., Soto-Gutierrez A., Feghali-Bostwick C. A., Wilson A. A., Kotton D. N., Nagaya M., Strom S. C., Roy-Chowdhury J., Stolz D. B., Perlmutter D. H., and Fox I. J. (2015) Induced pluripotent stem cells model personalized variations in liver disease resulting from alpha1-antitrypsin deficiency. Hepatology 62, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Back S. H., Lee K., Vink E., and Kaufman R. J. (2006) Cytoplasmic IRE1α-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 281, 18691–18706 [DOI] [PubMed] [Google Scholar]