

Abstract
Jaundice is a clinical manifestation of disorders of underlying bilirubin metabolism, hepatocellular dysfunction, or biliary obstruction. As clinical presentations of yellowing of eyes or skin can be somewhat nonspecific for the underlying etiology of disease, a stepwise approach to evaluation is necessary for accurate diagnosis and effective treatment plan. In this review, we discuss underlying mechanisms of cholestasis and jaundice as well as laboratory and imaging modalities needed to evaluate a patient presenting with hyperbilirubinemia. Jaundice occurs in settings of cholestasis or inability to effectively secrete bile as well as disorders of bilirubin metabolism and hepatocellular dysfunction. Clinical signs of jaundice occur when the serum bilirubin level exceeds 2.5 to 3 mg/dL. In all cases, evaluation begins with liver chemistry tests which include bilirubin (conjugated and unconjugated), alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and total protein. In patients with hepatobiliary causes of jaundice, the alkaline phosphatase is usually elevated. In these cases, evaluation of hepatic synthetic function is crucial to the formulation of a treatment plant. When serologic evaluation is combined with hepatobiliary imaging, underlying mechanism of disease can often be elucidated. A stepwise approach to evaluation can be cost and time saving as well as a framework to improve patient outcomes. In this review, we will outline a diagnostic approach to jaundice, beginning with pathophysiology of cholestasis followed by hyperbilirubinemia and markers of synthetic dysfunction.
Keywords: jaundice, hyperbilirubinemia, cholestasis
Objectives: Upon completion of this article, the reader will be able to identify the pathophysiology of hyperbilirubinemia, its etiology, clinical presentation, and diagnostic approach to assess and manage a patient with jaundice.
Accreditation: This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of Tufts University School of Medicine (TUSM) and Thieme Medical Publishers, New York. TUSM is accredited by the ACCME to provide continuing medical education for physicians.
Credit: Tufts University School of Medicine designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Cholestasis
Cholestasis occurs as a result of any condition which impairs the liver's ability to secrete bile. This may be due to decreased bile synthesis, defective bile secretion, or bile flow obstruction.1 The hallmark laboratory abnormality of cholestasis is elevation of the alkaline phosphatase. Concomitant elevation of the alkaline phosphatase and bilirubin is a hallmark of hepatobiliary dysfunction.
Alkaline Phosphatase
Alkaline phosphatase represents a group of zinc metalloenzymes that catalyze hydrolysis of esters.2 In the liver, alkaline phosphatase is predominantly found in the canalicular membrane of the hepatocytes3; however, it can also be found in osteoblasts of the bone, small intestinal brush border, the placenta, proximal tubules of the kidney, and white blood cells.4 The majority of serum alkaline phosphatase originates from the liver, bone, and intestine. Within the liver, bile acids induce translation of alkaline phosphatase mRNA. In a clinical setting of biliary obstruction, increased concentrations of bile acids promote expression of alkaline phosphatase, which results in translocation of the enzyme from the canalicular membrane to the basolateral or sinusoidal surface. High concentrations of alkaline phosphatase within the hepatic sinusoids can be measured as elevated levels in the serum.5 In healthy patients, alkaline phosphatase levels have been shown to have variability as a function of age, gender, race, and blood type.6 For example, mild elevations of alkaline phosphatase are commonly seen at times of bone growth such as in an adolescent during puberty and during pregnancy due to placental production of the enzyme.1 7 Decreased levels of alkaline phosphatase can also be seen in Wilson disease, zinc deficiency, malnutrition, hypothyroidism, vitamin C deficiency, pernicious anemia, and phosphorus deficiency.1 In most cases of pure cholestasis, alkaline phosphatase levels are proportionally higher than serum aminotransferases. Unlike hepatocellular injury, where the degree of aminotransferase elevation is indicative of certain mechanisms of injury, the magnitude of elevation is usually less helpful in distinguishing specific etiology of cholestasis.
5′-Nucleotidase
The function of 5′-nucleotidase (5′NT) is to convert extracellular nucleotides to nucleosides via catalyzing hydrolysis at the 5′ position on the pentose moiety. 5′NT is found in many tissues, including liver, brain, intestine, heart, blood vessels, and endocrine pancreas; however, for unclear reasons, serum levels become elevated only when originating from a hepatobiliary source.3 8 There can be discordance between serum alkaline phosphatase and serum 5′NT. In a study that used liver-specific alkaline phosphatase as the gold standard, 5′NT had a sensitivity of 47% and a specificity of 86% in detecting hepatobiliary disease.9 Given the relatively high specificity, 5′NT has the greatest clinical utility as a confirmatory test of hepatobiliary origin in patients with elevated alkaline phosphatase.
Gamma-Glutamyl Transpeptidase
Gamma-glutamyl transpeptidase (GGT) is a sialoglycoprotein that plays an important role in glutathione metabolism.10 Similar to alkaline phosphatase and 5′NT, GGT has a wide distribution throughout the body and can be found in the liver, kidney, spleen, brain, lungs, intestine, and prostate.11 Elevations of serum GGT are sensitive to reflect hepatobiliary disease; however, they are not specific and can be seen in diabetes, renal disease, myocardial infarction, rheumatic disease, neurologic disease, and pancreatitis.12 Many drugs, notably anticonvulsants, and alcohol use have been shown to cause increased GGT enzyme activity.13 Importantly, GGT levels are not elevated in musculoskeletal disease; therefore, this test is useful in ruling out a bone disease in a patient with an elevated alkaline phosphatase.
Evaluation of a Patient with Abnormal Alkaline Phosphatase Levels
Determining the source of alkaline phosphatase is the initial step in the evaluation of a patient with cholestasis (Fig. 1). In these patients, bilirubin may be normal or elevated. In a jaundiced patient, elevated alkaline phosphatase may represent diffuse hepatocellular dysfunction or significant bile duct obstruction which impedes adequate bile flow. A hepatobiliary source of alkaline phosphatase can be verified by an elevated 5′NT or GGT levels. Alternatively, fractionating alkaline phosphatase isoenzyme via electrophoresis will identify the origin of each component.1 14 However, in settings where alkaline phosphatase is elevated along with other liver chemistry abnormalities such as hyperbilirubinemia or elevated aminotransferases, confirmation of hepatobiliary source of alkaline phosphatase is usually not necessary. Once a hepatobiliary source of cholestasis is confirmed, the next step in evaluation is to determine whether the abnormality is intrahepatic or extrahepatic.
Fig. 1.
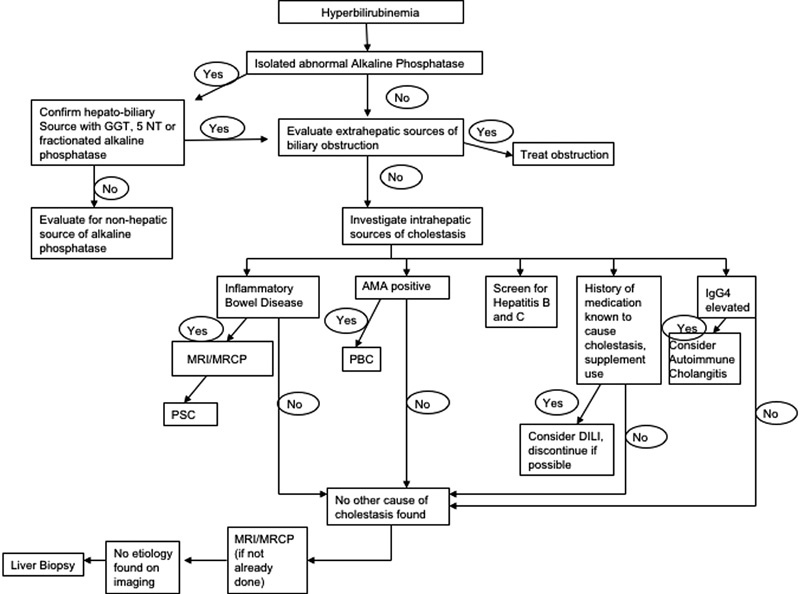
Evaluation of a patient with cholestasis.
Extrahepatic Source of Cholestasis
Patients with an extrahepatic source of cholestasis may present with fevers, right upper quadrant abdominal pain, or jaundice; however, they may also be asymptomatic. Initial evaluation of extrahepatic sources of cholestasis should begin with a right upper quadrant ultrasound to evaluate for the presence of biliary dilatation. Ultrasound is a safe and relatively inexpensive imaging modality and has a high specificity (95–100%) in the identification of common bile duct stones.15 Limitations of ultrasound include decreased sensitivity for obstruction, especially in obese patients, extensive overlying bowel gas, or in those with smaller culprit lesions.16 Computed tomography (CT) has been shown to be more accurate than ultrasound in the identification of both the level and the cause of biliary obstruction; however, this test is less sensitive in detecting choledocholithiasis and exposes patients to radiation.16 17 Finally, endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) are highly effective in diagnosing sources of extrahepatic cholestasis. Although ERCP has long been the gold standard, MRCP has been shown to be a safe noninvasive method with comparable sensitivity and specificity in diagnosing biliary obstruction.18
Choledocholithiasis is the most common cause of extrahepatic biliary obstruction; however, in patients with cholestasis, other sources of obstruction should also be considered (Table 1). In settings of predominant cholestasis, we recommend evaluation for extrahepatic causes of obstruction early in the evaluation, as many causes may be reversible and without intervention, it may pose a risk of infection or progressive hepatic fibrosis.
Table 1. Extrahepatic causes of cholestasis.
| Gallstones |
| Biliary cysts |
| Biliary stricture |
| Parasitic infection |
| Pancreatitis |
| Pancreatic pseudocyst |
| AIDS cholangiopathy |
| Immunoglobulin G4–related disease |
| Malignancy |
| Cholangiocarcinoma |
| Lymphoma |
| Pancreatic cancer |
| Ampullary cancer |
| Metastatic disease |
| Gallbladder cancer |
Intrahepatic Source of Cholestasis
Once extrahepatic causes of elevated alkaline phosphatase have been ruled out, intrahepatic causes should be considered. Jaundice is a common clinical finding in patients with intrahepatic sources of cholestasis, especially at late stages of disease. Patients with intrahepatic cholestasis may present with pruritus and fatigue.1 Laboratory evaluation may reveal hyperlipidemia19 and deficiencies of fat-soluble vitamins A, D, E, and K.20 A thorough history should be obtained which includes medication and supplement use to identify possible drug-induced cholestasis. Table 2 describes potential causes of intrahepatic cholestasis, which include viral, genetic, immune-mediated, and infiltrative etiologies. Table 3 describes the nonhepatic causes of elevated alkaline phosphatase. Serologic evaluation is often helpful to identify the specific etiology; however, at times, liver biopsy or imaging of the biliary system with MRCP is needed.
Table 2. Intrahepatic causes of cholestasis.
| Primary biliary cirrhosis |
| Primary sclerosing cholangitis |
| Viral causes |
| • Hepatitis B |
| • Hepatitis C |
| Infiltrative causes |
| • Amyloidosis |
| • Lymphoma |
| • Tuberculosis |
| • Sarcoidosis |
| • Other granulomatous diseases |
| Genetic causes |
| • Dubin-Johnson |
| • Rotor syndrome |
| • Benign recurrent intrahepatic cholestasis |
| • Progressive familial intrahepatic cholestasis |
| • Gilbert syndrome |
| • Alagille syndrome |
| Pregnancy |
| • Intrahepatic cholestasis of pregnancy |
| TPN |
| Sepsis |
| Vanishing bile duct syndrome |
| Cystic fibrosis |
| Medications |
Abbreviation: TPN, total parenteral nutrition.
Table 3. Nonhepatic causes of elevated alkaline phosphatase.
| Pregnancy |
| Bone disease |
| Chronic renal failure |
| Congestive heart failure |
| Lymphoma |
| Normal childhood growth |
| Infection |
| Inflammation |
Source: Adapted from Green and Flamm.3
Bilirubin
Bilirubin is derived from the catabolism of heme-containing proteins, the majority of which originates from senescent red blood cells.21 Initially, heme is oxidized within reticuloendothelial cells to form biliverdin, which is then reduced by biliverdin reductase to form water-insoluble unconjugated bilirubin. Within the liver, unconjugated bilirubin is taken up by the hepatocytes and conjugated by a group of enzymes called “uridine diphospho-glucuronosyltransferase” (UDP-glucuronosyltransferase). Once conjugated, bilirubin is water soluble and may be excreted by the kidneys. In addition, it is actively transported against concentration in an ATP-mediated step across the hepatocyte canalicular membrane into the bile. The half-life of bilirubin is approximately 4 hours; however, in settings of impairment of hepatic excretion of conjugated bilirubin, the conjugated bilirubin may become covalently bound to albumin and exhibit a half-life similar to that of albumin—approximately 21 days.22
Evaluation of a Patient with Hyperbilirubinemia
Liver disease that results in impairment of hepatic metabolism, transport of bilirubin, or injury to any portion of the hepatobiliary system may result in hyperbilirubinemia.4 23 Bilirubin is often elevated along with liver test abnormalities, in which case investigation of etiology of liver disease should be directed toward the predominant pattern: either a hepatocellular or cholestatic. When jaundice occurs with predominant hepatocellular pattern of injury diseases such as viral hepatitis, alcohol-related liver disease (including alcoholic hepatitis) and autoimmune diseases should be considered. When jaundice occurs with a predominantly cholestatic pattern of injury, biliary disorders are more likely the cause (Fig. 2). In cases of an isolated hyperbilirubinemia, determining whether this is predominantly due to unconjugated or conjugated hyperbilirubinemia is a key step in evaluation.
Fig. 2.
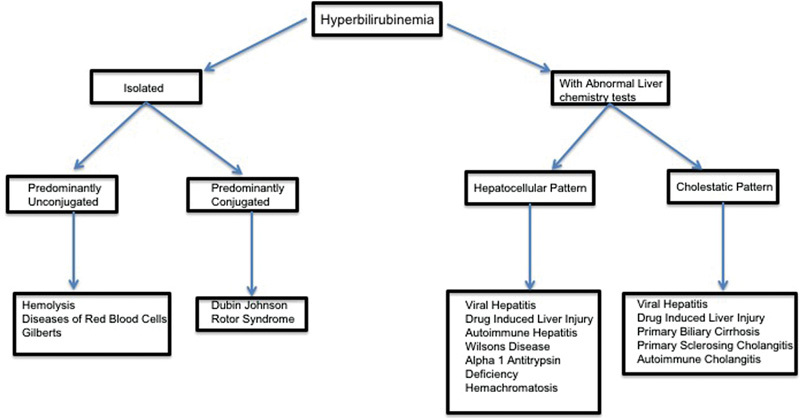
Overview of approach to hyperbilirubinemia.
Isolated Unconjugated Hyperbilirubinemia
Isolated unconjugated hyperbilirubinemia may be due to bilirubin overproduction, impaired hepatic uptake, or ineffective conjugation.4 Bilirubin overproduction is commonly seen in settings without any true hepatic injury such as hemolysis, hematoma resorption, or other diseases of red blood cells.24 In these cases, serum bilirubin is frequently less than 3 to 4 mg/dL. The most common nonhemolytic cause of isolated hyperbilirubinemia is Gilbert syndrome, which occurs in up to 8% of the population.25 26 In Gilbert syndrome, a genetic mutation leading to decrease expression of UDP-glucuronosyltransferase causes impaired glucuronidation of bilirubin, which causes a mildly increased unconjugated hyperbilirubinemia. Patients remain asymptomatic with the absence of clinical liver disease except for a mild jaundice which may be more apparent during fasting states or stress. Even in these states, serum bilirubin levels rarely exceed 3 mg/dL (Table 4). Although genetic testing is available in an asymptomatic patient without hemolysis and otherwise normal liver chemistry tests, a presumptive diagnosis of Gilbert syndrome can be made without genetic testing.
Table 4. Isolated unconjugated hyperbilirubinemia.
| Hematologic disorders |
| Sickle cell disease |
| Hereditary spherocytosis |
| Glucose-6-phosphate deficiency |
| Medication effects (i.e., ribavirin) |
| Microangiopathic hemolytic anemia |
| Immune-mediated hemolysis |
| Blood transfusion |
| Hematoma resorption |
| Ineffective erythropoiesis |
| Impaired uptake or conjugation |
| Gilbert syndrome |
| Crigler-Najjar syndrome |
Isolated Conjugated Hyperbilirubinemia
Elevation in serum-conjugated bilirubin levels is always indicative of some hepatic disorder. In the vast majority of cases, this elevation coincides with other perturbations of liver tests and usually supports a diagnosis of either hepatocellular or cholestatic disease. Isolated conjugated hyperbilirubinemia is far less common and is seen in Dubin-Johnson syndrome and Rotor syndrome. In both cases, there is impairment if bilirubin excretes into the bile,25 which may result in clinical jaundice. Treatment is usually not necessary because the clinical course is otherwise benign.
Tests of Hepatic Synthetic Function
Commonly measured laboratory tests such as albumin and prothrombin time can be a useful reflection of hepatic synthetic function. These tests are most helpful when prognosticating patients with cholestasis and jaundice.
Albumin
The adult liver synthesizes approximately 15 g of albumin per day. Hypoalbuminemia can be seen in conditions of poor hepatic function; however, there can be decreased production of albumin during environmental, toxic, or traumatic stress.27 Conditions such as nephrotic syndrome, malabsorption, and poor nutrition may also decrease serum albumin levels. In clinical settings of hypoalbuminemia especially with otherwise normal liver functions tests, nonhepatic etiologies should always be considered. The half-life of albumin is approximately 19 to 21 days; therefore, hypoalbuminemia is a more reliable marker for synthetic dysfunction seen in chronic liver diseases including cirrhosis, rather than acute hepatic injury.28
Prothrombin Time
As a part of the coagulation cascade, prothrombin is converted to thrombin utilizing the liver synthesized coagulation factors II, V, VII, and X. The prothrombin time measures this rate of conversion and becomes elongated in states of hepatic dysfunction when production of these coagulation factors is reduced. Since the half-life of these coagulation factors is relatively short, the prothrombin time can be a useful tool in measuring more acute changes in hepatic synthetic dysfunction and has been shown to have predictive capacity in determining the prognosis in fulminant hepatic failure.29 Caution is advised in interpreting prothrombin time results as a marker of hepatic dysfunction because elongation may also be seen in disseminated intravascular coagulation and vitamin K deficiency. In these cases, supplementation of vitamin K and measurement of individual coagulation factors can help differentiate these conditions from liver disease. The international normalized ratio (INR) is a standardized measurement of the prothrombin time that is often used as a marker of liver dysfunction. The INR is a key component of the Model of End-Stage Liver Disease (MELD) score which stratifies 3-month mortality risk in patients with cirrhosis and is currently used to prioritize patients on the liver transplant list.30
When to Refer to a Specialist
There are no evidence-based guidelines that specify criteria for referral to a gastroenterologist or a hepatologist. It is generally accepted that a referral should be considered when liver test abnormalities are unexplained and are persistently more than 1.5 to 2 times the upper limit of normal in more than one occasion. In addition, otherwise unexplained abnormalities in markers of synthetic function (such as albumin and prothrombin time) should also lead to consideration of referral. Finally, many practitioners choose to refer to a specialist when a liver biopsy may be needed. Liver biopsy can be helpful to obtain a diagnosis when serologic and radiographic evaluation is unrevealing as well as staging and prognosis when a diagnosis has already been made (Table 5).31
Table 5. Conditions that may require liver biopsy for diagnosis, staging, or prognosis31 .
| Abnormal liver tests of unknown etiology |
| Hepatitis B |
| Hepatitis C |
| Hemochromatosis |
| Wilson disease |
| Alpha-1-antitrypsin |
| Autoimmune hepatitis |
| Alcoholic liver disease |
| NAFLD/NASH |
| Infiltrative liver disease |
| Drug-induced liver disease |
| Acute liver failure |
| Post–liver transplantation |
| Primary biliary cirrhosis |
Abbreviations: NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Conclusion
Clinical signs of jaundice occur when serum bilirubin levels exceed 2.5 to 3.0 mg/dL. This finding itself is nonspecific and can represent disorders of bilirubin metabolisms, biliary obstruction, or hepatocellular dysfunction. Evaluation for cholestasis, characterized by an elevated alkaline phosphatase, is a key step in the differentiation of these disorders. Utilization of a standardized approach to patients with hyperbilirubinemia will minimize unnecessary testing and improve patient outcomes.
Authors' Contribution
Bilal Gondal, MD, MRCSI, is responsible for the analysis of data, preparation of the initial draft of the manuscript with electronic file, and literature review. He is also responsible as the corresponding author. Andrew Aronsohn, MD, is responsible for the analysis of data, collection of data, and the preparation of initial and final draft. He is also responsible for overall supervision of publication and final review of the article.
References
- 1.Siddique A, Kowdley K V. Approach to a patient with elevated serum alkaline phosphatase. Clin Liver Dis. 2012;16(2):199–229. doi: 10.1016/j.cld.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukherjee S, Gollan J L. Hoboken, NJ: Wiley-Blackwell; 2011. Assessment of liver function; pp. 20–35. [Google Scholar]
- 3.Green R M, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123(4):1367–1384. doi: 10.1053/gast.2002.36061. [DOI] [PubMed] [Google Scholar]
- 4.Woreta T A, Alqahtani S A. Evaluation of abnormal liver tests. Med Clin North Am. 2014;98(1):1–16. doi: 10.1016/j.mcna.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Franklin Herlon F H, Mitchell M C. Hoboken, NJ: Wiley Blackwell; 2012. Laboratory tests; pp. 17–43. [Google Scholar]
- 6.Manolio T A, Burke G L, Savage P J. et al. Sex- and race-related differences in liver-associated serum chemistry tests in young adults in the CARDIA study. Clin Chem. 1992;38(9):1853–1859. [PubMed] [Google Scholar]
- 7.Gordon T. Factors associated with serum alkaline phosphatase level. Arch Pathol Lab Med. 1993;117(2):187–190. [PubMed] [Google Scholar]
- 8.Goldberg D M. 5'Nucleotidase: recent advances in cell biology, methodology and clinical significance. Digestion. 1973;8(1):87–99. doi: 10.1159/000197303. [DOI] [PubMed] [Google Scholar]
- 9.Pagani F, Panteghini M. 5′-Nucleotidase in the detection of increased activity of the liver form of alkaline phosphatase in serum. Clin Chem. 2001;47(11):2046–2048. [PubMed] [Google Scholar]
- 10.Sotil E U Jensen D M Serum enzymes associated with cholestasis Clin Liver Dis 20048141–54., vi [DOI] [PubMed] [Google Scholar]
- 11.Goldberg D M. Structural, functional, and clinical aspects of gamma-glutamyltransferase. CRC Crit Rev Clin Lab Sci. 1980;12(1):1–58. doi: 10.3109/10408368009108725. [DOI] [PubMed] [Google Scholar]
- 12.Rosalki S B. Gamma-glutamyl transpeptidase. Adv Clin Chem. 1975;17:53–107. doi: 10.1016/s0065-2423(08)60248-6. [DOI] [PubMed] [Google Scholar]
- 13.Reichling J J, Kaplan M M. Clinical use of serum enzymes in liver disease. Dig Dis Sci. 1988;33(12):1601–1614. doi: 10.1007/BF01535953. [DOI] [PubMed] [Google Scholar]
- 14.Connell M D, Dinwoodie A J. Diagnostic use of serum alkaline phosphatase isoenzymes and 5-nucleotidase. Clin Chim Acta. 1970;30(2):235–241. doi: 10.1016/0009-8981(70)90108-7. [DOI] [PubMed] [Google Scholar]
- 15.Rogoveanu I, Gheonea D I, Saftoiu A, Ciurea T. The role of imaging methods in identifying the causes of extrahepatic cholestasis. J Gastrointestin Liver Dis. 2006;15(3):265–271. [PubMed] [Google Scholar]
- 16.Gossard A A, Talwalkar J A. Cholestatic liver disease. Med Clin North Am. 2014;98(1):73–85. doi: 10.1016/j.mcna.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Reddy S I Grace N D Liver imaging. A hepatologist's perspective Clin Liver Dis 200261297–310., ix [DOI] [PubMed] [Google Scholar]
- 18.Romagnuolo J, Bardou M, Rahme E, Joseph L, Reinhold C, Barkun A N. Magnetic resonance cholangiopancreatography: a meta-analysis of test performance in suspected biliary disease. Ann Intern Med. 2003;139(7):547–557. doi: 10.7326/0003-4819-139-7-200310070-00006. [DOI] [PubMed] [Google Scholar]
- 19.Longo M, Crosignani A, Podda M. Hyperlipidemia in chronic cholestatic liver disease. Curr Treat Options Gastroenterol. 2001;4(2):111–114. doi: 10.1007/s11938-001-0022-6. [DOI] [PubMed] [Google Scholar]
- 20.Sokol R J. Fat-soluble vitamins and their importance in patients with cholestatic liver diseases. Gastroenterol Clin North Am. 1994;23(4):673–705. [PubMed] [Google Scholar]
- 21.Lester R, Schmid R. Bilirubin metabolism. N Engl J Med. 1964;270:779–786. doi: 10.1056/NEJM196404092701507. [DOI] [PubMed] [Google Scholar]
- 22.Weiss J S, Gautam A, Lauff J J. et al. The clinical importance of a protein-bound fraction of serum bilirubin in patients with hyperbilirubinemia. N Engl J Med. 1983;309(3):147–150. doi: 10.1056/NEJM198307213090305. [DOI] [PubMed] [Google Scholar]
- 23.Longo D, Fauci A S. New York: McGraw-Hill Medical; 2010. Harrison's Gastroenterology and Hepatology; p. 738. [Google Scholar]
- 24.Molina E G, Reddy K R. Postoperative jaundice. Clin Liver Dis. 1999;3(3):477–488. doi: 10.1016/s1089-3261(05)70081-7. [DOI] [PubMed] [Google Scholar]
- 25.Erlinger S, Arias I M, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146(7):1625–1638. doi: 10.1053/j.gastro.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 26.Monaghan G, Ryan M, Seddon R, Hume R, Burchell B. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet. 1996;347(9001):578–581. doi: 10.1016/s0140-6736(96)91273-8. [DOI] [PubMed] [Google Scholar]
- 27.Rothschild M A, Oratz M, Schreiber S S. Serum albumin. Hepatology. 1988;8(2):385–401. doi: 10.1002/hep.1840080234. [DOI] [PubMed] [Google Scholar]
- 28.Rothschild M A, Oratz M, Zimmon D, Schreiber S S, Weiner I, Van Caneghem A. Albumin synthesis in cirrhotic subjects with ascites studied with carbonate-14C. J Clin Invest. 1969;48(2):344–350. doi: 10.1172/JCI105990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Grady J G, Alexander G J, Hayllar K M, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439–445. doi: 10.1016/0016-5085(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 30.Wiesner R, Edwards E, Freeman R. et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;124(1):91–96. doi: 10.1053/gast.2003.50016. [DOI] [PubMed] [Google Scholar]
- 31.Rockey D C Caldwell S H Goodman Z D Nelson R C Smith A D; American Association for the Study of Liver Diseases. Liver biopsy Hepatology 20094931017–1044. [DOI] [PubMed] [Google Scholar]