

Abstract
Introduction
Systemic Sclerosis (SSc) is a systemic autoimmune disease characterized by severe and often progressive cutaneous, pulmonary, cardiac and gastrointestinal tract fibrosis, cellular and humoral immunologic alterations, and pronounced fibroproliferative vasculopathy. There is no effective SSc disease modifying therapy. Patients with rapidly progressive SSc have poor prognosis with frequent disability and very high mortality.
Areas Covered
This paper reviews currently available therapeutic approaches for rapidly progressive SSc and discuss novel drugs under study for SSc disease modification.
Expert Opinion
The extent, severity, and rate of progression of SSc skin and internal organ involvement determines the optimal therapeutic interventions for SSc. Cyclophosphamide for progressive SSc-associated interstitial lung disease and mycophenolate for rapidly progressive cutaneous involvement have shown effectiveness. Methotrexate has been used for less severe skin progression and for patients unable to tolerate mycophenolate. Rituximab was shown to induce improvement in SSc-cutaneous and lung involvement. Autologous bone marrow transplantation is reserved for selected cases in whom poor survival risk outweighs the high mortality rate of the procedure. Novel agents capable of modulating fibrotic and inflammatory pathways involved in SSc pathogenesis, including tocilizumab, pirfenidone, tyrosine kinase inhibitors, lipid lysophosphatidic acid 1, and NOX4 inhibitors are currently under development for the treatment of rapidly progressive SSc.
Keywords: Pulmonary Fibrosis, Rapidly Progressive Systemic Sclerosis, Treatment
1. INTRODUCTION
Systemic sclerosis (SSc) is a systemic idiopathic autoimmune disease characterized by exaggerated extracellular matrix deposition in skin and various internal organs, severe fibro-proliferative vasculopathy and cellular and humoral immune response abnormalities [1–5]. These three pathogenic processes occur with variable severity in each particular SSc patient resulting in a highly heterogeneous clinical presentation that reflects SSc complex pathophysiology. The clinical heterogeneity of SSc and the scarcity of quantitative and objective assessment tools to evaluate SSc severity, extent of involvement, and rapidity of progression impose substantial limitations to the development of effective SSc disease-modifying therapies [6, 7]. To minimize these limitations various SSc clinical sub-sets have been defined according to the extent and rapidity of progression of skin and internal organ involvement, and the presence in the serum of specific autoantibodies [8–11]. One subset of patients considered to have “rapidly progressive SSc”, encompasses patients with diffuse cutaneous involvement with rapid progression of skin induration and evidence of target organ damage [12,13]. These patients also frequently present elevated erythrocyte sedimentation rate and C-reactive protein [14,15] as well as palpable tendon friction rubs [16–19]. Patients in this clinical subset have very poor prognosis and very high mortality [20–23]. Consequently, early and accurate identification of rapidly progressive SSc is paramount in order to decrease the mortality, halt the progression, and improve the prognosis of these patients.
Despite the remarkable enhancement in the overall survival of SSc patients in recent years, SSc associated mortality is still substantially higher than that of other inflammatory/autoimmune rheumatic diseases [22,24–26]. Furthermore, organ specific-related mortality in SSc has also changed over the last few decades. For example, SSc-associated acute renal involvement known as scleroderma renal crisis, a complication that represented the main cause of SSc mortality in prior decades, has declined markedly in its prevalence following the introduction of angiotensin converting enzyme inhibitors as the standard of care for treatment of this complication [27–30]. On the other hand, SSc-associated pulmonary involvement, either interstitial lung disease (ILD) or pulmonary arterial hypertension (PAH), has recently emerged as the leading cause of SSc mortality [31]. Furthermore, the occurrence of a high rate of cardiovascular events in SSc patients has been identified suggesting evidence of both large vessel and small vessel vasculopathy [32–35]. Despite substantial advances in the overall treatment of SSc and the notable increase in SSc survival accomplished recently, currently, there are no approved disease-modifying therapeutic interventions for SSc and there are no drugs that have been shown to reverse SSc-associated ILD. However, the recent development of novel antifibrotic and immunosuppressive therapies [36–39] offers unique opportunities to improve the outcomes of SSc patients, particularly of those with rapidly progressive disease manifestations who have the poorest prognosis and highest rates of disability and mortality. In this review we will discuss briefly the pathophysiology of SSc, will define SSc clinical subsets focusing on the identification of SSc patients with rapidly progressive disease, and will review currently used and promising investigational therapeutic strategies for the management of rapidly progressive SSc.
2. PATHOPHYSIOLOGY
The pathogenesis of SSc comprises three distinct processes: 1. Excessive deposition of extracellular matrix in skin and numerous internal organs; 2. Severe functional and structural fibroproliferative abnormalities in the microvasculature; and 3. Humoral and cellular immunologic abnormalities characterized by innate immunity alterations, involvement of macrophages and T-and B-lymphocytes, and the production of numerous disease-specific autoantibodies [1–5]. However, it has not been established which of these processes is of primary importance or how they are temporally related during the development and progression of SSc.
2.1. General hypothesis of SSc pathogenesis
A current hypothesis for SSc pathogenesis posits that unknown etiologic factors in a genetically receptive host [40–48] trigger microvascular injury characterized by structural and functional endothelial cell abnormalities [49–51]. These abnormalities result in the increased production and release of numerous and potent signaling molecules including cytokines, chemokines, polypeptide growth factors, and reactive oxygen species. The endothelial cell dysfunction triggers the attraction of specific inflammatory cellular elements from the bloodstream and bone marrow and their transmigration into the perivascular and parenchymal tissues [52]. These events lead to the establishment of a chronic inflammatory process with participation of macrophages and T- and B- lymphocytes, with further production and secretion of profibrotic cytokines and growth factors that induce the tissue accumulation of activated myofibroblasts, the effector cells ultimately responsible for the fibrotic process in SSc [53–56]. This sequence of events, diagrammatically illustrated in Figure 1, results in the development of a severe and often progressive fibroproliferative vasculopathy, and in the exaggerated and widespread accumulation of fibrotic tissue in the skin and multiple internal organs.
Figure 1.

Hypothetical sequence of events involved in tissue fibrosis and fibroproliferative vasculopathy in SSc. An unknown causative agent induces activation of immune and inflammatory cells in genetically predisposed hosts resulting in chronic inflammation. Activated inflammatory and immune cells secrete cytokines, chemokines, and growth factors which cause fibroblast activation, differentiation of endothelial and epithelial cells into myofibroblasts, and recruitment of fibrocytes from the bone marrow and the peripheral blood circulation. The activated myofibroblasts produce exaggerated amounts of ECM resulting in tissue fibrosis. Modified from [78] with permission.
2.2. Innate and cellular immunity alterations in SSc
In a population with genetic susceptibility an environmental triggering event has been postulated to initiate the complex pathophysiological changes leading to the establishment and expression of the disease clinical features. Several studies have proposed that viruses such as retroviruses, cytomegalovirus, Epstein-Barr virus or other infectious agents may be implicated in the early events of SSc pathogenesis [57–61].
Regardless of the initiating event or trigger, the extensive innate and cellular immune abnormalities in SSc [3–5,62–67] result in exaggerated Th2 response driving the increased expression and production of numerous and potent profibrotic cytokines and growth factors. Besides cellular immunity alterations, abnormal humoral immunity leading to the production of numerous autoantibodies is a nearly universal manifestation of SSc. Most circulating autoantibodies have not been shown to influence directly SSc pathogenesis and have been considered to represent autoimmune epiphenomena useful as disease diagnostic and clinical subset biomarkers, although certain autoantibodies correlate with specific clinical SSc patterns. For example, the presence of anti-RNA polymerase III antibodies indicates a higher risk of scleroderma renal crisis, whereas anti-centromere antibodies (ACA) are associated with limited skin involvement, and anti-topoisomerase I (Scl-70) antibodies are associated with increased risk of diffuse skin involvement and SSc-associated ILD [68–70]. The association of various circulating autoantibodies with SSc clinical subsets and certain clinical features are shown in Table 1.
Table 1.
Association of auto-antibodies with different SSc clinical phenotypes
| Antibody Type | Prevalence % | Disease subtype | Clinical Phenotype |
|---|---|---|---|
| Anti-centromere | 24–45 | IcSSc | CREST Syndrome Pulmonary arterial hypertension |
| Anti-Scl-70 | 15–42 | dcSSc | Rapidly Progressive SSc-ILD Cardiac Involvement |
| Anti-RNA polymerase III | 5–31 | dcSSc | Scleroderma Renal Crisis Tendon friction rubs, Joint contractures Myositis |
| Anti-U3RNP (Fibrillarin) | 2–16 | dcSSc | Scleroderma Renal Crisis Cardiac Involvement |
| Anti-PM-Scl | 4–11 | IcSSc | Myositis, arthritis, calcinosis Sicca syndrome SSc-ILD |
| Anti- Th/To | 1–18 | IcSSc | SSc-ILD Scleroderma Renal Crisis Gastrointestinal Involvement |
| Anti-U11/U12 RNP | 3.2 | - | SSc-ILD |
| Anti-U1-RNP | 2–14 | IcSSc | MCTD features |
| Anti-Ku | 2 | dcSSc | Myositis, arthritis, joint contractures |
| Anti-NOR 90 | 0.8–6 | dSSc | SSc-ILD |
| Anti-Ro52 | 20–27 | - | SSc-ILD |
lcSSc: Limited cutaneous SSc.
dcSSc: Diffuse cutaneous SSc.
SSc-ILD: SSc-associated Interstitial Lung Disease.
MCTD: Mixed connective tissue disease.
2.3. Myofibroblasts and tissue fibrosis in SSc
The cellular immune system alterations in SSc promote a prolonged and sustained pro-fibrotic state. Pro-fibrotic cytokines and growth factors including TGF-β, PDGF, endothelin-1, and CTGF, and other mediators known to induce increased proliferation of smooth muscle cells, subendothelial accumulation of fibrous tissue, and fibroblast activation in the skin and in the parenchyma of various organs, have been shown to participate in various aspects of SSc-associated fibrotic pathways [62–67,71–73].
In parallel, proliferation and accumulation of myofibroblasts in skin and target organs drives the excessive production and deposition of types I and III collagens and other extracellular matrix components such as COMP, glycosaminoglycans, and fibronectin in affected tissues. Myofibroblasts are the cells ultimately responsible for the establishment and progression of the fibrotic process. These cells are characterized by an increased expression of various interstitial collagens, the initiation of expression of α-smooth muscle actin (α-SMA), the reduction of production of extracellular matrix–degradative enzymes, and the acquisition of contractile and motile properties that allow them to migrate to non-affected tissues and to induce their contraction resulting in a marked increasing in tissue stiffness, a potent additional pro-fibrotic mechanism [54–56]. Thus, the accumulation of myofibroblasts in SSc-affected tissues and the uncontrolled persistence of their elevated biosynthetic functions are crucial determinants of the extent and rate of progression of the fibrotic process in SSc [72–74]. It is generally accepted that myofibroblasts are originated from the activation of quiescent tissue resident fibroblasts but other sources including transdifferentiation of epithelial cells (epithelial to mesenchymal transdifferentiation) or endothelial cells (endothelial to mesenchymal transdifferentiation), and of pericytes have been shown to be important sources of activated myofibroblasts [75–78]. These cellular phenotypic changes are induced by various profibrotic cytokines and growth factors including TGF-β, CTGF, PDGF, IL-6, IL-13, and others. The pathways and intracellular signaling events resulting in the profibrotic myofibroblast activation by TGF-β and PDGF responsible for the sustained stimulation by pro-fibrotic genes are illustrated in Figure 2.
Figure 2.
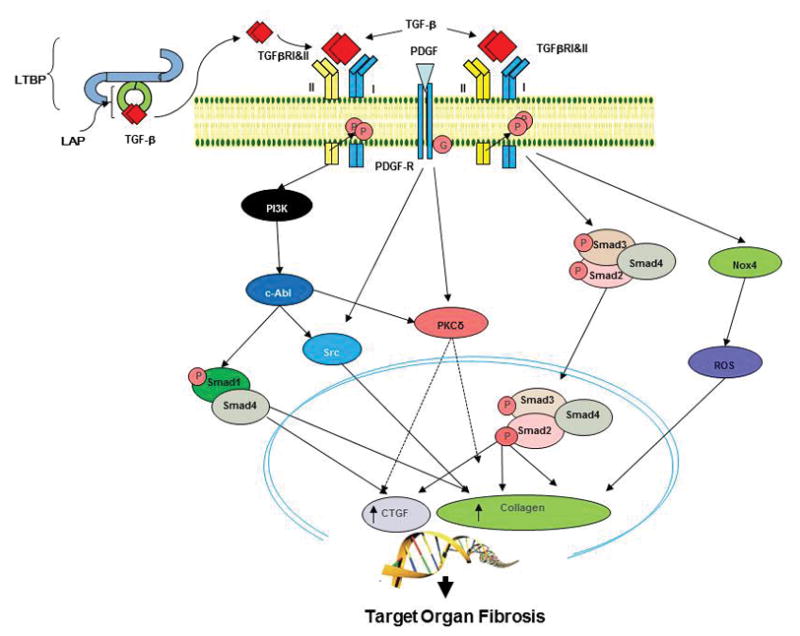
Schematic diagram of TGF-β and PDGF pathways involved in SSc-associated tissue fibrosis. TGF- β is released from the latent-TGF-β binding protein (LTBP) and after engaging its receptor (TGFR), induces heterodimerization between TGFRI and TGFRII forms. The activated receptor triggers stimulatory Smad pathways and non-Smad pathways such as the tyrosine kinases c-Abl, and Src and protein kinase Cδ (PKC-δ) that ultimately cause increased collagen and ECM production, resulting in tissue fibrosis. PDGF activation of its cognate receptor induces activation of Src and PKC-δ kinases contributing to the profibrotic effects of TGF-β. TGF-β also induces elevated expression of NOX-4 resulting in increased expression of profibrotic genes. Modified from [36] with permission.
3. CLINICAL PRESENTATION AND CLINICAL SUBSETS OF SSc
3.1. Clinical presentation
SSc is characterized by a complex and heterogeneous clinical phenotype ranging from limited skin disease to forms with diffuse skin sclerosis and progressive internal organ involvement. Some patients with diffuse skin involvement develop a rapidly progressive or occasionally a fulminant phenotype with severe internal organ damage and rapid functional organ failure resulting in a very high mortality rate [10–14].
3.2. Clinical subsets of SSc
SSc has been classified into two clinical subsets according to the degree of severity and extent of skin involvement: limited cutaneous and diffuse cutaneous SSc [10]. However, both limited and diffuse forms of SSc are systemic and, therefore, both can affect the respiratory, cardiovascular, gastrointestinal, renal, musculoskeletal, endocrine, and genitourinary systems. Limited cutaneous SSc is characterized by skin involvement distal to the elbows and knees but may involve the face and neck. The term CREST syndrome was coined to describe the high frequency of calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia [79] in patients with the limited cutaneous SSc phenotype. Diffuse cutaneous SSc refers to the form of SSc with skin thickening and induration affecting the trunk and/or the extremities proximal to the elbows and knees. Besides these two groups it has been recognized that there are infrequent cases displaying typical SSc internal organ alterations in the absence of clinically apparent cutaneous involvement. This subset is known as “scleroderma sine scleroderma” [80]. In addition, although not specifically recognized as a clinical subset, some SSc patients present a fulminant course (fulminant systemic sclerosis) with early mortality or very accelerated internal organ failure; a clinical presentation known as a “fulminant systemic sclerosis” [81–83].
3.3. SSc classification criteria
Recently, a joint committee of the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) published a revised classification criteria for SSc to improve the sensitivity of the widely used previous classification criteria [8, 9]. These criteria are shown in Table 2. Substantial differences in the clinical prognosis have been described between different subclasses. For example, one study found that patients with limited cutaneous SSc have better prognosis with average 5 year survival rates of about 90%, in contrast, patients with diffuse cutaneous SSc have a worse prognosis with average mortality rates of 78% and 62% at 5 and 10 years, respectively [84]. Prognosis in this group is strongly related to the rapidity of progression of skin induration and to the presence and severity of cardiac and pulmonary involvement.
Table 2.
ACR/EULAR Revised Systemic Sclerosis Classification Criteria
| Item | Sub-item(s) | Score |
|---|---|---|
| Skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints* | None | 9 |
| Skin thickening of the fingers** | Puffy fingers | 2 |
| Sclerodactyly (distal to the metacarpophalangeal joints but proximal to the interphalangeal joints) | 4 | |
| Fingertip lesions** | Digital tip ulcers | 2 |
| Fingertip pitting scars | 3 | |
| Telangiectasia | None | 2 |
| Abnormal nailfold capillaries | None | 2 |
| Pulmonary arterial hypertension and/or interstitial lung disease (maximum score is 2) | Pulmonary arterial hypertension and/or Interstitial lung disease | 2 |
| Raynaud’s phenomenon | None | 3 |
| Systemic sclerosis-related autoantibodies (maximum score is 3) | Anti-centromere and/or Anti-topoisomerase I and/or Anti-RNA polymerase III |
3 |
3.4. Assessment of SSc skin and internal organ involvement
The degree of skin induration is assessed employing the “Modified Rodnan Skin Score” (mRSS). The mRSS is a semi- quantitative scale based solely on the examiner’s subjective assessment of the severity/degree of skin induration at 17 pre-specified sites ranging from 0 to 3 at each site, thus providing a total score range from 0 to 51, depending of the degree of perceived skin thickening [85,86]. Whereas there is no consensus about the cut point for defining rapid progression in either skin or internal organ involvement, numerous studies have demonstrated that patients with rapidly increasing mRSS have substantially higher mortality than patients with slow progression [87,88]. In addition, older age at presentation, presence of tendon friction rubs and elevated erythrocyte sedimentation rates are factors related with worse clinical prognosis. An index of progression based on mRSS change over time termed “Skin Thickness Progression Rate” (STPR) has been proposed [13]. Patients with higher and intermediate STPR (indicating faster progression of mRSS) had higher morbidity and mortality than patients with lower STPR. However, despite the potentially useful predictive and prognostic value of the STPR, it has not been generally implemented.
3.5. Definition of rapidly progressive SSc
Although there is not an accepted and widely recognized clinical subset of rapidly progressive SSc, we have found it useful to identify patients in this clinical category to allow more extensive evaluation and close follow-up, including performance of more frequent testing and prompt initiation of more aggressive SSc disease modifying therapy. Based on the assessment of the clinical course of a large number of SSc patients followed at the Scleroderma Center of our Institution [81,87,89,90], and considering that SSc-associated ILD is not reversible but can only be stabilized with arrest in its progressive course, we have used of the following criteria for defining patients with rapidly progressive SSc:
Initial mRSS greater than 12 in SSc patients with recent onset skin disease (less than 12 months).
Increase in mRSS by greater than 12 points during a 6 month interval.
Decline in either FVC or TLC greater than 15% during a 6 month interval.
Other factors such as the occurrence of palpable tendon friction rubs, elevated erythrocyte sedimentation rate and C-reactive protein, and presence of anti-topoisomerase-1 (Scl-70) antibodies support the inclusion in the rapidly progressive SSc group.
4. TREATMENT OF RAPIDLY PROGRESSIVE SSc
We will review different drugs currently used in patients considered to have rapidly progressive SSc. The drugs currently being used or undergoing clinical trials are listed in Table 3. We should emphasize that owing to the difficulty in recruiting large numbers of patients with rapid SSc progression, most available studies have employed non-homogeneous inclusion criteria. The vast heterogeneity in patient populations included in the various studies should be taken in consideration when analyzing and evaluating the results of the different studies. The following are currently accepted therapeutic interventions for patients with rapidly progressive SSc:
Table 3.
Potential therapeutic targets in Rapidly Progressive SSc.
| Drug | Target | Mechanism |
|---|---|---|
| TGF-β pathway inhibitors | ||
| Anti-CTGF antibody | CTGF (CCN2) | CTGF (CCN2) is potently induced by TGF-β; Considered crucial in various fibrotic conditions. |
| Pirfenidone | TGF-β | |
| NOX-4 inhibitor | NOX-4 | CTGF inhibition has potential as an anti-fibrotic therapy |
| Exact mechanism of action unknown. | ||
| Suggested effect blocking TGF-β induced fibrosis pathways | ||
| NOX-4 is induced by TGF-β and displays potent profibrotic effects | ||
| Fresolimumab | TGF-β 1,2,3 | Inhibits the three TGF-β isoforms and blocks their profibrotic effects |
| Monoclonal Antibodies | ||
| Rituximab | B-cells | Chimeric monoclonal antibody recognizes B-cell surface protein CD20. Blocks B-cell role in SSc pathogenesis |
| Tocilizumab | IL-6 | Humanized monoclonal antibody against IL-6 receptor. IL-6 plays a profibrotic role in SSc pathogenesis |
| Belimumab | BAFF | Human monoclonal antibody that may modulate BAFF-mediated altered SSc immune responses |
| Lebrikizumab | IL-13Ra1 | Humanized monoclonal antibody against IL-13 receptors. IL-13 induced a profibrotic phenotype in normal dermal fibroblasts |
| Tralokinumab (CAT-354) | IL-13 | Human immunoglobulin Ig G4 monoclonal antibody. IL-13 induced a profibrotic phenotype in normal dermal fibroblasts |
| Tyrosine kinase inhibitors | ||
| Imatinib | c-Abl, PDGF | |
| Nilotinib | c-Abl, PDGF | Inhibit activity of tyrosine kinases mediating |
| Dasatinib | c-Abl, PDGF, Src | pro-fibrotic pathways |
| Nintedanib | VEGF, FGF, PDGF | |
| Non-receptor kinase inhibitors | ||
| Rho-kinase inhibitor | RhoA Kinase | Affects vascular homeostasis. May have direct antifibrotic effects |
| Statins | Rho-A | Inhibit prenylation of Rho-A kinase resulting in inhibition of profibrotic pathways |
| JAK-2 inhibitors | JAK-2 | Jak-2 is involved in stimulation of SSc fibroblasts by TGF-β; JAK-2 inhibition may have a role in antifibrotic therapy in SSc |
| Other signaling pathways | ||
| LPA-1 inhibitors | LPAR | Inhibits LPA-1 implicated in pulmonary fibrosis |
| Rosiglitazone, Pioglitazone | PPAR- γ | PPAR-γ inhibits the fibrogenic effects of TGF-β in vitro; agonists of PPAR-γ may suppress collagen synthesis, myofibroblast differentiation, and other TGF-β induced fibrotic responses |
| Hedgehog signaling | Shh | Has a role in tissue fibrosis through stimulating collagen release and myofibroblast differentiation in vitro and inducing fibrosis in vivo |
| Bortezomib | Proteasome | Proteasome inhibitor; may result in reduced collagen production and increased collagen degradation |
| Wnt inhibitors | Wnt | Wnt plays an important role in various profibrotic pathways |
| Rapamycin | FK binding protein 12 | Binds FK binding protein 12 inhibits mTOR resulting in a diminished SSc immune response in scleroderma. May have direct antifibrotic effects |
| Peptide inhibitors | ||
| Caveolin Scafolding-domain Peptide | CAV-1 | Restores caveolin-1 deficiency in SSc cells- Decreased CAV-1 is a potent pro-fibrotic |
| Endostatin Peptide | Endostatin | Shown to display potent antifibrotic effects in vitro |
| Anti-Thrombin | ||
| Dabigatran | Thrombin | Thrombin is a potential mediator of fibrosis because it is involved in ECM protein deposition, induction of profibrotic factors, and promotion of myofibroblast differentiation |
BAFF: B-cell activation factor
CAV: Caveolin
LPA: Lysophosphatidic Acid
Shh: Sonic hedgehog
4.1. Cyclophosphamide (CYC)
Following several early uncontrolled studies describing the efficacy of CYC in the treatment of SSc-associated ILD [91] two randomized controlled trials (RCT) comparing CYC with placebo have been conducted. In the Scleroderma Lung Study I (SLS I), 158 SSc patients with SSc-associated ILD of less than 7 years of duration were randomized to receive either oral CYC (1–2 mg/kg/d) or placebo for 1 year [92]. The statistical analyses chosen allowed to reduce the standard deviation driven by extreme values (Huber covariance estimation) and resulted in statistically significant differences. Although the mean absolute difference in an adjusted one year FVC percentage predicted between CYC and placebo group was only 2.53% in favor of the CYC group (p<0.03) the group receiving CYC treatment had significant improvement in quality of life parameters. However, the beneficial effects of CYC did not persist 12 months following cessation of CYC strongly suggesting the need for continued CYC maintenance treatment. The Fibrosing Alveolitis in Scleroderma Trial (FAST), randomized 45 SSc-associated ILD patients to be treated either with placebo or with 6-monthly infusions of CYC followed by 6 months of daily oral azathioprine (AZA) [93]. Owing to the small size of the cohort, statistical significance was not reached although the adjusted between-group difference in FVC of 4.2% showed a trend favoring the CYC treated group (p = 0.08). On the other hand, evidence from an unblinded trial comparing daily oral CYC to AZA for 1.5 years showed a significant difference favoring CYC in the FVC trend during the study period (+3% vs −11.1%; p<0.001). However, all available studies showed that CYC treatment was associated with statistically significantly higher frequency of adverse events including serious adverse events compared to the placebo groups. Other non-randomized trials of CYC in SSc-associated ILD have shown the same trend of stabilization or mild improvement in terms of lung volume measurements in pulmonary function tests [94,95]. It should be emphasized, however, that given the absence of a universally accepted definition for rapidly progressive lung disease in SSc patients and the lack of uniformity in the criteria employed for the inclusion of patients, the conclusions reached in these studies may not be fully applicable to all patients with SSc-associated ILD.
4.2. Mycophenolate mofetil (MMF)
There have not been any randomized control trials (RCT) employing MMF in patients with rapidly progressive SSc, although numerous non-randomized trials have shown consistent and significant reduction in the mRSS in patients with diffuse and progressive SSc [89,90,96,97]. For example, three different studies [89,90,97] found an average mRSS reduction of 14.1, 7.6 and 10.04 points, respectively. These results were significantly better than the spontaneous reduction in mRSS observed in some prior RCTs such as the relaxin and the bovine type I collagen trials [98,99]. Furthermore, an analysis of the subgroup of patients with recent skin changes [97] suggested that patients with shorter disease duration obtained greater benefit from MMF therapy, showing more pronounced improvement in the mRSS. It should be emphasized that those studies did not specifically include patients with SSc-associated ILD and most of the patients had only mild lung involvement. However, there are several uncontrolled studies that evaluated MMF effects on SSc-associated ILD. The results of these studies were encouraging showing estabilization of lung function in the majority of patients receiving long term (at least 1 year) MMF treatment [100–102]. However, one study that compared MMF to CYC showed that although both agents stabilized lung function to a comparable extent a deterioration of lung function was noticed at 2 years in the MMF group but not in the group receiving CYC. The authors, therefore, concluded that the study did not provide sufficient evidence to support replacing CYC for MMF for the treatment of SSc- associated ILD [103]. The results of the Scleroderma Lung Study II (SLS II) comparing prospectively MMF to CYC for the treatment of SSc-associated ILD are expected to be published in the near future.
4.3. Methotrexate (MTX)
Methotrexate has been recently recommended as a first line therapy drug for early SSc skin involvement by the EULAR expert consensus [104], ostensibly based on the interpretation of two small RCT trials published recently [105,106]. The first study described a small trial of 29 patients and evaluated the use of MTX as a putative disease modifying drug. The results showed a statistically significant improvement over placebo (p=0.03) at 24 weeks on a composite response index which included mRSS, DLCO, or VAS for general well-being [105]. However, the impact on mRSS alone was not significant (p=0.06). The second study was a RCT of 71 patients with diffuse SSc of recent onset (less than 3 years in duration) designed to evaluate MTX effects on skin involvement and physician global assessment. The results failed to show statistically significant difference between MTX and placebo arms [106]. However, subsequent analysis of the data applying a Bayesian approach and multiple imputations methods for missing data to reduce loss of subjects-associated bias suggested that subjects on MTX were more likely to attain a favorable mRSS response than subjects receiving placebo. Although the results of these two studies are encouraging, it can be argued that the statistical evidence presented may not be sufficiently strong to make a conclusive recommendation favoring MTX over other treatments for the cutaneous involvement in rapidly progressive SSc.
4.4. Rituximab
Rituximab is a monoclonal antibody directed against the CD20 B-cell antigen that has recently been utilized as a potential therapy for patients with SSc-ILD and cutaneous involvement. Several small and non-RTC controlled studies have been described [107–113]. One study that included 8 patients treated with rituximab found statistically significant improvement in FVC and DLCO and stabilization of HRCT alterations as well as improvement in the mRSS compared with a matched control group of 6 patients treated with different background medications [110]. The EUSTAR group recently evaluated rituximab treatment in a nested case-control designed study including 63 patients treated with rituximab [112]. The results showed a statistically significant improvement in skin fibrosis and an arrest in the progression of lung fibrotic changes in the rituximab-treated patients, whereas the matched control patients with SSc-ILD showed a decline in FVC resulting in significant differences between rituximab-treated and matched controls. There was also a statistically significant improvement in skin thickness assessed employing the mRSS in the rituximab-treated patients an effect that was substantially greater in a subgroup of 25 patients with severe diffuse SSc (mRSS greater than 16) in whom a decrease of mRSS from an average of 26.6 ± 1.4 to an average of 20.3 ± 1.8 was observed after 6–9 months of treatment. Of substantial importance were the observations that the side effect profile of Rituximab was acceptable without any serious adverse events [112]. These results clearly support the need for a prospective, double-blind RCT to assess the efficacy and safety of rituximab in patients with SSc-ILD.
4.5. Intravenous immunoglobulins (IVIG)
Following the paradigm that blockage of functional antibodies can halt SSc progression, IVIG has also been proposed for use in SSc refractory cases. Although some uncontrolled studies suggested that IVIG stabilized or improved mRSS, the data from the largest retrospective study was confounded by the concomitant use of MMF [114]. Furthermore, a sub-group analysis in a prior study conducted at the same institution comparing the effect of IVIG + MMF versus MMF alone did not show statistically significant differences between those groups [97]. Therefore, it will be important to perform controlled studies to determine whether IVIG is of therapeutic value for SSc. Indeed, a placebo randomized control trial is in progress to examine this possibility. We should emphasize, however, the good tolerability of IVIG in SSc patients and its potential role for the treatment of other SSc manifestations such as gastrointestinal involvement and myositis.
4.6. D-Penicillamine (D-Pen)
D-Penicillamine, once the cornerstone of treatment for diffuse SSc is not commonly used today. D-Pen exerts a strong anti-fibrotic effect by interfering with collagen cross-linking and by accelerating collagen turn-over even at low doses. Multiple open label trials have consistently shown improvement of skin score of SSc patients treated with D-Pen [115,116]. Furthermore, the observation of recurrence of diffuse SSc skin changes following discontinuation and subsequent improvement of the skin after resuming D-Pen, support its effectiveness [117]. In contrast with the results of these studies, a controlled study evaluating low dose vs. high dose D-Pen did not show statistical differences between the groups. However, since all patients received D-Pen the study design failed to exclude the possibility that even the low dose of D-Pen may have exerted a beneficial therapeutic effect. It should be emphasized that given the recent demonstration of potent pro-fibrotic effects induced by tissue stiffness resulting from tissue fibrosis and from the accumulation of relatively rigid interstitial collagen molecules [118], we believe that the addition of an antifibrotic agent capable of reducing fibrotic tissue stiffness should be considered a valid and useful alternative for treatment of SSc-associated tissue fibrosis. However, additional clinical studies should be conducted to appropriately evaluate this suggested approach.
4.7. Autologous hematopoietic stem cell transplantation (HSCT)
Numerous studies have proposed HSCT as treatment for severe cases of SSc following anecdotal evidence from the treatment of other severe autoimmune diseases with this approach [119–125]. Autologous transplantation has been preferred over allogeneic transplantation given its greater procedure related survival rates, the fact that donor search and matching procedures are not needed, and the absence of graft versus host disease. Initial pilot studies have modified the standard protocol introducing kidney and lung radiation shielding leading to a significant reduction in procedure related mortality. Three prospective trials were initiated to examine the role of HSCT in SSc treatment, including the ASTIS [121,122], ASSIST [123] and the Scleroderma: Cyclophosphamide or Transplant [SCOT]. These studies compared autologous HSCT to various CYC regimens. All studies included patients with mRSS greater than 14 and internal organ involvement (lung involvement largely accounted for the vast majority of the cases). Although there were substantial differences in the study design between the ASTIS (a phase II study allowing cross over from the cyclophosphamide to the HSCT group) [121], ASSIST [122] and SCOT trials (phase III study design recruiting larger number of patients), the inclusion and exclusion criteria were similar and the results of the three studies allow to draw valid conclusions regarding the effect of HSCT in patients with SSc progressive skin and target organ involvement. The following are the notable observations derived from these studies [123–125]: 1) Rapid improvement of skin involvement (reduction of mRSS) occurred uniformly following transplantation; 2) Improvement of pulmonary function tests was demonstrated in patients in the transplant arm, and 3) Although treatment related mortality was significantly higher in the HSCT group, the longer term and event-free survival favored the transplant group after 1 year.
Despite the highly encouraging results, it is not clear whether the improvement observed in the transplant groups is related to an antifibrotic/immunosuppressive effect of the high dose of cyclophosphamide used, or to the combination of other drugs used that included anti-thymocyte globulin and corticosteroids, or was the result of intensive lymphocyte ablation. However, despite of positive results, the cornerstone for applying this treatment is to identify SSc patients with very high risk of disease progression amenable to benefit from this approach and to carefully balance the potential benefits with the risk of serious side effects.
5. NOVEL SSc THERAPIES CURRENTLY UNDER EVALUATION
In 2014, the Food and Drug Administration approved 2 new drugs, pirfenidone and nintedanib, for the treatment of patients with IPF. Both drugs, which act by downregulating the expression or intracellular signaling by TGF-β, are now undergoing preliminary safety and efficacy studies in patients with SSc-ILD. Other potential SSc disease modifiers include modulators of profibrotic cytokines such as IL-6 inhibitors, anti- IL-13 antibodies, inhibitors of intracellular profibrotic pathways (monoclonal anti-CTGF antibodies, tocilizumab, endostatin-1-derived peptide, and caveolin scaffolding domain peptide), NOX4 inhibitors, and inhibitors of other pathways [36–39,126–133].
Also, multiple existing drugs that might be repurposed to treat fibrosis [134] such as the peroxisome proliferator-activated receptor-γ agonists, rosiglitazone [135], statins [136,137], fluoroquinolone antibiotics such as ciprofloxacin [138,139], and thrombin inhibitors such as dabigatran [140], are being considered. However, given that the pathogenic mechanisms of fibrotic diseases in SSc are quite complex with multiple and often redundant pathways, blocking a single molecule or pathway will likely not be sufficient. Therefore, it may be necessary to treat patients with multiple drugs that affect different pathways. We will briefly discuss some of the most promising drugs that are undergoing clinical trials or are under evaluation to initiate clinical trials for SSc in the near future.
5.1. Tocilizumab (TCZ)
Activation of fibroblasts by IL-6 in vitro and increased IL-6 production in the skin of SSc patients have been demonstrated and it has been suggested that IL-6 may play an important role in the pathogenesis of SSc-related fibrosis [141,142]. In this context, blockade of IL-6 receptor improved disease manifestations in a mouse model of SSc. Following these translational studies, two cases of SSc treated with TCZ were described [143]. In addition, TCZ showed efficacy to treat SSc related arthritis in a cohort of 15 patients [144]. A recently completed multicenter randomized controlled trial in patients with active/progressive SSc showed a trend to improvement in terms of mRSS, although the overall results did not achieve statistical significance at the end of a 48 week study period [145]. A phase III RCT is currently being conducted.
5.2. Tyrosine Kinase Inhibitors (TKIs)
Tyrosine kinases are small molecules mediating a vast number of intracellular processes. Since the discovery of imatinib, the first TKI, this large family of drugs have been extensively utilized for treating numerous malignancies with tyrosine kinase–dependent biology and a large number of TKIs have been developed targeting a variety of kinases with higher selectivity and specificity. All the TKIs act by occupying and blocking the kinase ATP-binding site, thus, halting the downstream intracellular cascades and associated molecular processes.
Numerous receptor activated and non-receptor tyrosine kinases including PDGF, c-Abl, and SRC play an important role in the pathogenesis of SSc and other fibrotic conditions mediating TGFβ and non-TGFβ pro-fibrotic effects [3,4]. According to the evidence from numerous animal models of tissue fibrosis demonstrating halting of the fibrotic process [146], and the results of encouraging case reports of the use of imatinib in SSc and other fibrotic diseases, an open-label trial was performed to evaluate the effects of imatinib on SSc. The results showed good tolerability and statistically significant improvement in the mRSS [147,148]. However, two subsequent small randomized control trials showed significant toxicity including fluid retention and volume overload, factors that would be expected to interfere in the quantification of mRSS as well as mimic disease-associated rapid lung function deterioration not allowing a clear evaluation of the disease progression indicators, thus, prompting discontinuation of the study drug [149]. A subsequent extension of the open label study used lower doses of imatininb (200 mg/d) improving its tolerability while showing continued improvement in mRSS. Therefore, it is quite likely that clinical studies employing low doses of imatinib and exclusion of patients with advanced lung disease who may be prone to develop pulmonary edema may achieve better outcomes.
Second generation TKIs such as nilotinib and dasatinib have also been evaluated in patients with SSc. Nilotinib showed efficacy in improving the mRSS over 12 months [150]. It is important to emphasize that TKI-specific toxicities should be considered in the design of future clinical trials with these drugs. For example nilotinib use may carry higher risk of heart conduction abnormalities and dasatanib may increase the risk of developing PAH in patients with SSc. Another TKI that may be effective as SSc disease modifying agent, is nintedanib, a multiple kinase inhibitor targeting vascular endothelial growth factor receptor, fibroblast growth factor receptor and PDGF. Nintedanib has been recently approved by the FDA for treatment of idiopathic pulmonary fibrosis and is currently undergoing a phase II placebo RCT evaluation for SSc-associated ILD [151,152].
5.3. Lipid lysophosphatidic acid 1 inhibitors (LPA-1)
LPA1 is a G protein-coupled receptor that upon binding to its respective receptor signaling molecule (LPAR), activates a variety of intracellular cascades [153,154]. Mice models deficient for the LPA-1 receptor, showed protection from development of fibrosis in the bleomycin-induced skin fibrosis model [155,156]. Similar protection from fibrosis was observed in normal mice treated with the LPA1 receptor antagonist AM095 [155]. The mechanism mediating the anti-fibrotic effects of LPA-1 inhibitors is not clear. However, based on pre-clinical evidence, a phase 2A trial was conducted and the results showed encouraging improvement in the mRSS with a reduction of 4 points in only 8 weeks of treatment and a more pronounced decline of 7.3 points at 24 weeks [Published in Abstract only].
5.4. Other novel therapeutic approaches
Stimulators of the soluble guanylate cyclase (sGC), currently approved for the treatment of pulmonary arterial hypertension have been recently shown to inhibit TGF-β signaling in vitro and in vivo in animal models of fibrosis [157], and early clinical trials are under way to examine their effects in SSc treatment. Similarly, other agents used for treating pulmonary arterial hypertension including phosphodiesterase inhibitors such as taladafil, endothelin receptor antagonists and prostanoid analogs may also have anti-fibrotic properties. However, it is not clear whether the potential antifibrotic effect of these agents would be clinically significant in SSc. Indeed, recent data in IPF showed no significant clinical improvement when using endothelin receptor antagonists.
6. CONCLUSIONS
Patients with rapid progression of skin and target organ involvement carry the worse prognosis among the wide clinical heterogeneity of patients within the SSc spectrum. Rapid and proactive identification of these patients and aggressive and prompt treatment are required to ameliorate this dismal prognosis. Organ involvement will influence the choice of the therapeutic agent. Early initiation of therapy with close monitoring is needed. Numerous drugs with different mechanisms of action targeting several components of the highly complex events of SSc pathogenesis are currently under development.
7. EXPERT OPINION
7.1. Approach to treatment
Patients with SSc and rapid disease progression should have a comprehensive evaluation of cutaneous, pulmonary, cardiovascular, and renal involvement including but not limited to pulmonary function tests, high resolution computerized tomography of the chest, transthoracic echocardiograms, complete serologic autoantibody assessment, and upper endoscopy and gastrointestinal transit studies. Clinical evaluation of skin involvement performed at baseline and at subsequent visits should allow to classify the clinical subset of the patient. To evaluate the rate of pulmonary involvement progression over time, pulmonary function tests are recommended to be performed every 2–3 months for the first 12 to 18 months following the initial evaluation. Although not universally accepted, it is our recommendation to perform high resolution computerized tomography every 6–8 months for the first 2 years following the first evaluation to detect progression of SSc-associated ILD at the earliest possible point.
Once rapid progression has been identified by documented evidence of lung or skin progression, prompt and aggressive treatment should be initiated. For rapidly progressive SSc-associated ILD, daily oral or monthly parenteral intravenous CYC is recommended. Although there are no clear data regarding the most appropriate time to initiate therapy for progressive lung disease a decline of FVC of 15% or more within 4–6 month period (performed at the same center and preferable employing the same equipment and performed by the same technologist), or the presence of greater than 20% ground glass appearance on high resolution CT scans at initial visit, a finding indicative of high likelihood of progressive fibrosis [158], have been utilized to indicate immediate treatment. While the duration of treatment in the Scleroderma Lung Study was 12 months with tolerable safety profile, we recommend 6 to12 months of treatment with intravenous monthly CYC followed by a maintenance dose of MMF minimize adverse events. In patients intolerant to CYC, MMF, or Rituximab should be considered as alternative choices based on open label studies showing SSc stabilization with these interventions.
Patients with rapidly progressive skin disease defined as described above, also should be treated promptly. Given the very promising open label trials with MMF for rapidly progressive cutaneous SSc involvement, MMF is recommended for treatment of skin involvement at an initial dose of 500mg twice daily increasing the dose every 4–6 weeks with a goal of reaching 2000 to 3000mg/d. It should be emphasized that in patients with rapid SSc progression, the skin score often increases during the early stages of treatment and does not reach a peak until few months following initiation of effective antifibrotic treatment leading sometimes to the premature discontinuation of an effective therapy. Usually a reduction of mRSS is detectable after 6 to 8 months of therapy although stabilization may be evident a few months earlier. For patients considered refractory to MMF (ie progression of skin disease after 6 months), the addition of a second agent such as MTX [104–106] or D-Penicillamine [81,159] or the use of IVIG therapy should be considered [114,160].
For patients with simultaneous progressive skin and pulmonary disease, CYC is preferred. MMF is an alternative in this case and results of the Scleroderma Lung study II comparing CYC vs MMF should be available soon. It should be emphasized, however, that given the potential lung toxicity of MTX and the fact that MTX lung toxicity cannot be differentiated from rapidly progressive SSc lung disease in a solely clinical basis, we recommend avoidance of MTX in this setting. On the other hand, Rituximab use can be considered for patients refractory to MMF or CYC as suggested per case series.
Selected patients with progressive disease failing the therapy mentioned above should be considered to receive autologous bone marrow transplant, although the high morbidity and mortality associated with the procedure itself (even if performed in very experienced centers) requires a very careful consideration of patient prognosis, perceived ability of the patient to tolerate the procedure, and a review of the potential risk and benefit profile for each patient. Once the decision to proceed with HSCT has been reached the patient should be referred to a specialized center with extensive experience in bone marrow transplantation for SSc patients. Although the goal of this review was not to cover the entire therapeutic spectrum for all SSc patients, it should be emphasized that an appropriate and optimal treatment of the gastrointestinal, vascular and musculo-skeletal SSc manifestations is a crucial and important component of the management of SSc. Furthermore, early recognition and management of emergent complications such as scleroderma renal crisis and SSc lung involvement are needed to improve SSc mortality and long term disability.
7.2. Perspective
The substantial and remarkable recent advancements in the understanding of the complex fibrosis pathways including the discovery of the profibrotic role of growth factors, cytokines, and other cellular mediators, the detailed identification of novel molecules involved in Smad and non-Smad intracellular pathways of TGF-β signaling, and the demonstration of an important role for other pro-fibrotic molecules such a PDGF and CTGF has allowed to identify novel targets for SSc-treatment. In this context some of the first and second generation TKIs have been used with variable and mixed clinical results, although it appears that their failure was mainly related to toxicity and side effects occurring when used at higher oncological doses. Promising novel drugs include pirfenidone and nintedanib, that were recently approved for the treatment of IPF, as well as numerous other drugs that are currently being evaluated in controlled clinical trials for SSc. One of these drugs, tocilizumab has shown very promising results in phase II/III studies and may become an FDA approved agent for use in SSc in the near future if the undergoing extension study confirms the trend of mRSS improvement and reaches statistical significance.
The poor outcome of SSc patients with rapidly progressive disease coupled with the unmet need for effective treatment and the recent designation of SSc as an orphan disease with vast potential for the development of effective antifibrotic drugs have promoted the development of novel drugs. Experience with prior agents has demonstrated that the potential toxicity profile is the main limitation to the development of new anti-fibrotic agents. Candidate drugs with significant potential gastrointestinal, fluid retention, lung and cardiac toxicity will certainly have limitations in their clinical development for SSc. Drugs with novel mechanism of action and tolerable toxicity profile will likely advance to phase II and III trials in the foreseeable future. It is also possible that the combination or sequential use of two or more drugs with different mechanisms of action may be required to improve the clinical outcomes and reduce the high mortality rates of patients with rapidly progressive SSc.
HIGHLIGHTS.
SSc pathophysiology is characterized by exaggerated extracellular matrix deposition in skin and various internal organs, severe fibro-proliferative vasculopathy and immune response abnormalities.
A clinical subset of SSc patients with rapidly progressive disease have very poor prognosis with frequent disability and a high mortality.
Treatment with Mycophenolate mofetil or Methotrexate is effective for treatment of skin involvement in patients with rapidly progressive SSc.
Use of Cyclophosphamide has been shown to be effective for treatment of rapidly progressive interstitial lung disease.
Novel anti-fibrotic and immunomodulatory drugs including tocilizumab, pirfenidone, tyrosine kinase inhibitors, lipid lysophosphatidic acid 1 inhibitors, specific NOX4 inhibitors, and several others are currently under evaluation for the treatment of rapidly progressive SSc.
Footnotes
Financial and competing interests disclosure
Supported by NIH/NIAMS grant AR 19616 to S Jimenez. F Mendoza is recipient of a NIH/NIAMS Research Supplement to Promote Diversity in Health Related Research (Parent Grant: 1-R21 AR 065638 to SAJ). The expert assistance of Ruth M. Johnson in preparation of the manuscript is gratefully acknowledged. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1*.Varga J, Abraham D. Systemic sclerosis: a protypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–67. doi: 10.1172/JCI31139. Classic review of the mechanisms of SSc-associated fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 3.Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annu Rev Pathol. 2011;6:509–37. doi: 10.1146/annurev-pathol-011110-130312. [DOI] [PubMed] [Google Scholar]
- 4*.Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of Systemic Sclerosis. Front Immunol. 2015;6:272. doi: 10.3389/fimmu.2015.00272. A comprehensive and updated review of SSc pathophysiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Stern EP, Denton CP. The pathogenesis of systemic sclerosis. Rheum Dis Clin North Am. 2015;41:367–82. doi: 10.1016/j.rdc.2015.04.002. A comprehensive and updated review of SSc pathophysiology. [DOI] [PubMed] [Google Scholar]
- 6.Mendoza FA, Keyes-Elstein LL, Jimenez SA. Systemic sclerosis disease modification clinical trials design: quo vadis? Arthritis Care Res. 2012;64:945–54. doi: 10.1002/acr.21667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pope JE, Khanna D, Johnson SR, et al. Disease modification and other trials in systemic sclerosis have come a long way, but have to go further. Arthritis Care Res. 2012;64:955–9. doi: 10.1002/acr.21673. [DOI] [PubMed] [Google Scholar]
- 8.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2013;72:1747–55. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 9.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737–47. doi: 10.1002/art.38098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 11.Steen VD, Powell DL, Medsger TA. Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 1988;31:196–203. doi: 10.1002/art.1780310207. [DOI] [PubMed] [Google Scholar]
- 12.Assassi S, del Junco D, Sutter K, McNearney TA, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum. 2009;61:1403–11. doi: 10.1002/art.24734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domsic RT, Rodriguez-Reyna T, Lucas M, et al. Skin thickness progression rate: a predictor of mortality and early internal organ involvement in diffuse scleroderma. Ann Rheum Dis. 2011;70:104–9. doi: 10.1136/ard.2009.127621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryan C, Knight C, Black CM, et al. Prediction of five-year survival following presentation with scleroderma: development of a simple model using three disease factors at first visit. Arthritis Rheum. 1999;42:2660–5. doi: 10.1002/1529-0131(199912)42:12<2660::AID-ANR23>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Muangchan C, Harding S, Khimdas S, et al. Association of C-reactive protein with high disease activity in systemic sclerosis: results from the Canadian Scleroderma Research Group. Arthritis Care Res (Hoboken) 2012;64:1405–14. doi: 10.1002/acr.21716. [DOI] [PubMed] [Google Scholar]
- 16.Shulman LE, Kurban AK, Harvey Am. Tendon friction rubs in progressive system sclerosis (scleroderma) Trans Assoc Am Physicians. 1961;74:378–88. [PubMed] [Google Scholar]
- 17.Steen VD, Medsger TA. The palpable tendon friction rub: an important physical examination finding in patients with systemic sclerosis. Arthritis Rheum. 1997;40:1146–51. doi: 10.1002/1529-0131(199706)40:6<1146::AID-ART19>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Khanna PP, Furst DE, Clements PJ, et al. Tendon friction rubs in early diffuse systemic sclerosis: prevalence, characteristics and longitudinal changes in a randomized controlled trial. Rheumatology (Oxford) 2010;49:955–9. doi: 10.1093/rheumatology/kep464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doré A, Lucas M, Ivanco D, et al. Significance of palpable tendon friction rubs in early diffuse cutaneous systemic sclerosis. Arthritis Care Res (Hoboken) 2013;65:1385–9. doi: 10.1002/acr.21964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Czirják L, Kumánovics G, Varjú C, et al. Survival and causes of death in 366 Hungarian patients with systemic sclerosis. Ann Rheum Dis. 2008;67:59–63. doi: 10.1136/ard.2006.066340. [DOI] [PubMed] [Google Scholar]
- 21.Ostojić P, Damjanov N. Different clinical features in patients with limited and diffuse cutaneous systemic sclerosis. Clin Rheumatol. 2006;25:453–7. doi: 10.1007/s10067-005-0041-0. [DOI] [PubMed] [Google Scholar]
- 22.Mendoza F, Derk CT. Systemic sclerosis mortality in the United States: 1999–2002 implications for patient care. J Clin Rheumatol. 2007;13:187–92. doi: 10.1097/RHU.0b013e318124a89e. [DOI] [PubMed] [Google Scholar]
- 23.Joven BE, Almodovar R, Carmona L, et al. Survival, causes of death, and risk factors associated with mortality in Spanish systemic sclerosis patients: results from a single university hospital. Semin Arthritis Rheum. 2010;39:285–93. doi: 10.1016/j.semarthrit.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Jacobsen S, Halberg P, Ullman S. Mortality and causes of death of 344 Danish patients with systemic sclerosis (scleroderma) Br J Rheumatol. 1998;37:750–5. doi: 10.1093/rheumatology/37.7.750. [DOI] [PubMed] [Google Scholar]
- 25.Mayes MD, Lacey JV, Jr, Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246–55. doi: 10.1002/art.11073. [DOI] [PubMed] [Google Scholar]
- 26.Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol. 2012;24:165–70. doi: 10.1097/BOR.0b013e32834ff2e8. [DOI] [PubMed] [Google Scholar]
- 27.Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29:315–33. doi: 10.1016/s0889-857x(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 28.Steen VD, Costantino JP, Shapiro AP, et al. Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990;113:352–7. doi: 10.7326/0003-4819-113-5-352. [DOI] [PubMed] [Google Scholar]
- 29.Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44:687–94. doi: 10.1016/j.semarthrit.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Guillevin L, Mouthon L. Scleroderma renal crisis. Rheum Dis Clin North Am. 2015;41:475–88. doi: 10.1016/j.rdc.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66:940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngian G, Sahhar J, Proudman SM, et al. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheumatic Dis. 2012;71:1980–3. doi: 10.1136/annrheumdis-2011-201176. [DOI] [PubMed] [Google Scholar]
- 33.Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis. 2013;72:1188–93. doi: 10.1136/annrheumdis-2012-202007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parks JL, Taylor MH, Parks LP, et al. Systemic sclerosis and the heart. Rheum Dis Clin North Am. 2014;40:87–102. doi: 10.1016/j.rdc.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Ungprasert P, Charoenpong P, Ratanasrimetha P, et al. Risk of coronary artery disease in patients with systemic sclerosis: a systematic review and meta-analysis. Clin Rheumatol. 2014;33:1099–104. doi: 10.1007/s10067-014-2681-4. [DOI] [PubMed] [Google Scholar]
- 36*.Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. 2013;1832:1088–103. doi: 10.1016/j.bbadis.2012.12.007. Comprehensive review of the biochemical and molecular mechanisms of tissue fibrosis focusing on the intracellular pro-fibrotic pathways as potential therapeutic targets. [DOI] [PubMed] [Google Scholar]
- 37.McMahan ZH, Wigley FM. Novel investigational agents for the treatment of scleroderma. Expert Opin Investig Drugs. 2014;23:183–98. doi: 10.1517/13543784.2014.848852. [DOI] [PubMed] [Google Scholar]
- 38.Volkmann ER, Furst DE. Management of systemic sclerosis-related skin disease: A review of existing and experimental therapeutic approaches. Rheum Dis Clin North Am. 2015;41:399–417. doi: 10.1016/j.rdc.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Dimitroulas T, Daoussis D, Garyfallos A, et al. Molecular and cellular pathways as treatment targets for biologic therapies in systemic sclerosis. Curr Med Chem. 2015;22:1943–55. doi: 10.2174/0929867322666150209161224. [DOI] [PubMed] [Google Scholar]
- 40.Broen JC, Radstake TR, Rossato M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat Rev Rheumatol. 2014;10:671–81. doi: 10.1038/nrrheum.2014.128. [DOI] [PubMed] [Google Scholar]
- 41.Salazar G, Mayes MD. Genetics, epigenetics, and genomics of systemic sclerosis. Rheum Dis Clin Am. 2015;41:345–66. doi: 10.1016/j.rdc.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bossini-Castillo L, López-Isac E, Mayes MD, et al. Genetics of systemic sclerosis. Semin Immunopathol. 2015 Jun; doi: 10.1007/s00281-015-0499-z. [DOI] [PubMed] [Google Scholar]
- 43.Feghali-Bostwick C, Medsger TA, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48:1956–63. doi: 10.1002/art.11173. [DOI] [PubMed] [Google Scholar]
- 44.Arnett FC, Cho M, Chatterjee S, et al. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001;44:1359–62. doi: 10.1002/1529-0131(200106)44:6<1359::AID-ART228>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 45.Frech T, Khanna D, Markewitz B, et al. Heritability of vasculopathy, autoimmune disease, and fibrosis in systemic sclerosis: a population-based study. Arthritis Rheum. 2010;62:2109–16. doi: 10.1002/art.27469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo Y, Wang Y, Wang Q, et al. Systemic sclerosis: genetics and epigenetics. Autoimmunity. 2013;41:161–7. doi: 10.1016/j.jaut.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Martin JE, Bossini-Castillo L, Martin J. Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012;131:1023–7. doi: 10.1007/s00439-011-1137-z. [DOI] [PubMed] [Google Scholar]
- 48.Bossini-Castillo L, López-Isac E, Martin J. Immunogenetics of systemic sclerosis: Defining heritablilty, functional variants and shared-autoimmunity pathways. J Autoimm. 2015 Jul 23; doi: 10.1016/j.jaut.2015.07.005. Pii: S0896–8411(15)30008-1 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 49.Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: vascular hypothesis. Semin Arthritis Rheum. 1975;4:351–68. doi: 10.1016/0049-0172(75)90017-7. [DOI] [PubMed] [Google Scholar]
- 50*.Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013;65:1953–62. doi: 10.1002/art.37988. Outstanding and up to date review of the crucial role of vascular alterations in SSc pathogenesis. [DOI] [PubMed] [Google Scholar]
- 51.Asano Y, Sato S. Vasculopathy in scleroderma. Semin Immunopathol. 2015;37:489–500. doi: 10.1007/s00281-015-0505-5. [DOI] [PubMed] [Google Scholar]
- 52.Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol. 2014;26:615–20. doi: 10.1097/BOR.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 53.Duffield JS, Lupher M, Thannickal VJ, et al. Host responsed in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–76. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinz B, Phan SH, Thannickal VJ, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–55. doi: 10.1016/j.ajpath.2012.02.004. Review of the central role of myofibroblasts in normal and pathologic fibrosis and tissue remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kramann R, DiRocco DP, Humphreys BD. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol. 2013;231:273–89. doi: 10.1002/path.4253. [DOI] [PubMed] [Google Scholar]
- 57.Hamamdzic D, Kasman LM, LeRoy EC. The role of infectious agents in the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2012;14:694–8. doi: 10.1097/00002281-200211000-00011. [DOI] [PubMed] [Google Scholar]
- 58.Randone SB, Guiducci S, Cerinic MM. Systemic sclerosis and infections. Autoimmune Rev. 2008;8:36–40. doi: 10.1016/j.autrev.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 59.Radić M, Martinivić Kaliterna D, Radić J. Infectious disease as aetiological factor in the pathogenesis of systemic sclerosis. Neth J Med. 2010;68:348–53. [PubMed] [Google Scholar]
- 60.Moroncini G, Mori S, Tonnini C, et al. Role of viral infections in the etipathogenesis of systemic sclerosis. Clin Exp Rheumatol. 2013;31(2 Suppl 76):3–7. [PubMed] [Google Scholar]
- 61.Fattal I, Shental N, Molad Y, et al. Epstein-Barr virus antibodies mark systemic lupus erythematosus and scleroderma patients negative for anti-DNA. Immunology. 2014;141:276–85. doi: 10.1111/imm.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boin F, Fanis U, de Bartlett SJ, et al. T cell polarization identifies distinct clinical phenotypes in scleroderma lung disease. Arthritis Rheum. 2008;58:1165–74. doi: 10.1002/art.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu YS, Kong J, Cheema GS, et al. The immunobiology of systemc sclerosis. Semin Arthritis Rheum. 2008;38:132–60. doi: 10.1016/j.semarthrit.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 64.O’Reilly S, Hügle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxford, England) 2012;51:1540–9. doi: 10.1093/rheumatology/kes090. [DOI] [PubMed] [Google Scholar]
- 65.Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol. 2015;37:511–7. doi: 10.1007/s00281-015-0503-7. [DOI] [PubMed] [Google Scholar]
- 66.Chizzolini C, Boin F. The role of the acquired immune response in systemic sclerosis. Semin Immunopathol. 2015;37:543–57. doi: 10.1007/s00281-015-0509-1. [DOI] [PubMed] [Google Scholar]
- 67.Raja J, Denton CP. Cytokines in the immunopathology of systemic sclerosis. Semin Immunopathol. 2015;37:543–57. doi: 10.1007/s00281-015-0511-7. [DOI] [PubMed] [Google Scholar]
- 68.Ho KT, Reveille JD. The clinical relevance of autoantibodies in scleroderma. Arthritis Res Ther. 2003;5:80–93. doi: 10.1186/ar628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69*.Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35–42. doi: 10.1016/j.semarthrit.2005.03.005. Classic review focused on the numerous serum autoantibodies present in SSc patients and their clinical associations. [DOI] [PubMed] [Google Scholar]
- 70.Mehra S, Walker J, Patterson K, et al. Autoantibodies in systemic sclerosis. Autoimmun Rev. 2013;12:340–54. doi: 10.1016/j.autrev.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 71.Jimenez SA, Castro SV, Piera-Velazquez S. Role of growth factors in the pathogenesis of tissue fibrosis in systemic sclerosis. Curr Rheumatol Rev. 2010;6:283–94. doi: 10.2174/157339710793205611. [DOI] [PubMed] [Google Scholar]
- 72.Krieg T, Abraham D, Lafyatis R. Fibrosis in connective tissue disease: the role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res Ther. 2007;9(Suppl 2):S4. doi: 10.1186/ar2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei J, Bhattacharyya S, Tourtellotte WG, et al. Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmun Rev. 2011;10:267–75. doi: 10.1016/j.autrev.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. 2013;15:215. doi: 10.1186/ar4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015;87:297–307. doi: 10.1038/ki.2014.287. [DOI] [PubMed] [Google Scholar]
- 76*.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–80. doi: 10.1016/j.ajpath.2011.06.001. Review of research studies supporting a role for the phenotypic change of endothelial cells into myofibroblasts in the pathogenesis of tissue fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Piera-Velazquez S, Jimenez SA. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair. 2012;5:S7. doi: 10.1186/1755-1536-5-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jimenez SA. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol. 2013:835948. doi: 10.1155/2013/835948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giordano M. The CREST acronym and some related problems. Arthritis Rheum. 1985;28:359. doi: 10.1002/art.1780280322. [DOI] [PubMed] [Google Scholar]
- 80.Rodnan GP, Fennell RH. Progressive systemic sclerosis sine scleroderma. JAMA. 1962;180:665–70. doi: 10.1001/jama.1962.03050210027006. [DOI] [PubMed] [Google Scholar]
- 81.Lally EV, Jimenez SA, Kaplan SR. Progressive systemic sclerosis: mode of presentation, rapidly progressive disease course, and mortality based on an analysis of 91 patients. Semin Arthritis Rheum. 1988;18:1–13. doi: 10.1016/0049-0172(88)90030-3. [DOI] [PubMed] [Google Scholar]
- 82.Clemson BS, Miller WR, Luck JC, et al. Acute myocarditis in fulminant systemic sclerosis. Chest. 1992;101:872–4. doi: 10.1378/chest.101.3.872. [DOI] [PubMed] [Google Scholar]
- 83.Marasovic-Krstulovic D, Jurisic Z, Perkovic D, et al. Fulminant diffuse systemic sclerosis following aortic valve replacement. Med Hypotheses. 2014;82:792–4. doi: 10.1016/j.mehy.2014.03.027. [DOI] [PubMed] [Google Scholar]
- 84.Scussel-Lonzetti L, Joyal F, Raynauld J, et al. Predicting mortality in systemic sclerosis: analysis of a cohort of 309 French Canadian patients with emphasis on features at diagnosis as predictive factors for survival. Medicine. 2002;81:154–67. doi: 10.1097/00005792-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 85.Clements PJ, Lachenbruch PA, Ng SC, et al. Skin score. A semiquantitative measure of cutaneous involvement that improves prediction of prognosis in systemic sclerosis. Arthritis Rheum. 1990;33:1256–63. doi: 10.1002/art.1780330828. [DOI] [PubMed] [Google Scholar]
- 86.Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol. 1995;22:1281–5. [PubMed] [Google Scholar]
- 87.Derk Ct, Huaman G, Littlejohn J, et al. Predictors of early mortality in systemic sclerosis: a case-control study comparing early versus late mortality in systemic sclerosis. Rheumatol Int. 2012;32:3841–4. doi: 10.1007/s00296-011-2301-4. [DOI] [PubMed] [Google Scholar]
- 88.Maurer B, Graf N, Michel BA, et al. Prediction of worsening of skin fibrosis in patients with diffuse cutaneous systemic sclerosis using the EUSTAR database. Ann Rheum Dis. 2015;74:1124–31. doi: 10.1136/annrheumdis-2014-205226. [DOI] [PubMed] [Google Scholar]
- 89.Derk CT, Grace E, Shenin M, et al. A prospective open-label study of mycophenolate mofetil for the treatment of diffuse systemic sclerosis. Rheumatology (Oxford) 2009;48:1595–9. doi: 10.1093/rheumatology/kep295. [DOI] [PubMed] [Google Scholar]
- 90*.Mendoza FA, Nagle SJ, Lee JB, et al. A prospective observational study of mycophenolate mofetil treatment in progressive diffuse cutaneous systemic sclerosis of recent onset. J Rheumatol. 2012;39:1241–7. doi: 10.3899/jrheum.111229. One of the earliest prospective studies supporting the beneficial role of Mycophenolate mofetil in the treatment of cutaneous involvement in rapidly progressive SSc. [DOI] [PubMed] [Google Scholar]
- 91.Silver RM, Warrich JH, Kinsella MB. Cyclophosphamide and low dose prednisone therapy in patients with systemic sclerosis (Scleroderma) with interstitial lung disease. J Rheum. 1993;20:838–44. [PubMed] [Google Scholar]
- 92.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–66. doi: 10.1056/NEJMoa055120. Results of this study supports the role of CYC in SSc-ILD. [DOI] [PubMed] [Google Scholar]
- 93.Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–70. doi: 10.1002/art.22204. [DOI] [PubMed] [Google Scholar]
- 94.Giacomelli R, Valentini G, Salsano F, et al. Cyclophosphamide pulse regimen in the treatment of alveolitis in systemic sclerosis. J Rheumatol. 2002;29:731–6. [PubMed] [Google Scholar]
- 95.Domiciano DS, Bonfá E, Borges CTL, et al. A long-term prospective randomized controlled study of non-specific interstitial pneumonia (NSIP) treatment in scleroderma. Clin Rheumatol. 2011;30:223–9. doi: 10.1007/s10067-010-1493-4. [DOI] [PubMed] [Google Scholar]
- 96.Nihtyanova SI, Brough GM, Black CM, et al. Mycophenolate mofetil in diffuse cutaneous systemic sclerosis-a retrospective analysis. Rheumatology (Oxford) 2007;46:442–5. doi: 10.1093/rheumatology/kel244. [DOI] [PubMed] [Google Scholar]
- 97*.Le EN, Wigley FM, Shah AA, et al. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2011;70:1104–7. doi: 10.1136/ard.2010.142000. Large retrospective study describing improvement in skin involvement in patients with diffuse SSc following treatment with Mycophenolate mofetil. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khanna D, Clements PJ, Furst DE, et al. Recombinant human relaxin in the treatment of systemic sclerosis with diffuse cutaneous involvement: a randomized, double–blind, placebo-controlled trial. Arthritis Rheum. 2009;60:1102–11. doi: 10.1002/art.24380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Postlethwaite AE, Wong WK, Clements P, et al. A multicenter, randomized, double-blind, placebo-controlled trial of oral type I collagen treatment in patients with diffuse cutaneous systemic sclerosis: I. oral type I collagen does not improve skin in all patients, but may improve skin in late-phase disease. Arthritis Rheum. 2008;58:1810–22. doi: 10.1002/art.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest. 2008;133:455–60. doi: 10.1378/chest.06-2861. [DOI] [PubMed] [Google Scholar]
- 101.Simeón-Aznar CP, Fonollosa-Plá V, Tolosa-Vilella C, et al. Effect of mycophenolate sodium in scleroderma-related interstitial lung disease. Clin Rheumatol. 2011;30:1393–8. doi: 10.1007/s10067-011-1823-1. [DOI] [PubMed] [Google Scholar]
- 102.Omair MA, Alahmadi A, Johnson SR. Safety and effectiveness of mycophenolate in systemic sclerosis: A Systematic review. PLoS One. 2015;10:e012425. doi: 10.1371/journal.pone.0124205. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Panopoulos ST, Bournia VK, Trakada G, et al. Mycophenolate versus cyclophosphamide for progressive interstitial lung disease associated with systemic sclerosis: a 2-year case control study. Lung. 2013;191:483–9. doi: 10.1007/s00408-013-9499-8. [DOI] [PubMed] [Google Scholar]
- 104.Pope J, Harding S, Khimdas S, et al. Agreement with guidelines from a large database for management of systemic sclerosis: results from the Canadian Scleroderma Research Group. J Rheumatol. 2012;39:524–31. doi: 10.3899/jrheum.110121. [DOI] [PubMed] [Google Scholar]
- 105*.van den Hoogen FH, Boerbooms AM, et al. Comparison of methotrexate with placebo in the treatment of systemic sclerosis: a 24 week randomized double-blind trial, followed by a 24 week observational trial. Br J Rheumatol. 1996;35:364–72. doi: 10.1093/rheumatology/35.4.364. Description of one of the earliest placebo-controlled randomized double blind trial showing a beneficial effect of low dose Methotrexate for the treatment of SSc skin involvement. [DOI] [PubMed] [Google Scholar]
- 106*.Pope JE, Bellamy N, Seibold JR, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum. 2001;44:1351–8. doi: 10.1002/1529-0131(200106)44:6<1351::AID-ART227>3.0.CO;2-I. Randomized control study describing the potential therapeutic benefit of Methotrexate in diffuse SSc. [DOI] [PubMed] [Google Scholar]
- 107.Lafyatis R, Kissin E, York M, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009;60:578–83. doi: 10.1002/art.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith V, Van Praet JT, Vandooren B, et al. Rituximab in diffuse cutaneous systemic sclerosis: an open-label clinical and histopathological study. Ann Rheum Dis. 2010;69:193–7. doi: 10.1136/ard.2008.095463. [DOI] [PubMed] [Google Scholar]
- 109.Bosello S, De Santis M, Lama G, et al. B cell depletion in diffuse progressive systemic sclerosis: safety, skin score modification and IL-6 modulation in an up to thirty-six months follow-up open-label trial. Arthritis Res Ther. 2010;12:R54. doi: 10.1186/ar2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daoussis D, Liossis SN, Tsamandas AC, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (Oxford) 2010;49:271–80. doi: 10.1093/rheumatology/kep093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McQueen FM, Solanki K. Rituximab in diffuse cutanueous systemic sclerosis: should we be using it today? Rheumatology (Oxford) 2015;54:757–67. doi: 10.1093/rheumatology/keu463. [DOI] [PubMed] [Google Scholar]
- 112*.Jordan S, Distler JHW, Maurer B, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74:1188–94. doi: 10.1136/annrheumdis-2013-204522. Nested case control analysis of the EUSTAR experience with a large cohort of severe diffuse SSc describing improvement of skin fibrosis and stabilization of lung involvement. [DOI] [PubMed] [Google Scholar]
- 113.Giuggioli D, Lumetti F, Colaci M, et al. Rituximab in the treatment of patients with systemic sclerosis. Our experience and review of the literature. Autoimmun Rev. 2015 Jul 22; doi: 10.1016/j.autrev.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 114.Poelman CL, Hummers LK, Wigley FM, et al. Intravenous immunoglobulin may be an effective therapy for refractory, active diffuse cutaneous systemic sclerosis. J Rheumatol. 2015;42:236–42. doi: 10.3899/jrheum.140833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Steen VD, Medsger TA, Jr, Rodnan GP. D-Penicillamine therapy in progressive systemic sclerosis (scleroderma): a retrospective analysis. Ann Intern Med. 1982;97:652–9. doi: 10.7326/0003-4819-97-5-652. [DOI] [PubMed] [Google Scholar]
- 116.Jimenez SA, Sigal SH. A 15-year prospective study of treatment of rapidly progressive systemic sclerosis with D-penicillamine. J Rheumatol. 1991;18:1496–503. [PubMed] [Google Scholar]
- 117.Medsger TA, Jr, Lucas M, Wildy KS, et al. D-penicillamine in systemic sclerosis? Yes! Scand J Rheumatol. 2001;30:192–4. doi: 10.1080/030097401316909503. [DOI] [PubMed] [Google Scholar]
- 118.Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–35. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nash RA, McSweeney PA, Crofford LJ, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for severe systemic sclerosis: long-term follow-up of the US multicenter pilot study. Blood. 2007;110:1388–96. doi: 10.1182/blood-2007-02-072389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Farge D, Passweg J, van Laar JM, et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004;63:974–81. doi: 10.1136/ard.2003.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121*.van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311:2490–8. doi: 10.1001/jama.2014.6368. Randomized study comparing the therapeutic effects of Cyclophosphamide with autologous hematopoietic stem cell transplantation for severe SSc. [DOI] [PubMed] [Google Scholar]
- 122.van Laar JM, Farge D, Tyndall A. Autologous Stem cell Transplantation International Scleroderma (ASTIS) trial: hope on the horizon for patients with severe systemic sclerosis. Ann Rheum Dis. 2005;64:1515. doi: 10.1136/ard.2005.043240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123*.Burt RK, Shah SJ, Dill K, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomized phase 2 trial. Lancet. 2011;378:498–506. doi: 10.1016/S0140-6736(11)60982-3. This study describes one center experience with autologous stem cell transplantation on selected SSc patients. [DOI] [PubMed] [Google Scholar]
- 124*.Tyndall A, Furst DE. Adult stem cell treatment of scleroderma. Curr Opin Rheumatol. 2007;19:604–10. doi: 10.1097/BOR.0b013e3282e6f534. Outstanding review of adult stem cell transplantation treatment of SSc. [DOI] [PubMed] [Google Scholar]
- 125.Van Laar JM, Naraghi K, Tyndall A. Haematopoietic stem cell transplantation for poor-prognosis systemic sclerosis. Rheumatol (Oxford) 2015 doi: 10.1093/rheumatology/kev117. pii: kev117 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 126.Asano T. Future treatments in systemic sclerosis. J Dermatol. 2010;37:54–70. doi: 10.1111/j.1346-8138.2009.00758.x. [DOI] [PubMed] [Google Scholar]
- 127.Rice LM, Padilla CM, McLaughlin SR, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015;125:2795–807. doi: 10.1172/JCI77958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ikawa Y, Ng PS, Endo K, et al. Neutralizing monoclonal antibody to human connective tissue growth factor ameliorates transforming growth factor-beta-induced mouse fibrosis. J Cell Physiol. 2008;216:680–7. doi: 10.1002/jcp.21449. [DOI] [PubMed] [Google Scholar]
- 129.Yamaguchi Y, Takihara T, Chambers RA, et al. A peptide derived from endostatin ameliorates organ fibrosis. Sci Transl Med. 2012;4:136ra71. doi: 10.1126/scitranslmed.3003421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2008;20:713–9. doi: 10.1097/bor.0b013e3283103d27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meyer C, Liu Y, Dooley S. Caveolin and TGF-β entanglements. J Cell Physiol. 2013;228:2097–102. doi: 10.1002/jcp.24380. [DOI] [PubMed] [Google Scholar]
- 132.Jiang F, Liu GS, Dusting GJ, et al. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014;1:267–72. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Piera-Velazquez S, Makul A, Jimenez SA. Increased expression of NADPH oxidase 4 (NOX4) in systemic sclerosis dermal fibroblasts: regulation by transforming growth factor β. Arthritis Rheumatol. 2015 Jun 19; doi: 10.1002/art.39242. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nagaraja V, Denton CP, Khanna D. Old medications and new targeted therapies in systemic sclerosis. Rheumatology (Oxford) 2014 Jul 26; doi: 10.1093/rheumatology/keu285. Pii: keu285. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wei J, Ghosh AK, Sargent JL, et al. PPARδ downregulation by TGF–β in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PloS One. 2010;5(11):e13778. doi: 10.01371/journal.pone.0013778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Derk CT, Jimenez SA. Statins and the vasculopathy of systemic sclerosis: potential therapeutic agents? Autoimmun Rev. 2006;5:25–32. doi: 10.1016/j.autrev.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 137.Timár O, Szekanecz Z, Kerekes G, et al. Rosuvastin improves impaired endothelial function, lowers high sensitivity CRP, complement and immuncomplex production in patients with systemic sclerosis-a prospective case-series study. Arthritis Res Ther. 2013;15:R105. doi: 10.1186/ar4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rubén EC, Manuel VR, Agustin OR, et al. Ciprofloxacin utility as antifibrotic in the skin of patients with scleroderma. J Dermatol. 2010;37:323–9. doi: 10.1111/j.1346-8138.2010.00826.x. [DOI] [PubMed] [Google Scholar]
- 139.Bujor AM, Haines P, Padilla C, et al. Ciprofloxacin has antifibrotic effects in scleroderma fibroblasts via downregulation of Dnmt1 and upregulation of Fli1. Int J Mol Med. 2012;30:1473–80. doi: 10.3892/ijmm.2012.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bogatkevich GS, Ludwika-Bradley A, Silver RM. Dabigatran, a direct thrombin inhibitor, demonstrates antifibrotic effects in lung fibroblasts. Arthritis Rheum. 2009;60:3455–64. doi: 10.1002/art.24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mihara M, Moriya Y, Ohsugi Y. IL-6-soluble IL-6 receptor complex inhibits the proliferation of dermal fibroblasts. Int J Immunopharmacol. 1996;18:89–94. doi: 10.1016/0192-0561(95)00106-9. [DOI] [PubMed] [Google Scholar]
- 142.Takemura H, Suzuki H, Fujisawa H, et al. Enhanced interleukin 6 production by cultured fibroblasts from patients with systemic sclerosis in response to platelet derived growth factor. J Rheumatol. 1998;25:1534–9. [PubMed] [Google Scholar]
- 143.Shima Y, Kuwahara Y, Murota H, et al. The skin of patients with systemic sclerosis softened during the treatment with anti-IL-6 receptor antibody tocilizumab. Rheumatology (Oxford, England) 2010;49:2408–12. doi: 10.1093/rheumatology/keq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elhai M, Meunier M, Matucci-Cerinic M, et al. Outcomes of patients with systemic sclerosis-associated polyarthritis and myopathy treated with tocilizumab or abatacept: a EUSTAR observational study. Ann Rheum Dis. 2013;72:1217–20. doi: 10.1136/annrheumdis-2012-202657. [DOI] [PubMed] [Google Scholar]
- 145.Khanna D, Denton CP, Juhreis A, et al. Safety and efficiency of subcutaneous tocilizumab in adults with systemic sclerosis: week 48 data from the fascinate trial. Ann Rheum. 2015 doi: 10.1136/annrheumdis-2015-eular.2281. [DOI] [Google Scholar]
- 146.Akhmetshina A, Venalis P, Dees C, et al. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum. 2009;60:219–24. doi: 10.1002/art.24186. [DOI] [PubMed] [Google Scholar]
- 147.Spiera RF, Gordon JK, Mersten JN, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. 2011;70:1003–9. doi: 10.1136/ard.2010.143974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gordon J, Udeh U, Doobay K, et al. Imatinib mesylate (Gleevec™) in the treatment of diffuse cutaneous systemic sclerosis: results of a 24-month open label, extension phase, single-centre trial. Clinical Exp Rheumatol. 2014;32:S-189–93. [PubMed] [Google Scholar]
- 149.Khanna D, Sagger R, Mayes MD, et al. A one year phase I/II, open label pilot trial of imanitib mesylate in the treatment of systemic sclerosis associated active interstitial lung disease. Arthritis Rheum. 2011;63:3540–6. doi: 10.1002/art.30548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gordon JK, Martyanov V, Magro C, et al. Nilotinib (Tasigna) in the treatment of early diffuse systemic sclerosis: an open-label pilot clinical trial. Arthritis Res Ther. 2015;17:213. doi: 10.1186/s13075-015-0721-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 152.Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 153.Swaney JS, Chapman C, Correa LD, et al. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J Pharmacol Exp Ther. 2011;336:693–700. doi: 10.1124/jpet.110.175901. [DOI] [PubMed] [Google Scholar]
- 154.Yung YC, Stoddard NC, Mirendil H, et al. Lysophosphatidic Acid signaling in the nervous system. Neuron. 2015;85:669–82. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Swaney JS, Chapman C, Correa LD, et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br J Pharmacol. 2010;160:1699–713. doi: 10.1111/j.1476-5381.2010.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pattanaik D, Postlethwaite AE. A role for lysophosphatidic acid and sphingosine 1-phosphate in the pathogenesis of systemic sclerosis. Discov Med. 2010;10:161–7. [PubMed] [Google Scholar]
- 157.Dees C, Beyer C, Distler A, et al. Stimulators of soluble guanylate cyclase (sGC) inhibit experimental skin fibrosis of different aetiologies. Ann Rheum Dis. 2015;74:1621–5. doi: 10.1136/annrheumdis-2014-206809. [DOI] [PubMed] [Google Scholar]
- 158.Hoffmann-Vold AM, Aaløkken TM, Lund MB, et al. Predictive value of serial high-resolution computed tomography analyses and concurrent lung function tests in systemic sclerosis. Arthritis Rheumatol. 2015;67:2205–12. doi: 10.1002/art.39166. [DOI] [PubMed] [Google Scholar]
- 159.Derk CT, Huaman G, Jimenez SA. A retrospective randomly selected cohort study of D-penicillamine treatment in rapidly progressive diffuse cutaneous systemic sclerosis of recent onset. Br J Dermatol. 2008;158:1063–8. doi: 10.1111/j.1365-2133.2008.08452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cantarini L, Rigante D, Vitale A, et al. Intravenous immunoglobulins (IVIG) in systemic sclerosis: a challenging yet promising future. Immunol Res. 2015;61:326–37. doi: 10.1007/s12026-014-8615-z. [DOI] [PubMed] [Google Scholar]