

SUMMARY
Our understanding of the mechanisms that regulate hematopoietic stem/progenitor cells (HSPCs) has been advanced by the ability to genetically manipulate mice; however, germline modification is time-consuming and expensive. Here we describe fast, efficient, and cost-effective methods to directly modify the genomes of mouse and human HSPCs using the CRISPR/Cas9 system. Using plasmid and virus-free delivery of guide RNAs alone into Cas9-expressing HSPCs, or Cas9-guide-RNA ribonucleoprotein (RNP) complexes into wild-type cells, we have achieved extremely efficient gene disruption in primary HSPCs from mouse (>60%) and human (~75%). These techniques enabled rapid evaluation of the functional effects of gene loss of Eed, Suz12, and DNMT3A. We also achieved homology-directed repair in primary human HSPCs (>20%). These methods will significantly expand applications for CRISPR/Cas9 technologies for studying normal and malignant hematopoiesis.
Keywords: HSC, genome editing, CRISPR/Cas9
Graphical Abstract
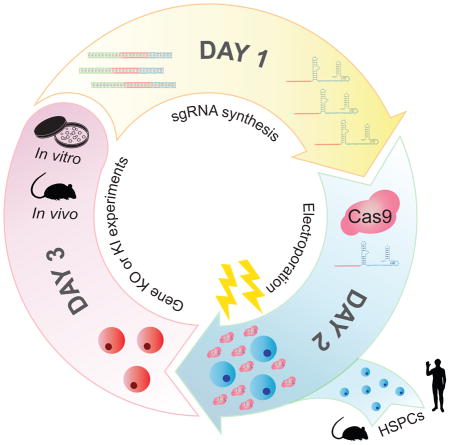
INTRODUCTION
The ability to genetically manipulate the genomes of animal models or isolated cells has driven hematopoietic stem cell (HSC) research, revealing key mechanisms that control HSC self-renewal and differentiation. Numerous genetically engineered mouse models have contributed to our understanding of the pathways that control HSC maintenance and regeneration in physiological settings (Rossi et al., 2012). Although this approach produces invaluable insights and still remains a gold standard in studying HSC biology, large time- and cost-commitments are required to generate new mouse models, and the approach is generally not amenable to high-throughput studies. On the other hand, somatic engineering of HSPC genomes has been achieved, in large, using retroviral vectors to either overexpress or to knock-down the expression of genes of interest (Riviere et al., 2012). This approach has been used to screen for both positive and negative regulators of HSC function from up to 100 candidate genes (Deneault et al., 2009; Hope et al., 2010). Although this approach allows investigators to quickly assess the function of multiple genes, retroviral transduction negatively impacts HSC function during in vitro culture, and retroviral genome integration is a serious concern when these engineered cells are used clinically (Hacein-Bey-Abina et al., 2003). A new method to edit the genomes of hematopoietic stem/progenitor cells (HSPCs) should not only accelerate gene discovery research, but also open up new clinical opportunities in using engineered HSPCs for gene therapy.
Among the several engineered nucleases enabling site specific genome editing, the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) system (Jinek et al., 2012) stands out since it does not require cumbersome engineering of nucleases for each target but only requires a 20 nucleotide RNA sequence contained within a chimeric single-guide RNA (sgRNA) to drive the endonuclease Cas9 to its target sequence. Thus, CRISPR/Cas9 provides a versatile, modular, and cost-effective means to edit the genomes of multiple model systems (Hsu et al., 2014; Sternberg and Doudna, 2015). Several delivery methods have been used to perform CRISPR/Cas9-mediated gene editing of HSPCs, including lentiviral transduction (Heckl et al., 2014), plasmid DNA transfection (Mandal et al., 2014), or chemically modified RNA (Hendel et al., 2015), achieving up to 48% gene disruption in human HSPCs. While these studies have shown the enormous potential of HSPC gene editing by CRISPR/Cas9, a method that is highly efficient, simple without the need of any cloning and nucleotide modifications, and addresses clinical concerns of retroviral genome insertion, is still lacking. We sought to develop simple strategies to perform CRISPR/Cas9-mediated gene editing in HSPCs with minimal manipulations and while avoiding viral integration into the HSPC genome. Here, we describe fast, efficient, and cost-effective methods of CRISPR/Cas9-mediated gene-editing in primary murine and human HSPCs, and demonstrate that this method can be used to directly examine gene function.
RESULTS
Efficient gene disruption in mouse HSPCs
We reasoned that transfecting HSPCs isolated from Cas9-expressing mice (Platt et al., 2014) with sgRNA would be an efficient method to edit the genome of HSPCs, since only the small RNA molecules would need to be introduced. To test this idea, we designed small guide RNAs to target the GFP gene (GFP-sg1) co-expressed in the Cas9-expressing mice. When we electroporated c-kit+ HSPCs with in vitro transcribed GFP-sg1, we observed highly efficient loss of GFP expression by flow cytometry, compared to cells electroporated with sgRNA against Rosa26 (R26-sg) (Figure 1A). Although electroporation reduced the survival of HSPCs approximately 20% immediately after electroporation, cells maintained at least 80% viability throughout the experiment for up to 96 hours post electroporation (Figure 1B). In this condition maintaining high viability, we found that 67±4% of HSPCs lost GFP expression upon electroporation of GFP-sg1 (Figure S1A–B), demonstrating efficient gene editing with high cell viability. The frequency of GFP ablation exhibited an sgRNA dose-dependent increase, plateauing at 1 μg of GFP-sg1 for 105 HSPCs per transfection (Figure 1C).
Figure 1. Gene editing in murine HSPCs.

(A) A representative flow cytometry histogram showing efficient ablation of GFP by electroporating GFP-sg1 into Cas9-expressing HSPCs. Black histogram represents GFP− HSPCs, and green and red histograms represents Rosa26 (R26) and GFP disrupted HSPCs, respectively (n=3). (B) Survival of HSPCs was determined by trypan blue staining of cells cultured without electroporation, cells mock electroporated without sgRNA, and cells electroporated with R26 or GFP sgRNA. 1 μg of sgRNA was used to electroporate 105 cells (n=3). (C) Deletion efficiencies of GFP exhibiting sgRNA dose-dependent response. A plateau in gene editing efficiency was reached by 1 μg of sgRNA per 105 cells (n=3). (D) A brief culture of murine HSPCs for 1 to 3 hours increased gene-editing frequency, while overnight (O/N) culture did not further increase gene editing (n=3). (E) After electroporating c-kit+ HSPCs with GFP-sg1, HSCs were sorted clonally into methylcellulose media. Most (40 out of 48) HSC colonies exhibited loss of GFP expression, as shown by the representative flow cytometric histograms for 3 HSC-derived colonies from one donor mouse (n=3 independent experiments). (F) A representative histogram demonstrating efficient ablation of GFP expression by electroporating Cas9/GFP-sg1 RNP into GFP expressing HSPCs (n=3). (G) Quantification of results in (F). Even as little as 200 ng of GFP-sg1 efficiently ablated GFP upon delivery with Cas9 protein (1 μg). (H) T7E1 assays performed with GFP amplicon derived from R26- or GFP-disrupted HSPCs. PCR amplicons were either treated (+) or untreated (−) with T7E1. Arrowheads indicate the bands with expected size assuming small indels, based on the Cas9 cleavage site. 1 μg of sgRNA was used to electroporate 105 Cas9-expressing cells unless otherwise noted. All data represent mean ± standard deviation; *, p<0.05; **, p<0.01; and ***, p<0.001 by Student’s t-test. See also Figure S1.
Because retroviral transduction is enhanced by culturing with cytokines to stimulate the cell cycle of quiescent HSPCs, we also tested whether brief in vitro exposure to cytokines in culture before sgRNA electroporation increased GFP gene editing. We electroporated Cas9-expressing HSPCs after varying the duration of culture. We found that a brief culture (1 to 3 hours) increased the frequency of GFP negative cells from around 60% in fresh progenitors to a maximum of 85±1% after 3h, without further increase after 12 hours (Figure 1D). In the context of this gene-editing strategy, incorporation of an optimized scaffold sequence previously shown to improve Cas9-mediated imaging (Chen et al., 2013) did not significantly increase the frequencies of gene disruption further (Figure S1C).
Because the c-kit+ population contains various progenitors besides HSCs, we tested whether HSCs themselves undergo successful gene disruption upon transfection of sgRNAs. After electroporation of Cas9-expressing c-kit+ cells with GFP-sg1, we sorted CD150+CD48−lineage−Sca-1+ cells (c-kit expression was attenuated within 1 hour of pre-culture before electroporation, Figure S1D) into semi-solid media and monitored colonies arising from these single HSCs for GFP ablation. Flow cytometry of individual colonies revealed that most HSC-derived colonies (40 out of 48: 83%) lost GFP expression (representative 3 colonies shown in Figure 1E), while electroporation with or without sgRNA did not affect the clonality of HSCs compared to cultured, un-electroporated cells (Figure S1E). Thus, our method efficiently ablates genes in HSCs with minimum impact on HSC survival.
With the optimized sgRNA delivery method in hand, we next considered whether Cas9 protein pre-complexed with sgRNA to generate a ribonucleoprotein particle (RNP) (Kim et al., 2014; Lin et al., 2014; Schumann et al., 2015) could also be used to edit genes in murine HSPCs. Different amounts of GFP-sg1 were mixed with Cas9 protein and the RNP complex was electroporated into HSPCs isolated from a mouse strain that ubiquitously expresses GFP (Schaefer et al., 2001). As shown in Figure 1F–G, approximately 70% of HSPCs lost GFP expression upon co-delivery of Cas9 protein and GFP-sg1. Two other sgRNAs against GFP also attenuated GFP expression in significant fractions of HSPCs (Figure S1G). Under this condition, HSPCs electroporated with Cas9 RNP maintained viability of >80% for up to 96 hours after electroporation (Figure S1F). Since loss of GFP expression is an indirect measure of genome editing, we directly examined indel frequencies at the genomic level. First we performed T7 endonuclease I (T7E1) assays and found that a significant fraction of HSPCs accrued indels (Figure 1H). We note that the T7E1 assay often underestimates the rate of indel formation, potentially due to self-hybridization of the alleles that carry the indel, incomplete duplex melting, and inefficient cleavage of single nucleotide indels (Schumann et al., 2015). To accurately determine the nature of the indels, we next performed high-throughput sequencing, which revealed that approximately 60% of HSPCs accrued small indels (Figure S1H and Table S2). These genomic analyses corroborated the flow cytometry data demonstrating that GFP alleles were mutated by the Cas9 RNP approach. Thus, this strategy allows us to study gene function directly in HSPCs in any genetic background.
Functional assessment of gene disruption in mouse HSPCs
We then tested whether targeted gene editing in HSPCs by CRISPR/Cas9 could be used to alter HSPC function. We chose to disrupt two polycomb repressive complex 2 (PRC2) components Eed and Suz12, which are both found mutated in human leukemias (Shih et al., 2012). It has been shown that monoalleleic loss, but not biallelic loss, of Eed or Suz12 confers proliferative advantages to HSPCs (Lee et al., 2015; Xie et al., 2014). We transfected wild-type HSPCs with Cas9-RNP complexes against Rosa26, Eed, or Suz12, plated them on semi-solid media, and serially replated the cells. Whereas most Rosa26-targeted HSPCs lost proliferative capacity upon the fourth replating, Eed- and Suz12-targeted HSPCs exhibited extensive proliferative capacity at the fourth passage (Figure 2A). T7E1 assays performed on HSPC cultures shortly (48 hours) after electroporation revealed that a substantial fraction of cells accrued indels in Eed and Suz12 (Figure 2B). Sanger sequencing and TIDE analysis (Brinkman et al., 2014) revealed that approximately 50% of the sequence reads accrued small indels between 4bp deletion and 2bp insertion (Figure S2A, B). We also cloned individual colonies at later passages and examined the editing status. T7E1 assay revealed that Eed- and Suz12-edited colonies acquired indels, and sequencing verified that they all acquired monoallelic loss of Eed or Suz12 (Figure 2C–F), consistent with the haploinsufficient function of Eed and Suz12 (Lee et al., 2015; Xie et al., 2014). These results establish that Cas9 RNPs can be used to perform gene editing in primary murine HSPCs regardless of genetic background, resulting in clear loss-of-function phenotypes.
Figure 2. Editing endogenous genes in murine HSPCs.

(A) Gene editing of Eed or Suz12 using Cas9-sgRNA RNP increased the ability of murine HSPCs to serially replate in culture. 1 μg Cas9 protein and 1 μg of sgRNA were used (n=4). (B) T7E1 assays performed with Eed (upper panel) or Suz12 (lower panel) amplicon derived from Rosa26- (R26), Eed-, or Suz12- disrupted HSPCs 48 hours after electroporation. The numbers below the gel image represents the cleavage efficiency determined by densitometric analysis (n=3). (C, D) T7E1 assays performed with Eed (C) or Suz12 (D) amplicons derived from Rosa26- (R26), Eed-, or Suz12-disrupted colonies, with (+) or without (−) the nuclease (n=4). Arrowheads indicate the bands with expected size based on the Cas9 cleavage site, while asterisks indicate non-specific bands. (E, F) Sequencing results of representative 4 clones each (out of 12) after electroporating with Eed (E) or Suz12 (F) sgRNA. All colonies analyzed (Eed: 12/12, Suz12: 12/12) acquired indels. Red line represents the position of the PAM sequence. All data represent mean ± standard deviation; ***, p<0.001 by Student’s t-test. See also Figure S2.
Efficient gene disruption in human HSPCs
Encouraged by the results obtained in mice, we assessed the feasibility and efficiency of our protocol in human hematopoietic cells. We first sought to target human CD45 (hCD45) in HL-60 cells, a human acute myeloid leukemia cell line. We electroporated HL-60 cells with Cas9/hCD45-sg RNPs testing three different CD45 guides. Strikingly, all three guides displayed very high efficiencies of disrupting CD45 expression (sg1: 98%, sg2: 91% and sg3: 74%) as assessed by flow cytometry (Figure S3A). Two additional AML cell lines, OCI-AML2 and Kasumi, exhibited similar editing efficiencies (Figure 3A).
Figure 3. Efficient CD45 knock-out in human hematopoietic cells.

(A) Flow cytometry analysis of hCD45 expression in three AML cell lines and activated primary T cells 96 hours following electroporation with Cas9 only (blue) or Cas9/hCD45-sg1 RNP (red). (B) Effects of pre-culture before electroporation on gene disruption efficiency. hCD45 loss were examined in CD34+ cells cultured for 0, 24 and 48 hours in presence of cytokines before electroporation with Cas9/hCD45-sg1 RNP. hCD45 expression was evaluated 4 days after electroporation. Each experiment (n=8) was performed on CD34+ cells isolated from single donors. (C) Flow cytometry analysis of CD45 and CD34 expression in CD34+ cells 96 hours following electroporation with Cas9 only (left panel) or Cas9/hCD45-sg1 RNP (right panel). (D–E) Cell viability examined by trypan blue staining (D, n=8) or flow cytometry (E, n=6) of CD34+ cells electroporated either with Cas9 only (black) or with Cas9/hCD45-sg1 RNP (grey) relative to non-electroporated cells at the indicated time points. The cell counts (D) or viability (E) of Cas9 only and Cas9/hCD45-sg1 transfected cells were compared to the viable cell counts of non-electroporated cells. *, p<0.05; **, p<0.01; and ***, p<0.001 by non parametric t-test.
We also tested whether our protocol was capable of editing peripheral blood mononuclear cell (PBMC)-derived primary T-lymphocytes. While resting T-cells were resistant to CD45 editing (data not shown), activated T-cells targeted with hCD45-sg1 exhibited efficient (86±2%; n=3) loss of CD45 (Figure 3A), with some residual CD45bright and CD45mid cells (Figure S3B). We performed high-throughput sequencing of unfractionated T-cells as well as cells sorted based upon CD45 expression (CD45bright, CD45mid, CD45dim, and CD45neg cells) and discovered that while the unfractionated cells had an indel frequency of 83%, more than 95% of alleles in CD45neg and CD45dim cells, and 40% of alleles in CD45mid cells had acquired indels (Figure S3B). Interestingly, CD45dim cells harbored 51% in-frame and 49% out-of-frame indels (Figure S3C), which may explain the residual CD45 expression (Figure S3C). Importantly, low indel frequencies (0.2%, 1.7%, 0.4%; read depth: 924, 301, 527) were observed at the top three predicted off-target (OT) sites (Figure 4B and Supplemental Methods).
Figure 4. CRISPR-Cas9 mediated gene disruption and HDR in human HSPCs.

(A) Table showing indel frequencies at targeted loci. When multiple sgRNAs were used in the same experiment, sg1 indicates alleles with disruption of sg1 only, sg2 indicates alleles with disruption of sg2 only and Δ indicates alleles with a deletion between sg1 and sg2. Tables containing raw allele counts for each indel/deletion are found in Table S2. (B) Indel frequencies at 3 predicted hCD45-sg1 off-target sites. Off-target sites (OT1-3) were predicted using CRISPRscan (Moreno-Mateos et al., 2015). (C) Plots showing percentages of human cells (left) in the bone marrow and the spleen of 16 NSG recipient mice (8 Cas9 only and 8 Cas9/hCD45-sg1 RNP) and the fraction of engrafted human cells that have lost hCD45 (right) in Cas9 only (black) and Cas9/hCD45-sg1 RNP (red). Human CD34+ cells from individual cord blood donors were electroporated with Cas9 only and Cas9/hCD45-sg1 RNP and transplanted into sublethally irradiated NSG mice. Engraftment was analyzed 8 weeks post-transplant. (D) Flow cytometry analysis of two engrafted NSG mice. Upper panels show the engraftment of normal human cells (CD45posHLA-ABCpos). Lower panels show the presence of hCD45 knock-out cells (highlighted in red) both in the bone marrow and the spleen. (E) Western blot analysis of DNMT3A expression in CD34+ cord blood cells 96 hours after electroporation with Cas9 only, Cas9/hCD45-sg1 RNP, Cas9/DNMT3A exon 7/8-sg (4 sgRNAs) RNP, or Cas9/DNMT3A exon 7-sg (2 sgRNAs) RNP. (F) Schematic representation of the CRISPR-mediated knock-in (top). Three single nucleotide changes, two of which (red) result in the formation of a BsiWI restriction site (italics), were introduced into hCD45 exon 25. The most common observed alleles from a representative sample (bottom), which include both precise (HDR; all three single nucleotide changes) and imprecise (HDR*; two out of three nucleotide changes) knock-in events, were assessed by high-throughput sequencing and their allele frequencies are displayed. The numbers in red represent frequencies of reads containing the BsiWI restriction site. (G) A gel image of BsiWI digested PCR amplicon prepared from CD34+ cord blood cells targeted with Cas9/hCD45-sg1 (1 μg each) RNP and different single-stranded DNA oligonucleotides (ssODN) containing BsiWI sites. Both symmetric (S) and asymmetric (A) homology arms were tested. BsiWI digests the 282 bp amplicon with HDR editing into 172 and 110 bp fragments. The numbers below the gel represent efficiency of restriction site knock-in (HDR+HDR*) as determined by high-throughput sequencing.
We then tested whether primary human CD34+ HSPCs could be gene edited. To search for the optimal conditions, we first electroporated primary CD34+ cells derived from umbilical cord blood with Cas9/hCD45-sg1 RNP using 9 different electroporation parameters and performed flow cytometry 96 hours later to measure CD45 expression levels together with cell viability (Figure S3D). The optimized electroporation parameter (Condition 9) was used for all further experiments. Since short-term ex-vivo expansion is routinely exploited to increase transfection and transduction efficiency, we explored the impact of short cytokine exposure on transfection and gene disruption efficiency. We electroporated CD34+ cells from single donors (n=5) with mRNA encoding for GFP-fused Nucleophosmin 1 (GFP-NPM1) immediately after isolation, after 24 or 48 hours of cytokine exposure and analyzed GFP expression 24 hours later. While fresh cells showed only 28±13% GFP positivity, cells cultured for either 24 or 48 hours displayed transfection efficiencies higher than 75% (24h: 81%±12; 48h: 78%±10) (Figure S3E). To assess whether CRISPR-mediated gene disruption efficiency was influenced by exposure to cytokines, we performed a similar experiment electroporating CD34+ from single donors (n=8) with Cas9/hCD45-sg1 RNP again at 0, 24 and 48 hours of culture and analyzed CD45 expression 4 days later. As expected, freshly isolated CD34+ cells not exposed to cytokines exhibited limited loss of CD45 cell surface expression (8±4%). However, gene disruption efficiency was significantly higher in cells cultured for 48 hours compared to 24 hours (73±16% vs 41±12%, p=0.003) (Figure 3B), implying that the efficiency of gene disruption is dependent upon more than transfection efficiency. Importantly, CD34 expression remained unchanged even following efficient CD45 knock-out (Figure 3C). We also determined the impact of CRISPR-Cas9 delivery on human HSPCs viability. CD34+ cells from single donors were electroporated with Cas9 only or Cas9/hCD45-sg1 RNP or left untreated in culture, and viable cell counts were recorded by trypan blue staining at 24, 60 and 96 hours after electroporation. Non-electroporated cells expanded in vitro, reaching 2.1×105 cells, 5×105 cells, and 1.1×106 cells at 24, 60, and 96 hours of culture, respectively, starting with 105 cells. Electroporated cells exhibited significant but acceptable cell loss in Cas9/hCD45-sg1 RNP-treated cells compared to non-electroporated cells (24h: 69±31%, 60h: 40±13%, 96h: 34±17%; n=8) (Figure 3D). Cell viability assessed by flow cytometry at 48 and 96 hours corroborated these results (48h: 48±18%, 96h: 45±11%; n=6) (Figure 3E). These results are consistent with those reported by others using electroporation of site-directed nucleases (De Ravin et al., 2016; Genovese et al., 2014).
CD45 knock-out efficiency measured by flow cytometry (73±16%) was confirmed by high-throughput sequencing of the hCD45 locus (75±10%) (Figure 4A and Table S2). As in T-cells, CD34+ cells electroporated with Cas9/hCD45-sg1 (n=3) displayed minimal off-target (OT) cleavage (1.0%, 7.0% and 0.1%; average read depth: 5319, 5164, 5089) at OT1, OT2, OT3, respectively (Figure 4B). To verify that the edited CD34+ HSPCs maintained engraftment and multilineage differentiation capacity, we transplanted Cas9 only (n=8) and Cas9/hCD45-sg1 RNP edited cells (n=8) into sub-lethally irradiated NOD scid gamma (NSG) mice. To avoid possible donor-dependent bias, each experimental pair (i.e. one Cas9 only replicate and one Cas9/hCD45-sg1 RNP treated replicate) was performed on cells derived from a single cord blood. Bone marrow of 16/16 recipients and spleens of 13/16 recipients were successfully engrafted with human cells (Figure 4C). Importantly, we observed significant levels of engraftment by hCD45neg cells in the bone marrow of 7/8 mice and in the spleen of 5/8 mice transplanted with Cas9/hCD45-sg1 RNP edited cells (Figure 4C and D). Sequencing of the amplified human CD45 locus from bone marrow cells confirmed the presence of indels with frequencies consistent with flow cytometry data (Figure S4B). As expected for this time point (Goyama et al., 2015; McDermott et al., 2010), engrafted human HSPCs gave rise mainly to B- and myeloid cells in the bone marrow in both Cas9 only and Cas9/hCD45-sg1 RNP engrafted mice, with no differences between these two groups (Figure S4C). The Cas9/hCD45-sg1 RNP edited samples displayed no significant difference in CD45 gene disruption between B- and myeloid cells (Figure S4D). We also analyzed human CD34+ HSPCs in one bone marrow sample in which 37% of engrafted human cells were CD45 negative. CD45 loss was evident in 40% of CD34+ cells (Figure S4E) and 36% of the CD34+CD38− population, which contains the most immature progenitor cells. These results establish that our method allows for efficient gene disruption of human CD34+ HSPCs while retaining multilineage reconstitution capacity and the ability of these cells to engraft and expand in recipient mice. Given the known limitations of NSG mice to support robust generation of cells from all human hematopoietic lineages, such as T-cells and erythroid cells, further assessment in additional models and longer time points will reveal whether the most long-term HSCs are successfully modified and viable after these treatments.
Multiple guide approach allows for rapid deletion of genes of interest
To ensure functional ablation of a target gene, and to facilitate rapid assessment of gene editing by PCR, larger deletions are often desirable. Thus, we synthesized guide RNA pairs targeting exons 7–14 of DNMT3A. We tested combinations of up to four guides per electroporation in HL-60 cells, assessing the frequency and spectra of deletions after 12 hours, and the level of DNMT3A protein after 96 hours. All tested combinations of guides demonstrated efficient deletions and significantly diminished DNMT3A protein (Figure S4G).
We then targeted DNMT3A in primary CD34+ cord blood cells. Guides targeting exons 7 and 8 were tested and the expected deletions were observed by PCR (Figure S4F). Importantly, the DNMT3A protein was nearly absent 96 hours after disruption of exon 7 or exons 7+8 (Figure 4E). To verify this result, DNMT3A exon 7 was targeted in CD34+ cells enriched from an additional five cord blood samples and high-throughput sequencing confirmed a gene disruption frequency of 69±4% (Figure 4A and Table S2), suggesting that most cells likely experience loss of at least one allele, while many cells lose both alleles. To further validate the flexibility and efficiency of our approach, we targeted exon 10 of DNMT3A (n=10) and exon 3 of NR3C1 (n=5) in CD34+ cells and high-throughput sequencing showed allelic disruption frequencies of 86±14% and 75±6%, respectively (Figure 4A, S4H, and Table S2).
Efficient HDR-mediated gene editing in human HSPCs
Finally, we considered whether these editing strategies could be used to introduce specific point mutations into primary human HSPCs using Cas9-mediated homology directed repair (HDR). HDR would enable specific lesions to be introduced in HSPCs, potentially correcting deleterious mutations or mimicking cancer-associated mutations. Single-stranded oligonucleotide HDR templates (ssODNs) were designed with symmetric or asymmetric homology arms (Richardson et al., 2016) to introduce three basepair changes, two of which result in the generation of a BsiWI site near the hCD45-sg1 spacer sequence (Figure 4F). After 48 hours of culture with cytokines, CD34+ HSPCs were electroporated with the Cas9/hCD45-sg1 RNP along with either 10 or 30 pmol of ssODNs. Addition of 10 or 30 pmol of ssODN did not significantly affect the viability (Figure S3F, G), although a trend toward lower viability after electroporation with a higher dose of ssODN was observed. Following BsiWI digestion of genomic DNA 24 hours after transfection, all samples with a donor template displayed a digested band indicating successful HDR (Figure 4G). High-throughput sequencing performed on these samples revealed efficient precise knock-in (22%; range: 19–25%) of the mutant allele (Figure 4F and Table S2). Additionally, many reads (1–2%) displayed imprecise or partial knock-in of the mutant allele (1 out of 3 or 2 out of 3 bases) (Figure 4F and Table S2). Samples electroporated with only the donor template displayed no detectable mutant allele by high-throughput sequencing (not shown). Thus, our method allows homology-directed gene editing at a substantial frequency in human HSPCs. Additional testing in multiple mouse and other animal models will be required to establish whether the most long-term HSCs are successfully edited.
Discussion
The strategies we describe herein enable efficient gene editing, particularly loss-of-function studies, in both murine and human HSPCs directly. With the RNP-based methods, it is possible to generate gene-edited HSPCs within a week starting with conception of the study, including downtime for oligonucleotide synthesis. This is substantially shorter than editing HSPCs using lentiviral transduction, which requires cloning sgRNAs into lentiviral vectors, generation and quality control of lentiviral particles, and results in variable HSPC transduction efficiencies and risks of lentiviral integration in undesirable sites. Furthermore, electroporation of RNPs is a transient “hit-and-run” approach that obviates the need for a special mouse strain expressing Cas9, and reduces concerns of constitutive Cas9 expression and off-target cleavage. The costs of generating gene-edited HSPCs with these methods are substantially lower than that based on lentiviral methods. While commercially available Cas9 protein makes gene ablation with RNPs feasible, we also envision this strategy could be used for other Cas9 derivatives where transient effects may be desirable, such as CRISPR interference (Konermann et al., 2015; Qi et al., 2013).
One limitation of our protocol is that it lacks the ability to mark the cells that were successfully electroporated, unlike lentiviral transduction methods in which transduced cells can be marked by fluorescent proteins or by antibiotic resistance. However, with a gene editing efficiency approaching 90% as we show for some targets, our method makes it possible to examine the molecular or phenotypic changes without selection for transfected cells. We have shown that deletion of DNMT3A from primary human CD34+ cells leads to precipitous reduction in DNMT3A protein levels in the population within 96 hours after electroporation.
We have observed that gene editing efficiency is slightly more efficient in human than murine HSPCs. It is possible that this difference reflects developmental states, since the human cells are derived from cord blood rather than bone marrow. Additional optimization using murine HSPCs from the fetal liver or after mobilization may further increase the gene editing efficiency.
Perhaps the most significant finding we observed was efficient HDR in human HSPCs. The ability to introduce specific mutations in HSPCs will enable the expanded study of cancer-driver mutations in AML and other hematologic diseases. More importantly, the repair of inherited mutations in both common and rare blood disorders using CRISPR may now be feasible without integration of viral vectors or delivery of plasmid DNA, representing a major stepping-stone toward therapeutic gene editing. We have not yet achieved detectable HDR frequencies in mouse HSPCs and it is possible that a longer culture time prior to electroporation is necessary to activate this repair process in murine cells.
These remarkable knockout and HDR efficiencies in human HSPCs may suggest the possibility of broad and immediate utility. However, most ex vivo manipulations have ultimately been shown to impact function of the most long-term HSCs (Genovese et al., 2014; Hoban et al., 2015), and all of the available assays have limitations in their ability to assess bona fide HSC function. Therefore, caution in application of these strategies is still warranted.
In conclusion, we describe a fast, efficient, and cost-effective method to edit the genomes of both murine and human HSPCs based on the CRISPR/Cas9 system. The ability to quickly and efficiently edit primary HSPCs makes it possible to test the function of genetic variants identified in association with hematologic diseases such as leukemia or bone marrow failure. Moreover, the high efficiency offers the possibility to perform large-scale combinatorial gene editing in HSPCs to model complex mutational landscapes.
EXPERIMENTAL PROCEDURES
Production of sgRNA and electroporation
Protospacer sequences for each target gene were identified using the CRISPRscan algorithm (www.crisprscan.org) (Moreno-Mateos et al., 2015). DNA templates for sgRNAs were made using the protocol described by Li et al (Li et al., 2013) using primers listed in Table S1. 1 μg of sgRNA was electroporated into 1 × 105 Cas9-expressing c-kit+ murine HSPCs after 1–3 hours of culture. The optimized electroporation condition for murine HSPCs is 1700V, 20ms, 1 pulse using Neon transfection system (Thermo Fisher Scientific). To electroporate Cas9-sgRNA RNPs, 200 ng to 1 μg of sgRNA was incubated with 1 μg Cas9 protein (PNA Bio) for 10–15 minutes at room temperature, and electroporated as above. Human CD34+ cells were isolated by AutoMACS (Miltenyi Biotec) using CD34 microbeads, and electroporated as described for murine cells except the optimized electroporation condition was 1600V, 10ms, 3 pulses. See Supplemental Experimental Procedures for details.
Statistical analysis
For comparisons involving two groups, unpaired Student’s t tests (two-tailed) or non-parametric tests were used. See Figure Legends for details.
Supplementary Material
Acknowledgments
This work was supported by the Cancer Prevention and Research Institute of Texas (RP16028, RP140001, R1201), the Gabrielle’s Angel Foundation for Cancer Research, the Evans Foundation, and the National Institutes of Health (DK092883, CA183252, CA125123, P50CA126752, CA193235 and DK107413). MCG is supported by Baylor Research Advocates for Student Scientists. Flow-cytometry was partially supported by the NIH (NCRR grant S10RR024574, NIAID AI036211 and NCI P30CA125123) for the BCM Cytometry and Cell Sorting Core. The authors declare no competing financial interests.
Footnotes
AUTHOR CONTRIBUTIONS
MCG, LB, AL, AEM, MAG, and DN designed and discussed experiments. MCG, LB, AL, AEM, AK, DW and JH performed experiments. MCG, LB, KAH analyzed the sequencing data. CR provided reagents. All authors analyzed data. MCG, LB, AL, MAG, and DN wrote and edited the paper with input from all authors.
ACCESSION NUMBERS
The NCBI Sequence Read Archive accession number for the data reported in this paper is PRJNA339433.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ravin SS, Reik A, Liu PQ, Li L, Wu X, Su L, Raley C, Theobald N, Choi U, Song AH, et al. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol. 2016;34:424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneault E, Cellot S, Faubert A, Laverdure JP, Frechette M, Chagraoui J, Mayotte N, Sauvageau M, Ting SB, Sauvageau G. A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell. 2009;137:369–379. doi: 10.1016/j.cell.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, Moi D, Mazzieri R, Bonini C, Holmes MC, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. 2015;125:2630–2640. doi: 10.1182/blood-2014-11-570218. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (New York, NY. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, Ebert BL. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, Ho M, Lumaquin D, Gray D, Lill GR, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope KJ, Cellot S, Ting SB, MacRae T, Mayotte N, Iscove NN, Sauvageau G. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell. 2010;7:101–113. doi: 10.1016/j.stem.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, NY. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Miller S, Hyland C, Kauppi M, Lebois M, Di Rago L, Metcalf D, Kinkel SA, Josefsson EC, Blewitt ME, et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood. 2015;126:167–175. doi: 10.1182/blood-2014-12-615898. [DOI] [PubMed] [Google Scholar]
- Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016 doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- Riviere I, Dunbar CE, Sadelain M. Hematopoietic stem cell engineering at a crossroads. Blood. 2012;119:1107–1116. doi: 10.1182/blood-2011-09-349993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, Goodell MA. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11:302–317. doi: 10.1016/j.stem.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cellular immunology. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, Doudna JA. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol Cell. 2015;58:568–574. doi: 10.1016/j.molcel.2015.02.032. [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80. doi: 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.