

Abstract
Key points
The inheritance of two defective alleles of SLC4A4, the gene that encodes the widely‐expressed electrogenic sodium bicarbonate cotransporter NBCe1, results in the bicarbonate‐wasting disease proximal renal tubular acidosis (pRTA).
In the present study, we report the first case of compound‐heterozygous inheritance of pRTA (p.Arg510His/p.Gln913Arg) in an individual with low blood pH, blindness and neurological signs that resemble transient ischaemic attacks.
We employ fluorescence microscopy on non‐polarized (human embryonic kidney) and polarized (Madin–Darby canine kidney) renal cell lines and electrophysiology on Xenopus oocytes to characterize the mutant transporters (R510H and Q913R).
Both mutant transporters exhibit enhanced intracellular retention in renal cells, an observation that probably explains the HCO3 − transport deficit in the individual.
Both mutants retain a close‐to‐normal per molecule Na+/HCO3 − cotransport activity in Xenopus oocytes, suggesting that they are suitable candidates for folding‐correction therapy. However, Q913R expression is uniquely associated with a depolarizing, HCO3 − independent, Cl−‐conductance in oocytes that could have pathological consequences if expressed in the cells of patients.
Abstract
Proximal renal tubular acidosis (pRTA) is a rare, recessively‐inherited disease characterized by abnormally acidic blood, blindness, as well as below average height and weight. pRTA is typically associated with homozygous mutation of the solute carrier 4 family gene SLC4A4. SLC4A4 encodes the electrogenic sodium bicarbonate cotransporter NBCe1, a membrane protein that acts to maintain intracellular and plasma pH. We present the first description of a case of compound‐heterozygous inheritance of pRTA. The individual has inherited two mutations in NBCe1: p.Arg510His (R510H) and p.Gln913Arg (Q913R), one from each parent. In addition to the usual features of pRTA, the patient exhibits unusual signs, such as muscle spasms and fever. We have recreated these mutant transporters for expression in model systems. We find that both of the mutant proteins exhibit substantial intracellular retention when expressed in mammalian renal cell lines. When expressed in Xenopus oocytes, we find that the R510H and Q913R‐mutant NBCe1 molecules exhibit apparently normal Na+/HCO3 − cotransport activity but that Q913R is associated with an unusual HCO3 − independent anion‐leak. We conclude that a reduced accumulation of NBCe1 protein in the basolateral membrane of proximal‐tubule epithelia is the most probable cause of pRTA in this case. We further note that the Q913R‐associated anion‐leak could itself be pathogenic if expressed in the plasma membrane of mammalian cells, compromising the benefit of strategies aiming to enhance mutant NBCe1 accumulation in the plasma membrane.
Keywords: acid‐base, epithelial transport, pH
Key points
The inheritance of two defective alleles of SLC4A4, the gene that encodes the widely‐expressed electrogenic sodium bicarbonate cotransporter NBCe1, results in the bicarbonate‐wasting disease proximal renal tubular acidosis (pRTA).
In the present study, we report the first case of compound‐heterozygous inheritance of pRTA (p.Arg510His/p.Gln913Arg) in an individual with low blood pH, blindness and neurological signs that resemble transient ischaemic attacks.
We employ fluorescence microscopy on non‐polarized (human embryonic kidney) and polarized (Madin–Darby canine kidney) renal cell lines and electrophysiology on Xenopus oocytes to characterize the mutant transporters (R510H and Q913R).
Both mutant transporters exhibit enhanced intracellular retention in renal cells, an observation that probably explains the HCO3 − transport deficit in the individual.
Both mutants retain a close‐to‐normal per molecule Na+/HCO3 − cotransport activity in Xenopus oocytes, suggesting that they are suitable candidates for folding‐correction therapy. However, Q913R expression is uniquely associated with a depolarizing, HCO3 − independent, Cl−‐conductance in oocytes that could have pathological consequences if expressed in the cells of patients.
Abbreviations
- AE
anion exchanger
- DIDS
4,4′‐diisothiocyano‐2,2′‐stilbenedisulphonic acid
- ECl
equilibrium potential for Cl−
- EGFP
enhanced green fluorescent protein
- ENa
equilibrium potential for Na+
- Erev
reversal potential
- Gm
membrane conductance
- HEK
human embryonic kidney
- MDCK
Madin–Darby canine kidney
- NBCe1
sodium bicarbonate cotransporter (electrogenic) 1
- NMDG
N‐methyl‐d‐glucamine
- pRTA
proximal renal tubular acidosis
- PT
proximal tubule
- SLC4
solute carrier family 4
- Vm
membrane potential
- WT
wild‐type
- ZO‐1
zona occludens protein 1
Introduction
Carbon dioxide and bicarbonate are the main physiological buffers for the daily load of dietary and metabolic acids and serve to maintain plasma pH within a narrow range (7.35–7.45), preserving normal physiological functions.
To maintain the pH‐buffering capacity of plasma, plasma [CO2] is maintained at ∼1. 2 mm by equilibration with CO2 in the airspaces of the lungs whereas plasma [HCO3 −] is maintained at 22–24 mm, mainly by the action of epithelial cells in the renal proximal tubule (PT) (Boron, 2012; Giebisch & Windhager, 2012). These cells generate new HCO3 −, to replace that lost to acid titration, and reabsorb HCO3 − from the glomerular filtrate to prevent its loss to the urine. The renal NBCe1 variant NBCe1‐A (encoded by the solute carrier 4 family gene SLC4A4) is expressed in the basolateral membrane of PT epithelia, and comprises the main pathway by which new and filtered HCO3 − exit PT epithelia for the interstitial fluid and hence the bloodstream (Boron & Boulpaep, 1983; Romero et al. 1997). It has been calculated that NBCe1‐A must operate with a cotransport stoichiometry of 1 Na+ to 3 HCO3 − to support HCO3 − efflux, given the predicted prevailing electrochemical gradients across the PT cell basolateral membrane of the PT cell (Yoshitomi et al. 1985; Sasaki et al. 1987). However, most measurements of the action of NBCe1‐A in model systems disclose a stoichiometry of 1 Na+ to 2 HCO3 − (or 1 CO3 2−) equivalents (Seki et al. 1993; Heyer et al. 1999; Sciortino & Romero, 1999; Grichtchenko & Boron, 2002; Lee et al. 2011; Zhu et al. 2013).
Defective HCO3 − handing by the PT results in HCO3 − wasting in the urine, as well as drastically lowered plasma [HCO3 −] and pH. These are characteristic signs of proximal renal tubular acidosis (pRTA) (Alper, 2010). Patients with genetic defects in NBCe1 exhibit classic signs of pRTA (plasma [HCO3 −] = 5–15 mm, plasma pH 7.1–7.2) often accompanied by developmental issues, such as short stature and mental retardation, as well as ocular defects, such as band keratopathy, cataracts and glaucoma (Igarashi et al. 1999; Seki et al. 2013). Non‐renal SLC4A4 gene products (NBCe1‐B to NBCe1‐E) are expressed throughout the body (e.g. neurons, astrocytes, secretory epithelia, corneal endothelia, lens epithelia, myocytes) and play direct and critical roles in support of processes such as neuronal excitability, intestinal fluid secretion and the maintenance of vision (Choi et al. 1999; Bevensee et al. 2000; Bok et al. 2001; Li et al. 2005 b; Liu et al. 2011). Metabolic acidosis, developmental issues and glaucoma appear to be specific consequences of renal NBCe1 dysfunction because they are exhibited by an individual with an NBCe1‐A‐specific pRTA mutation (Igarashi et al. 2001). Global slc4a4‐null mice exhibit pRTA and do not thrive well, dying shortly after weaning with impacted intestines, although HCO3 − administration can prolong their lifespan by several months (Gawenis et al. 2007; Lo et al. 2011; Fang et al. 2015). A strain of mouse that specifically lacks renal NBCe1‐A exhibits metabolic acidosis but otherwise appears to thrive normally (Chen et al. 2014; Handlogten et al. 2015).
Thirteen SLC4A4 mutations have been described in individuals with pRTA. In each case, the inheritance of pRTA is recessive. Affected individuals are homozygous for each mutation and the usually consanguineous parents are heterozygous carriers that do not exhibit signs of pRTA. These NBCe1‐A mutations (reported in the context of GenBank Accession NP_0037570) fall into two groups: missense and nonsense. Missense mutations make up the greatest proportion and include p.Arg298Ser (Igarashi et al. 1999, 2002), p.Ser427Leu (Dinour et al. 2004), p.Thr485Ser (Horita et al. 2005), p.Gly486Arg (Suzuki et al. 2008), p.Arg510His (Igarashi et al. 1999; Shiohara et al. 2000; Suzuki et al. 2010), p.Leu522Pro (Demirci et al. 2006), p.Ala799Val (Horita et al. 2005) and p.Arg881Cys (Horita et al. 2005; Suzuki et al. 2010). The remainder include two nonsense mutations, p.Gln29X (Igarashi et al. 2001) and p.Trp516X (Lo et al. 2011); two frame‐shift mutations, p.Asn721ThrfsX30 (Inatomi et al. 2004) and p.Ser982AsnfsX4 (Suzuki et al. 2010); and one deletion mutation, p.Leu738del (Kari et al. 2014).
We report a novel case of compound‐heterozygous inheritance of pRTA. One of the patient's alleles encodes the previously reported p.Arg510His (R510H) mutation, whereas the other encodes the novel missense mutation p.Gln913Arg (Q913R). In addition to describing the clinical features of the patient, we use heterologous systems to examine the nature of the molecular defects that underlie the disease using a combination of fluorescence microscopy, electrophysiology, western blotting and a 36Cl‐accumulation assay.
Methods
Ethical approval
Written informed consent of agreement on biochemical and genetic analysis and publication were obtained from the patient and his parents, in compliance with the principles of the Declaration of Helsinki. The present study was approved by the ethics committee of Peking Union Medical College Hospital. The extraction of ovaries from Xenopus laevis frogs was performed in accordance with the rules and recommendations of the Institutional Animal Care and Use Committee (IACUC) at the University at Buffalo.
cDNA clones
NBCe1‐A.pcDNA3.1 was a gift from Dr Ashley M. Toye (University of Bristol, Bristol, UK).
NBCe1‐A‐EGFP.pGH19 was a gift from Dr Walter F. Boron (Case Western Reserve University, Cleveland, OH, USA).
NBCe1‐A‐EGFP.pcDNA3.1 was generated from NBCe1‐A.pcDNA3.1 using the following procedures. (1) An EcoRI site was introduced immediately upstream of the NBCe1‐A termination codon using a QuickChange mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) in accordance with the manufacturer's instructions using the primer 5′‐CGCCACACATCATGCGAATTCTGAAAGGGCAATTCTG‐3′ and its reverse complement (the introduced EcoRI site is underlined). (2) An enhanced green fluorescent protein (EGFP) open reading frame was amplified from NBCe1‐A‐EGFP.pGH19 using the forward primer 5′‐CGAAGGAATTCTCACCGGTCGCCACCATG‐3′ (which includes a non‐complementary 5′ EcoR1 site, underlined) and the reverse primer 5′‐CGAAGCTCGAGCTACTTGTACAGCTCGTCC‐3′ (which includes a non‐complementary 3′ XhoI site). (3) The vector construct and EGFP insert were both restricted with EcoRI and XhoI (in the vector construct the XhoI site is located 38 bp downstream of the NBCe1‐A stop‐codon) and the EGFP insert was ligated into the vector construct to produce NBCe1‐A‐EGFP.pcDNA3.1.
R510H and Q913R mutations were introduced to NBCe1‐A‐EGFP.pcDNA3.1 and NBCe1‐A‐EGFP.pGH19 using a QuikChange site‐directed mutagenesis kit (Agilent) and the primer pairs 5′‐GACTATTTGGAGTTTCACCTTTGGATTGGCC‐3′ and its reverse complement (for R510H, base change underlined) or 5′‐GTTCACTTTCCTGCGGGTGTTGTGTCTGGC‐3′ and its reverse complement (for Q913R).
Synthesis of oligonucleotide primers and sequencing of cDNA clones was performed by Eurofins Genomics (Huntsville, AL, USA).
Mammalian cell culture and transfection
Human embryonic kidney (HEK)293 cells (passage number 6–10) were maintained in Dulbecco's modified Eagle's medium/F‐12 (Invitrogen, Carlsbad, CA, USA) with 10 % fetal bovine serum, 100 units mL–1 of penicillin and 100 μg mL−1 streptomycin. Cells were grown at 37°C in a humidified atmosphere with 5% CO2/95% air. Cells were maintained in T‐75 flasks and liberated using 0.25% trypsin‐EDTA prior to seeding at 4 × 104 cells per well in CELLview 35‐mm glass bottom culture dishes (VWR, Radnor, PA, USA). Cells were seeded 2 days prior to transfection. NBCe1‐A‐EGFP.pcDNA3.1 constructs were transfected into HEK293 cells (at ∼70% confluence) using Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer's instructions.
We chose Madin–Darby canine kidney (MDCK)‐II cells (passage number 2–4; Sigma‐Aldrich, St Louis, MO, USA) as our model of a polarized renal epithelial cell (Dukes et al. 2011). MDCK‐II cells were maintained in minimal essential medium with 5% fetal bovine serum but without antibiotics. Cells were grown at 37°C in a humidified atmosphere with 5% CO2/95% air. Cells were maintained in T‐75 flasks and liberated using 0.05% trypsin‐EDTA (Thermo Fisher Scientific, Waltham, MA, USA) prior to seeding at 1 × 105 cells per well in Lab‐Tek® II CC2™ four‐chambered slides (Electron Microscopy Sciences, Hatfield, PA, USA). One day after seeding (at ∼80–90% confluence), cells were bathed for 30 min in 2 mm EGTA at 37°C (Zuhorn et al. 2007) and NBCe1‐A‐EGFP.pcDNA3.1 constructs were transfected into the cells using Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer's instructions. Cells were processed for microscopy after 48 h.
Fluorescence microscopy
Live HEK293 cells were washed twice with warm Hank's buffered salt solution (pH 7.4) and stained with 5 ng mL−1 red‐fluorescent AlexaFluor®594 wheat germ agglutinin (Invitrogen) in Hank's buffered salt solution for 10 min at 37°C. Cells were washed twice with Buffer A (in mm: 138 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4, 1 Na2HPO4, 5 d‐glucose and 20 Hepes, pH 7.4) and 250 μl of buffer was added to each well for imaging. Images were acquired with an Axiovert 200 fluorescence microscope equipped with an Axiocam MRC camera (Carl Zeiss, Jena, Germany).
MDCK‐II cell monolayers were washed in PBS and fixed and permeabilized with a 5 min incubation in 60% methanol/40% acetone mixture at −20°C. The monolayers were washed for 2 × 5 min with PBS, blocked for 15 min with PBS that included 4% BSA, and washed for 3 × 5 min in PBS. For EGFP/zona occludens protein 1 (ZO‐1) co‐immunolocalization, a rabbit anti‐GFP‐tag antibody #A‐11122 (Thermo Fisher Scientific) was applied at a dilution of 1:100, together with an Alexa594‐conjugated mouse‐anti‐ZO‐1 antibody ‘ZO1‐1A12’ #3339194 (Thermo Fisher Scientific) at a dilution of 1:1000, in PBS + 4% BSA for 1 h, followed by 3 × 5 min washes in PBS. An Alexa488‐conjugated goat‐anti‐rabbit secondary antibody #A‐11034 (Thermo Fisher Scientific) was applied at a dilution of 1:500 in PBS + 4% BSA for 1 h, followed by 3 × 5 min washes in PBS. Finally, the monolayer was mounted beneath a coverslip using Vectashield medium (Vector Laboratories, Burlingame, CA, USA). For Na,K‐ATPase immunolocalization, a mouse anti‐Na,K‐ATPase antibody C464.6 (EMD Millipore, Billerica, MA, USA) was used at a dilution of 1:1000 followed by an Alexa594‐conjugated goat‐anti‐mouse secondary antibody #A‐11032 (Thermo Fisher Scientific) used at a dilution of 1:200. Images were acquired with an Axio Imager 2 fluorescence microscope equipped with an AxioCam MRm camera and an Apotime.2 with Zen 2 Pro software to allow optical sectioning (Carl Zeiss). Optical slices were 0.24 μm apart and acquisition parameters were identical for all images.
Preparation of oocytes
De‐folliculated oocytes were isolated from tricaine‐anaesthetized X. laevis frogs (Xenopus Express, Brooksville, FL, USA) as described previously (Musa‐Aziz et al. 2010). In brief, frogs were anaesthetized by immersion in a 0.2% tricaine solution for 15 min (or until unresponsive to toe pinch) and ovaries were surgically extracted. The frog was subsequently terminated by exsanguination. The extracted ovaries were cut into small pieces into a Ca2+‐free wash buffer (in mm: 82 NaCl, 2 KCl, 20 MgCl2 and 5 Hepes, adjusted to pH 7.45 with NaOH). After three washes in this solution, the ovary pieces were transferred into the Ca2+‐free buffer containing 2 mg mL−1 type‐IA collagenase (Sigma‐Aldrich) and digested for 30 min. Two more washes in Ca2+‐free buffer followed, and a second collagenase treatment was performed, until individual oocytes were liberated and judged by light microscopy to be free of their follicular membranes. The oocytes were rinsed in Ca2+‐free NRS twice more, then once in our ‘ND96’ solution (see below), and finally resuspended and maintained in OR3 medium at 18°C. OR3 medium contains 14 g L−1 of Leibovitz's L‐15 medium (Thermo Fisher Scientific), 100 units mL–1 penicillin, 100 μg mL−1 streptomycin and 5 mm Hepes, adjusted to pH 7.5 using NaOH, and then adjusted to 195 ± 5 mosmol kg−1 osmolality with H2O. Osmolality was measured using a Vapro vapor pressure osmometer (Wescor, Logan, UT, USA).
NBCe1‐A expression in oocytes
cRNA was generated from NotI linearized NBCe1‐A‐EGFP.pGH19 cDNA using a T7 mMessage mMachine transcription kit (Life Technologies, Grand Island, NY, USA) and purified using an RNeasy MinElute cleanup kit (Qiagen, Valencia, CA, USA). Except where specifically stated otherwise, de‐folliculated oocytes were injected with 25 nl of 1000 ng nl−1 cRNA or 25 nl of H2O. Injected oocytes were maintained in OR3 medium for 2–5 days prior to experimentation.
Biotinylation and western blotting
Groups of 12 oocytes were subjected to a biotinylation protocol using the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific) in accordance with the manufacturer's instructions, with the exception that solutions applied to intact cells were diluted to 195 ± 5 mosmol kg−1. Total and biotinylated protein extracts were resolved on 3–8% Tris‐acetate protein gels (Thermo Fisher Scientific) and transferred onto polyvinylidene fluoride membranes using an iBlot™ dry blotting system (Invitrogen). EGFP‐immunoreactivity was detected using the JL‐8 anti‐EGFP monoclonal antibody (Clontech Laboratories, Mountain View, CA, USA), a horseradish peroxidase‐conjugated goat anti‐mouse secondary antibody (#55563; MP Biomedicals, Solon, OH, USA) and Pierce ECL 2 western‐blotting substrate (Thermo Fisher Scientific). Images were acquired using a Pierce MyECL™ imager (Thermo Fisher Scientific).
Electrophysiology
Oocytes were placed in an oocyte recording chamber (#RC‐3Z; Warner Instruments, Hamden, CT, USA) and impaled with sharp microelectrodes that had been pulled from Clark borosilicate capillary glass (#GC200TF‐10; Warner Instruments) using a P‐1000 micropipette puller (Sutter Instrument, Novato, CA, USA) and filled with saturated KCl (#SP138‐500; Thermo Fisher Scientific). The microelectrode half‐cells were connected to an OC‐725C oocyte clamp (Warner Instruments) and the oocytes were subjected to voltage clamp protocols as the composition of the superfusing solution was altered. Following each solution change, under the control of the pClamp, version 10.4 (Molecular Devices, Sunnyvale, CA, USA), the oocyte potential was clamped to its spontaneous membrane potential (V m) prior to initiating the protocol in which the potential was stepped to −160 mV for 100 ms, returned to spontaneous V m for 100 ms and then stepped to −140 mV, and so on, at +20 mV intervals. Solution changes were effected using a six channel perfusion pinch‐valve control system (#VC‐6; Warner Instruments). Oocytes were removed from clamp immediately prior to any solution change and were reclamped 1 min after solution change. If the incoming solution included 4,4′‐diisothiocyano‐2,2′‐stilbenedisulphonic acid (DIDS), tenidap or niflumic acid, this period was extended to 2 min to allow each drug sufficient time to exert its action.
Electrophysiology solutions
The ‘ND96’+Na/0HCO3 solution contained (in mm): 93.5 NaCl, 2 KCl, 1.8 MgCl2, 1 CaCl2 and 5 Hepes, pH 7.5 with fresh NaOH solution.
The +Na/+HCO3 solution contained (in mm): 60.5 NaCl, 33 NaHCO3, 2 KCl, 1.8 MgCl2, 1 CaCl2 and 5 Hepes. Note that the NaCl, KCl, and Hepes are added first and the solution is adjusted to pH 7.5 with fresh NaOH solution. Next, the NaHCO3 is added and the pH is adjusted back to pH 7.5 by sparging with 5% CO2/21% O2/balance N2 generated from component gases using a Series 4000 gas mixing system (Environics, Tolland, CT, USA). The CaCl2 and MgCl2 were added last.
The 0Na/+HCO3 solution contains (in mm): 60.5 N‐methyl‐d‐glucamine (NMDG).Cl, 33 NMDG.HCO3, 2 KCl, 1.8 MgCl2, 1 CaCl2 and 5 Hepes. Note that 60.5 NMDG base is added first and the pH adjusted to 7.5 using HCl to generate NMDG.Cl. KCl and Hepes are added next and the solution is adjusted to pH 7.5 with NMDG solution. Next, 33 more NMDG base is added and the pH is adjusted back to 7.5 by sparging with 5% CO2/21% O2/balance N2. The CaCl2 and MgCl2 are added last.
In Cl‐replacement protocols, gluconate salts substitute for Cl− salts and we doubled the concentration of Mg2+ and Ca2+ salts to compensate for divalent‐cation chelation. DIDS (Sigma‐Aldrich) was dissolved directly into the working solution, used on the same day as preparation, and stored in a foil covered glass bottle protecting it from light. Niflumic acid and tenidap were prepared as 50 mm stock solutions in DMSO and diluted (1:500) to a working concentration in ND96 solution that was also protected from light using foil. For these experiments only, our ND96 solution without niflumic acid or tenidap also included DMSO at a dilution of 1:500 (v/v).
36Cl influx assays
In total, 8 μL of 16 mCi/gCl Na36Cl (American Radiolabeled Chemicals, St. Louis, MO, USA) was added to 500 μL of ND96 solution. Groups of 30 oocytes were bathed in this 36Cl solution for 2 h, washed in 2 × 50 mL ND96, and separated into batches of five cells in Eppendorf tubes. Cells were triturated in 100 μL of distilled water prior to the addition of 100 μL of 10% SDS. The solubilized oocyte mixture was added to 5 mL of Ecoscint cocktail (National Diagnostics, Atlanta, GA, USA) in 20 mL scintillation vials. Each vial was counted for 10 min using a Wallac 1409 liquid scintillation counter (Perkin Elmer, Waltham, MA, USA).
Data analysis
DNA Electropherograms were prepared using SeqMan Pro, version 11.2.1 (DNASTAR Inc., Madison, WI, USA). Western blot densitometry was performed using ImageJ, version 1.48 (NIH, Bethesda, MD, USA), merged images and z‐axis projections of polarized MDCK‐II cells were generated using ImageJ, version 1.50e (Schneider et al. 2012), and the brightness of images was increased, as indicated, using Powerpoint (Microsoft, Redmond, WA, USA). Pearson's coefficients for co‐localization were calculated using the ‘Just Another Localization Plugin’ (JAcoP) for ImageJ. Briefly, for each cell, a 4–12 μm2 area of interest was selected around a section of lateral membrane (marked by Na,K‐ATPase immunofluorescence, resolution = 95 pixels/μm2) in the x–y‐plane and the co‐incidence of NBCe1‐A‐EGFP immunofluorescence was reported for each pixel in that area through the entire z‐stack. Current–voltage relationships were generated from voltage clamp data using Clampfit, version 9.0 (Molecular Devices).
Statistical analysis
Statistical analyses were performed using Minitab, version 17 (Minitab Inc., State College, PA, USA), with the exception of t tests, which were performed using Excel 2013 (Microsoft Corp.). Data were analysed either by one‐way ANOVA (multiple comparisons), a general linear model (multiple comparisons with multiple factors) or by a t test (select comparisons) as indicated. P < 0.05 was considered statistically significant. When paired data were analysed by a t test, the P value was determined by Bonferroni correction for the number of groups being subjected to the same comparison to control for false‐positive results (e.g. when three sets of t tests were performed, P = 0.05/3 = 0.017).
Results
Description of patient
The patient is a male of Han Chinese descent, with normal stature, blood pH and HCO3 − levels; he is the only child of parents who do not identify as being consanguineous within at least three generations. He was diagnosed with pRTA (serum [HCO3 −] = 11 mm, pH 7.27) without hypokalaemia at the age of 5 years. He has subsequently received intermittent alkali‐therapy. At the age of 6 years, he was diagnosed with bilateral glaucoma, cataracts and band keratopathy. The patient presented again at age 25 years; at which stage he was completely blind (Fig. 1 A) with short stature (>3 SD below mean), pRTA (without regular alkali treatment, serum [HCO3 −] = 13 mm, pH 7.26) and enamel hypoplasia (Fig. 1 B). Computed tomography scans of the patient's head over the last three consecutive years have revealed a persistent bilateral calcification in the basal ganglia, which appears most prominently in the left hemisphere (Fig. 1 C).The patient has had numerous episodes that resemble transient ischaemic attacks, the signs of which include aphasia, delirium, dyskinesia, hemianaesthesia, hypermyotonia and fever. Neither the patient, nor his parents report a history of migraine.
Figure 1. Features of the patient .
A, the patient is blind and exhibits bilateral band keratopathy, cataracts and glaucoma. B, the patient also exhibits signs of enamel hypoplasia and poor teeth alignment. C, computed tomography scan of the patient's head; arrows indicate the location of calcifications. D–I, electropherograms showing the sequence of genomic DNA (prepared from peripheral blood samples) in the vicinity of the mutations for the patient and his parents. The predicted amino acid translation is shown beneath each trace, with mutations indicated in red. J, pedigree of the compound‐heterozygous patient. K, cartoon depicting the predicted topological location of the R510H and Q913R mutations in NBCe1. Note that, in the patient, the mutations are carried on separate SLC4A4 alleles.
Mutation screening
Sequencing across the SLC4A4 gene locus of the patient revealed two heterozygous mutations. The first, in exon 13 (c.1529G>A: GenBank Nucleotide Accession # AF007216) (Fig. 1 D), is predicted to produce a p.Arg510His substitution (R510H) in NBCe1‐A (GenBank Protein Accession # AAC51645). This mutation has been described previously in homozygous form in individuals with pRTA (Igarashi et al. 1999; Shiohara et al. 2000; Suzuki et al. 2010). The second, in exon 21 (c.2738A>G) (Fig. 1 E), is predicted to produce a novel p.Gln913Arg substitution (Q913R). Neither of these nucleotide substitutions were detected in the genomes of 100 unrelated, healthy Chinese individuals. The patient's mother is heterozygous for the R510H mutation (Fig. 1 F) but does not carry the Q913R mutation (Fig. 1 G). The patient's father does not carry the R510H mutation (Fig. 1 H) but is heterozygous for the Q913R mutation (Fig. 1 I). The pedigree of the compound‐heterozygous patient is shown in Fig. 1(J) and the predicted topological location of these mutations within NBCe1‐A is mapped onto the cartoon shown in Fig. 1(K).
Expression of NBCe1‐A constructs in renal epithelial cell lines
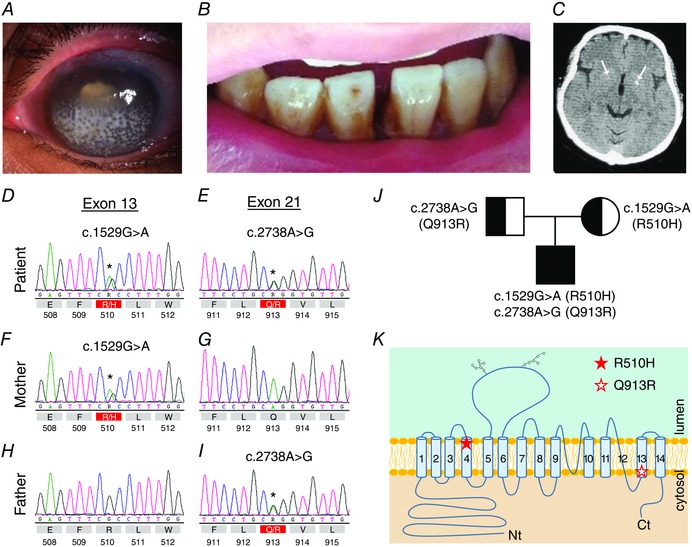
We transfected HEK293 cells with cDNAs encoding wild‐type (WT)‐EGFP, R510H‐EGFP or Q913R‐EGFP (collectively ‘NBCe1‐A‐EGFP constructs’). R510H‐EGFP has already been shown to exhibit enhanced intracellular retention in renal cells and serves as our negative control (Li et al. 2005 a; Suzuki et al. 2010). Figure 2 shows the EGFP fluorescence as a result of NBCe1‐A‐EGFP construct expression in living HEK293 cells in which the location of the plasma membrane is labelled with AlexaFluor®594 wheat germ agglutinin (red in Fig. 2). Cells expressing WT‐EGFP exhibit enhanced EGFP fluorescence that is especially intense at the cell perimeter, consistent with plasma membrane localization (Fig. 2 A). On the other hand, cells expressing R510H‐EGFP (Fig. 2 B) or Q913R‐EGFP (Fig. 2 C) exhibit enhanced EGFP fluorescence throughout.
Figure 2. The distribution of NBCe1‐A‐EGFP in living HEK293 cells .
A, left: EGFP fluorescence (green) discloses the location of WT‐EGFP in HEK cells; centre: Alexa‐594‐tagged‐wheat‐germ‐agglutinin fluorescence (red) outlines the cell perimeter; right: overlay of WT‐EGFP and WGA‐594 fluorescence showing WT‐EGFP enriched at cell perimeters. B, overlay of R510H‐EGFP and WGA‐594 fluorescence showing R510H‐EGFP distributed throughout the cytoplasm. C, overlay of Q913R‐EGFP and WGA‐594 fluorescence showing Q913R‐EGFP distributed throughout the cytoplasm. Images in (A) to (C) are representative of the distribution of NBCe1‐A as observed in three replicate transfections. The brightness of all images in (A) to (C) has been increased by 20% for clarity.
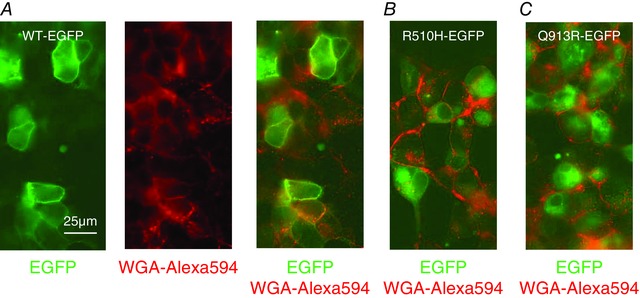
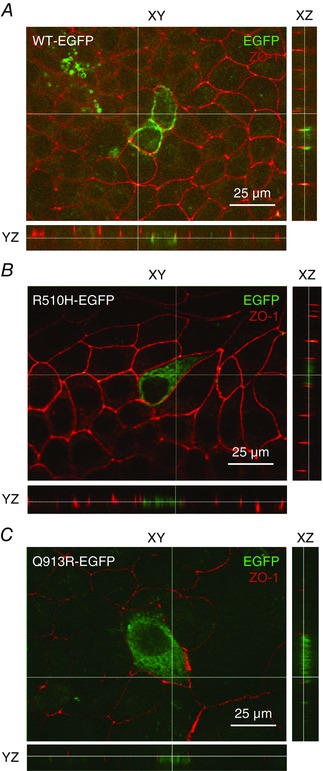
HEK293 cells did not form well‐polarized monolayers in our hands; thus, we visualized the polarized distribution of NBCe1‐A‐EGFP constructs in the better‐behaved canine, renal cell line MDCK‐II. A demonstration of polarity in MDCK‐II cells required that we fixed and permeabilized the cells to allow immunolocalization of the tight junction protein ZO‐1. Thus, in contrast to our handling of HEK cells in Fig. 2, we indirectly visualized NBCe1‐A‐EGFP using an anti‐EGFP antibody in conjunction with an Alexa488‐conjugated secondary antibody. Figure 3 shows the distribution of NBCe1‐A‐EGFP constructs in polarized MDCK‐II cells compared to that of ZO‐1. In x–y sections, the distribution of NBCe1‐A‐EGFP constructs exhibits a pattern reminiscent of that observed in living HEK cells in Fig. 2. The projections in Fig. 3 A reveal that WT‐EGFP exhibits similar x–y co‐ordinates to ZO‐1 but in a lower (i.e. closer to the basal membrane) z‐plane. This compartment probably comprises the lateral membrane. On the other hand, the location of R510H‐EGFP and Q913R‐EGFP in polarized MDCK‐II cells is very different from that of WT‐EGFP. Both mutants exhibit different x–y co‐ordinates to ZO‐1, being predominantly located in compartments between the lateral membranes of each cell (e.g. Fig. 3 B and C). Figure 4 shows the distribution of NBCe1‐A‐EGFP constructs in polarized MDCK‐II cells compared to that of the endogenous Na,K‐ATPase, a marker of the basolateral membrane. By contrast to WT‐EGFP whose distribution substantially overlaps with that of the Na,K‐ATPase (Fig. 4 A), R510H‐EGFP (Fig. 4 B) and Q913R‐EGFP (Fig. 4 C) show the now familiar pattern of increased intracellular retention. This observation is supported by the Pearson's coefficients that report a significantly greater extent of co‐localization between Na,K‐ATPase and WT‐EGFP than with either R510H‐EGFP or Q913R‐EGFP (Fig. 4 D).
Figure 3. The distribution of NBCe1‐A‐EGFP vs. ZO‐1 in polarized MDCK‐II cells .
The location of NBCe1‐A‐EGFP is disclosed using an anti‐GFP primary antibody followed by an Alexa488‐conjugated secondary antibody. The distribution of the polarized‐cell tight‐junction marker ZO‐1 is disclosed using an Alexa594‐conjugated anti‐ZO‐1 antibody. A, cells transfected with WT‐EGFP exhibit lateral distribution of NBCe1‐A‐EGFP. B, cells transfected with R510H‐EGFP exhibit a cytoplasmic distribution of NBCe1‐A‐EGFP. C, cells transfected with Q913R‐EGFP also exhibit a cytoplasmic distribution of NBCe1‐A‐EGFP. Images in (A) to (C) are representative of the distribution of NBCe1‐A‐EGFP as observed in more than four cells in each of three independent transfections. The brightness of all images in (A) to (C) has been increased by 50% for clarity. The thin white lines in each indicate the location of the optical slices displayed in the other planes shown in each panel.
Figure 4. The distribution of NBCe1‐A‐EGFP vs. the Na,K‐ATPase in polarized MDCK‐II cells .
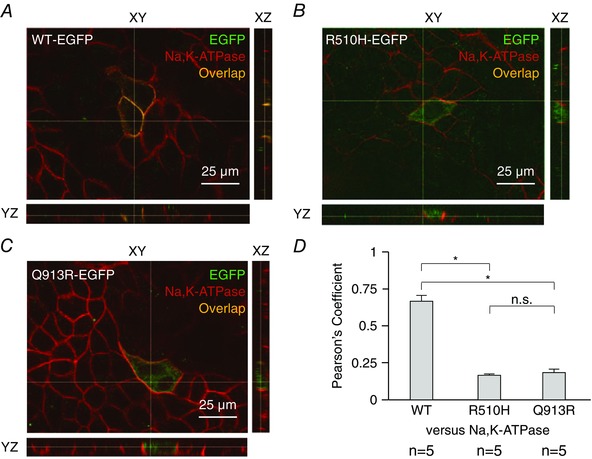
The location of NBCe1‐A‐EGFP is disclosed using an anti‐EGFP primary antibody followed by an Alexa488‐conjugated secondary antibody. The distribution of the basolateral marker Na,K‐ATPase is disclosed using an anti‐Na,K‐ATPase primary antibody followed by an Alexa594‐conjugated secondary antibody. A, cells transfected with WT‐EGFP exhibit lateral distribution of NBCe1‐A‐EGFP. B, cells transfected with R510H‐EGFP exhibit cytoplasmic distribution of NBCe1‐A‐EGFP. C, cells transfected with Q913R‐EGFP also exhibit cytoplasmic distribution of NBCe1‐A‐EGFP. Images in (A) to (C) are representative of the distribution of NBCe1‐A‐EGFP as observed in more than four cells in each of three independent transfections. The brightness of all images in (A) to (C) has been increased by 50% for clarity. The thin white lines in each indicate the location of the optical slices displayed in the other planes shown in each panel. D, Pearson's coefficients report the degree of co‐localization of EGFP and Na,K‐ATPase immunoreactivity in five images equivalent to those shown in (A) to (C). *Significance vs. WT‐EGFP with 95% confidence using an ANOVA with Tukey's comparison test. n.s., no significant difference.
Electrogenic Na+/HCO3 − cotransport activity of NBCe1‐A constructs in Xenopus oocytes
To determine whether the mutants retained electrogenic Na+/2HCO3 − cotransport activity, we injected control oocytes with H2O and experimental oocytes with cRNAs encoding either WT‐EGFP, R510H‐EGFP or Q913R‐EGFP. We injected a fourth group of experimental oocytes with a 1:1 mixture of R510H‐EGFP and Q913R‐EGFP cRNAs as a model of the compound‐heterozygous state of the patient. Table 1 shows the spontaneous V m of these oocytes as they were exposed to a HCO3 −‐free and a HCO3 −‐containing solution. All groups of oocytes exhibited resting membrane potentials (V m) similar to oocytes expressing WT‐EGFP in HCO3 −‐free solution (95% confidence, One‐way ANOVA with Tukey's comparison test). The V m of H2O‐injected cells did not change when we superfused the cells with a HCO3 −‐containing solution. The same manoeuvre caused the V m of oocytes expressing WT‐EGFP to hyperpolarize to a value more negative than −110 mV as a result of the net influx of negative charge towards the predicted reversal potential of a Na+/2HCO3 − cotransporter in oocytes (Sciortino & Romero, 1999; Parker et al. 2012). Oocytes expressing R510H‐EGFP behaved the same as WT‐EGFP in this regard, although the V m of oocytes expressing Q913R‐EGFP or the R510H‐EGFP/Q913R‐EGFP mixture did not hyperpolarize to the same extent as WT‐EGFP or R510H‐EGFP expressing oocytes (95% confidence, one‐way ANOVA with Tukey's comparison test), as if Na+/2HCO3 − cotransport activity does not dominate oocyte V m in these cells.
Table 1.
Spontaneous V m of oocytes at the instant of voltage clamp for the cells shown in Fig. 5
| + Na, 0HCO3 | + Na, +HCO3 | n | |
|---|---|---|---|
| H2O | −41 ± 3 mV | −41 ± 3 mV* | 8 |
| WT‐EGFP | −37 ± 2 mV | −115 ± 3 mV | 8 |
| R510H‐EGFP | −37 ± 1 mV | −121 ± 5 mV | 6 |
| Q913R‐EGFP | −30 ± 1 mV* | −75 ± 1 mV* | 8 |
| R510H‐EGFP/Q913R‐EGFP | −31 ± 2 mV | −84 ± 5 mV* | 7 |
*Significant difference from equivalent value in cells expressing WT‐EGFP (P < 0.01 in two‐tailed, unpaired t test).
To investigate the Na+ and HCO3 −‐dependent conductance conferred upon the oocyte membrane by NBCe1‐A‐EGFP construct expression, we subjected the cells to a voltage clamp protocol. In control experiments, we determined that the membrane conductance (G m) of a H2O‐injected oocyte (example in Fig. 5 A) is unresponsive to alterations in extracellular [Na+] or [HCO3 −], whereas the membrane of a WT‐EGFP‐expressing cell (example in Fig. 5 B) exhibits a Na+ and HCO3 −‐dependent increase in conductance that is characteristic of the action of NBCe1 (Sciortino & Romero, 1999; Parker et al. 2012). Oocytes expressing R510H‐EGFP (Fig. 5 C), Q913R‐EGFP (Fig. 5 D) or the R510H‐EGFP/Q913R‐EGFP mixture (Fig. 5 E) all exhibit Na+‐and HCO3 − dependent conductances consistent with the action of NBCe1 (P < 0.01, paired, one‐tailed t tests) (Fig. 5 F). A general linear model with Tukey's comparison test reports with 95% confidence that G m for WT‐EGFP in NaHCO3‐containing solution is significantly greater than that exhibited by both mutants and that G m for Q913R‐EGFP in NaHCO3‐containing solution is significantly greater than that exhibited by R510H‐EGFP.
Figure 5. Na+‐dependence of wild‐type and mutant NBCe1‐A activity in oocytes .
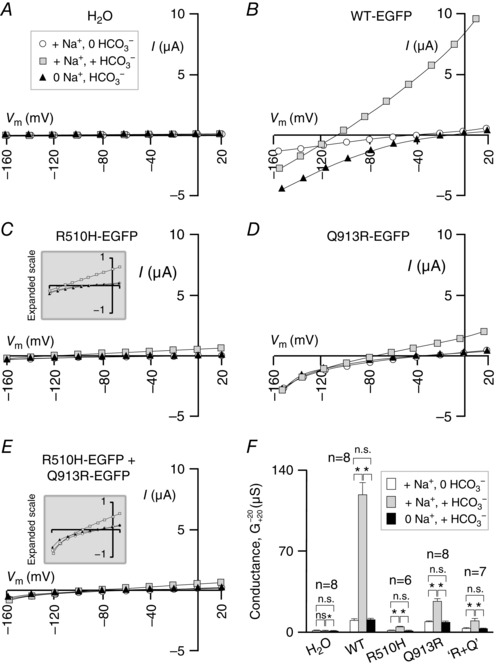
A, current–voltage relationships from a representative H2O‐injected oocyte during sequential exposure to a ‘+Na/0HCO3 ’, a +’Na/+HCO3 ’ and a ‘0Na/+HCO3 ’ solution. Note that the data fell on top of each other and thus the sets have been offset from each other by 3 mV for clarity. Equivalent representative data are shown for oocytes expressing (B) WT‐EGFP, (C) R510H‐EGFP, (D) Q913R‐EGFP and (E) a mixture of R510H‐EGFP and Q913R‐EGFP. F, averaged membrane conductances for each group of cells in each solution, measured between −20 mV and +20 mV, where the outward currents elicited by electrogenic Na/HCO3 import in our protocol are the largest. * P < 0.01 in a paired one‐tailed t test between groups indicated by a horizontal bracket. n.s., no significant difference.
Plasma membrane abundance of NBCe1‐A constructs in Xenopus oocytes
We compared the relative abundance of each EGFP‐tagged mutant vs. WT‐EGFP in the oocyte plasma membrane to determine a normalization factor for the functional data presented in Fig. 5. We detected no anti‐NBCe1‐A‐EGFP immunoreactivity in western blots of biotinylated protein extracts from H2O‐injected oocytes (Fig. 6 A, H2O) but oocytes expressing WT‐EGFP and Q913R‐EGFP exhibit a robust immunoreactive band consistent with the predicted molecular mass (∼160 kDa) of complex‐glycosylated NBCe1‐A‐EGFP (Fig. 6 A, WT). A less‐prominent band (∼330 kDa) evident in some lanes probbaly represents dimeric NBCe1‐A‐EGFP (Choi et al. 2003; Parker et al. 2012). An overexposure of the same blot (Fig. 6 A, inset) demonstrates the presence of R510H‐EGFP protein in this protein fraction. Figure 6 B shows the averaged intensities of the immunoreactive bands from each lane from four or more blots similar to those shown in Fig. 6 A, normalized to the intensity of WT‐EGFP on each blot. Despite each group of oocytes being injected with identical quantities of cRNA, the abundance of mutant NBCe1‐A‐EGFP molecules is significantly less than that of WT‐EGFP (95% confidence, one‐way ANOVA with Tukey's comparison test) (Fig. 6 B). Among the mutants, we detected significantly more NBCe1‐A‐EGFP in the plasma membrane of oocytes expressing Q913R‐EGFP than in cells expressing R510H‐EGFP or the R510H‐EGFP/Q913R‐EGFP mixture (95% confidence, one‐way ANOVA with Tukey's comparison test) (Fig. 6 B). We note that the pattern of variant abundance exhibits a trend similar to that of the magnitude of the conductances elicited by the expression of each in Fig. 5 F.
Figure 6. NBCe1‐A‐EGFP protein expression in Xenopus oocytes .
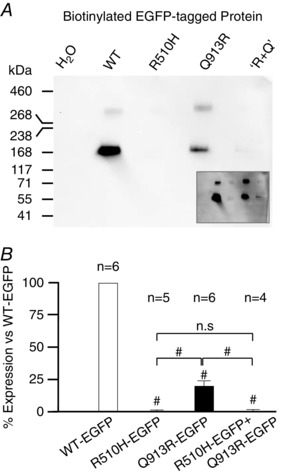
A, representative anti‐EGFP western blot of biotinylated (i.e. extracellular accessible, plasma membrane resident) protein extracts from Xenopus oocytes. ‘R+Q’ denotes cells injected with a 1:1 mixture of R510H‐EGFP + Q913R‐EGFP cRNAs. NBCe1‐A‐EGFP immunoreactivity is barely detectable in lanes 3 and 5 but was evident with longer exposures (A, inset) and exhibits an electrogenic Na/HCO3 cotransport activity that is readily detected by voltage clamp (Fig. 5 C, inset). Protein ladder is HiMark™ pre‐stained protein standard (Thermo Fisher Scientific). B, averaged intensities of immunoreactive bands from blots such as that shown in (A), normalized to the intensity of WT‐EGFP. #Significance vs. WT‐EGFP with 95% confidence using an ANOVA with Tukey's comparison test. #Significance in the same test between the groups indicated. n.s., no significant difference.
The reversal potential of Q913R‐EGFP Na/HCO3 cotransport activity in oocytes
Oocytes expressing Q913R‐EGFP exhibit a deficit in their ability to hyperpolarize compared to oocytes expressing WT‐EGFP or R510H‐EGFP (Table 1), which is not explained by inadequate HCO3 −‐dependent G m (Fig. 5) or plasma membrane abundance of Q913R‐EGFP (Fig. 6). Thus, we aimed to determine whether the reversal potential (E rev) of the transport process (i.e. the value of V m at which the transport process reaches electrochemical equilibrium) was altered by the Q913R mutation. The point of intersection of the I–V plots gathered in our NaHCO3‐containing solution ± the NBCe1‐inhibitor DIDS reports the E rev of the DIDS‐inhibited transport process (Sciortino & Romero, 1999). The application of 200 μm DIDS (K i ∼40 μm; Lu & Boron, 2007) to oocytes expressing NBCe1‐A‐EGFP reduced the HCO3 −‐stimulated currents (measured at +20 mV) by 89 ± 1% in the case of WT‐EGFP (n = 6) (Fig. 7 A) and 107 ± 6% in the case of Q913R‐EGFP (n = 6) (Fig. 7 B). Thus, the Na/HCO3 cotransport activity of Q913R‐EGFP is appropriately DIDS‐sensitive. Figure 7 shows the E rev values reported by the two clones. These values are statistically indistinguishable from each other (95% confidence, unpaired, two‐tailed t test). Thus, the dampened hyperpolarization of oocytes expressing Q913R‐EGFP does not appear to be a consequence of a change in E rev caused by the Q913R mutation, although it is consistent with a depolarizing influence in the membranes of cells expressing Q913R‐EGFP that prevents the apparently normal Na+/2HCO3 − cotransport process from dominating V m.
Figure 7. DIDS‐sensitivity of WT‐EGFP and Q913R‐EGFP activity in oocytes .
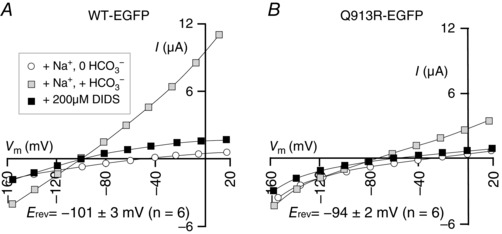
A, I–V relationships from a representative WT‐EGFP‐expressing oocyte during sequential exposure to a Na/0HCO3, a Na/HCO3 solution and the same Na/HCO3 solution including 200 μm DIDS. B, equivalent data are shown for oocytes expressing Q913R‐EGFP. The reversal potential (E rev) for the DIDS‐sensitive transport process is reported by voltage at which the I–V plots ± DIDS (lines with grey vs. black squares) intersect (i.e. where the transporter is neither promoting influx, nor efflux and appears the same as the inhibited transporter) Note that, using this same protocol, the application of DIDS to H2O‐injected oocytes did not influence the membrane conductance (P = 0.50, paired, two‐tailed t test; data not shown).
HCO3 − independent conductance in Q913R‐EGFP‐expressing oocytes
Even in the absence of HCO3 −, oocytes expressing Q913R‐EGFP tend on average to be more depolarized than cells expressing other NBCe1‐A constructs (P < 0.01 in unpaired, one‐tailed t tests vs. WT‐EGFP or vs. R510H‐EGFP) (Table 1). These features are consistent with the presence of a depolarizing ion permeability such as might be associated with the expression of Na+ or Cl− permeable pathways that are independent of Na+/2HCO3 − cotransport activity. Moreover, as is evident in the examples shown in Fig. 5 and Fig. 7, Q913R‐EGFP expressing oocytes exhibit an unusual rectification in G m (note the bending in the I–V plots for Q913R‐EGFP that is not evident for WT‐EGFP or R510H‐EGFP). This feature is most pronounced when these cells are voltage clamped at extremely negative values where the driving force for Na+ and Cl− leaks is expected to be large (equilibrium potential for Na+ (E Na) +50 mV, equilibrium potential for Cl− (E Cl) −20 mV: Weber, 1999).
To investigate the possible presence of a Na+ leak in the cell membrane, we voltage clamped cells as they were serially exposed to ND96 ([Na+] = 96 mm) and a modified version of ND96 in which [Na+] = 10 mm with the balance replaced by the substituting cation NMDG+ (Fig. 8 A–C). E Na has a positive value. Thus, we measured currents at −160 mV (Fig. 8 D), the value in our protocol at which currents should be greatest as a result of Na+ influx through a leak pathway. In ND96, cells expressing Q913R‐EGFP exhibit significantly greater currents at −160 mV than both H2O‐injected and WT‐EGFP‐expressing cells (95% confidence, general linear model with Tukey's post hoc analysis) (Fig. 8 D). Lowering [Na+] to 10 mm resulted in a significant decrease in current at −160 mV in all three cell populations (P < 0.017, paired one‐tailed t tests) (Fig. 8 D). However, even when [Na+] = 10 mm, Q913R‐EGFP‐expressing cells maintained a significantly greater current at −160 mV than both H2O‐injected and WT‐EGFP‐expressing cells (95% confidence, general linear model with Tukey's comparison test), as if the leak still persists.
Figure 8. The influence of Na+ on the bicarbonate independent action of WT‐EGFP and Q913R‐EGFP in oocytes .
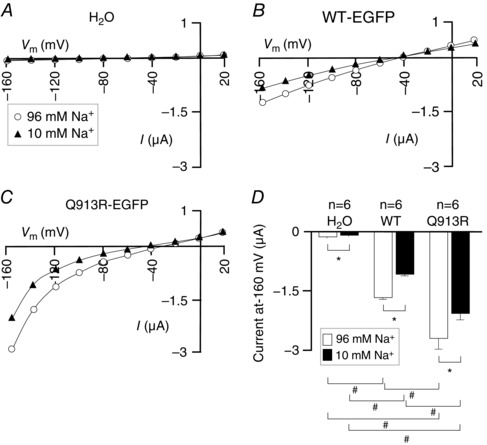
A, I–V relationships from a representative H2O‐injected oocyte during sequential exposure to a solution containing 96 mm Na+ and a solution containing 10 mm Na+. Equivalent data are shown for oocytes expressing (B) WT‐EGFP or (C) Q913R‐EGFP. D, average currents at −160 mV gathered from a greater number of replicates such as those in (A) to (C). * P < 0.017 between the groups indicated using a paired, one‐tailed t test. #Significance with 95% confidence between the groups indicated using a general linear model with Tukey's comparison test.
To investigate the possible presence of a Cl− leak in the cell membrane, we voltage clamped cells as they were serially exposed to ND96 ([Cl−] = 113 mm) and a modified version of ND96 in which [Cl−] = 13 mm with the balance replaced by the substituting anion gluconate. Predicted E Cl for an oocyte is −20 mV; thus, currents resulting from Cl− influx through a leak pathway should be greatest over the positive voltage range. Therefore, for these experiments, we expanded the voltage range over which we clamped (Fig. 9 A–C) and measured currents at +120 mV (Fig. 9 D). This allows us to observe the leak at a driving force (+120 − E Cl ≅ 150 mV) similar to that operating on Na/2HCO3 transport by NBCe1 at +20 mV (+20 − E rev ≅ 120 mV) (Fig. 5). When clamped at positive voltages, H2O‐injected cells exhibit substantial endogenous currents (Fig. 9 A). Thus, for reference, the insets in Fig. 9 B and Fig. 9 C show example I–V plots for WT‐EGFP and Q913R‐EGFP expressing cells with the average endogenous current subtracted. The data shown in Fig. 9 D are uncorrected for the endogenous currents. We find that cells expressing Q913R‐EGFP exhibit significantly greater currents at +120 mV than H2O‐injected cells and WT‐EGFP‐expressing cells (95% confidence, general linear model with Tukey's comparison test) (Fig. 9 D). Lowering [Cl−] to 13 mm causes a significant decrease in current at +120 mV in all three cell types (P < 0.017, paired one‐tailed t test with Bonferroni correction). Importantly, when [Cl−] is lowered to 13 mm, the current at +120 mV for Q913R‐EGFP‐expressing cells is indistinguishable from that of H2O‐injected cells and WT‐EGFP‐expressing cells (95% confidence, general linear model with Tukey's comparison test), as if the leak is no longer evident in the positive voltage range. To determine whether Q913R‐EGFP‐expressing oocytes exhibit increased permeability to Cl− at rest (i.e. not under voltage clamp), we measured the uptake of 36Cl during a 2 h incubation in an ND96‐like solution. Figure 10 shows that oocytes expressing Q913R‐EGFP accumulated significantly more Cl than H2O‐injected cells or cells expressing WT‐EGFP.
Figure 9. The influence of Cl− on the bicarbonate independent action of WT‐EGFP and Q913R‐EGFP in oocytes .
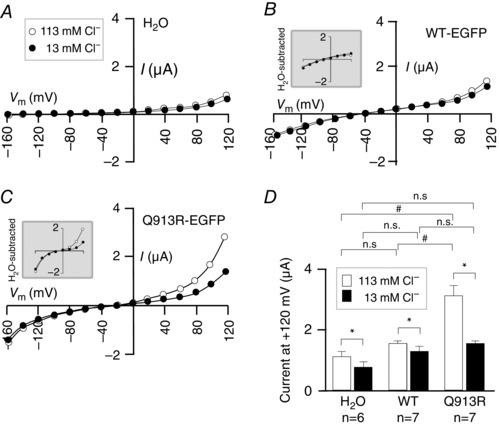
A, I–V relationships from a representative H2O‐injected oocyte during sequential exposure to a solution containing 113 mm Cl− and a solution containing 13 mm Cl−. Equivalent data are shown for oocytes expressing (B) WT‐EGFP or (C) Q913R‐EGFP. Insets are provided for (B) and (C) to show what the I–V plots look like with the averaged currents from H2O‐injected cells subtracted. D, average currents at +120 mV gathered from a greater number of replicates such as those in (A) to (C). * P < 0.017 between the groups indicated using a paired, one‐tailed t test. #Significance with 95% confidence between the groups indicated using a general linear model with Tukey's comparison test. n.s., no significant difference.
Figure 10. Chloride uptake by NBCe1‐A‐EGFP expressing oocytes .
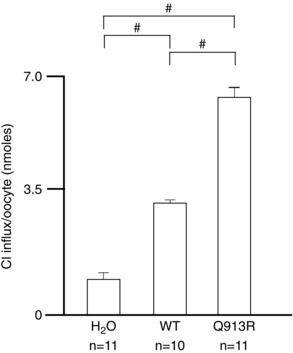
Oocytes were incubated for 2 h in a solution that included Na36Cl. We used the specific activity of the bathing solution to convert the measure of accumulated 36Cl into an estimate of net Cl influx over the same period. #Significance with 95% confidence between the groups indicated using a one‐way ANOVA with Tukey's comparison test. Each ‘n’ represents one group of five oocytes.
To further investigate the nature of the Q913R‐EGFP‐associated conductance, we examined the influence of niflumic acid (Fig. 11), a blocker of the Ca2+‐activated Cl−‐channel that is endogenous to oocytes, (White & Aylwin, 1990) and of NBCe1‐mediated Na+/HCO3 − cotransport (Liu et al. 2007). Niflumic acid exerted a small but significant inhibitory influence upon the currents exhibited by all three populations of cells (P < 0.017, paired, one‐tailed t tests) (Fig. 11 D), although the extent of inhibition was not different between the groups (95% confidence, one‐way ANOVA of the current inhibited by niflumic acid in each cell). Finally, we note that application of 200 μm DIDS or 100 μm tenidap (Fig. 11 E and F), which are both NBCe1‐interacting molecules that block the Na/HCO3 cotransport activity of WT NBCe1‐A (Lu & Boron, 2007), resulted in a small inhibitory effect on the HCO3 − independent conductance in cells expressing WT‐EGFP and Q913R‐EGFP.
Figure 11. The influence of niflumic acid, DIDS and tenidap on Q913R‐EGFP in oocytes .
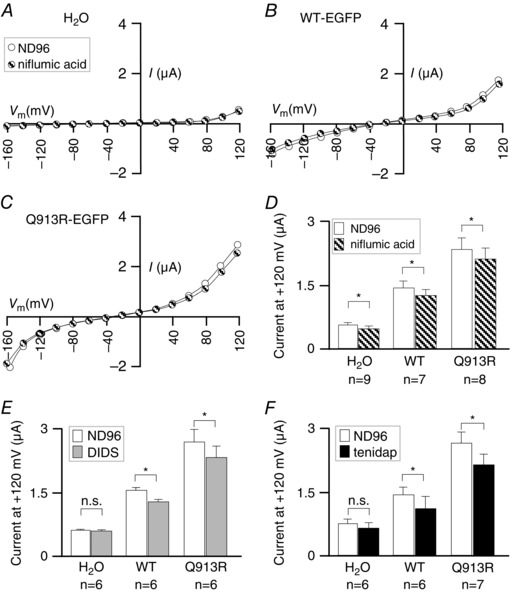
A, I–V relationships from a representative H2O‐injected oocyte during sequential exposure to ND96 followed by ND96 + 100 μm niflumic acid. Equivalent data are shown for oocytes expressing (B) WT‐EGFP or (C) Q913R‐EGFP. D, average currents at +120 mV gathered from a greater number of replicates such as those in (A) to (C). E, data similar to that shown in (D) for 200 μm DIDS. F, data similar to that shown in (D) for 100 μm tenidap. * P < 0.017 between the groups indicated using a paired, one‐tailed t test. Note that, in Fig. 9 D, the endogenous current in H2O‐injected cells at +120 mV was larger than that observed in experiments shown in (D), representing the batch dependence (among oocytes isolated from different batches of frogs) of endogenous currents.
Discussion
The disease‐causing mutations
The present study is the first to describe a patient with pRTA resulting from the compound‐heterozygous inheritance of two non‐identical SLC4A4 mutations. The patient has inherited one mutation from each parent and thus the mutations probably do not reside on the same allele. Furthermore, the heterozygous parents of individuals with pRTA do not exhibit signs of pRTA (Igarashi et al. 1999) and so the patient has probably not retained a WT SLC4A4 allele. Most of the signs of disease in this individual (metabolic acidosis, small stature, band keratopathy, cataracts, glaucoma, dentition defects, calcification of the basal ganglia) are typical of other cases of pRTA. The exception are the episodes similar to transient ischaemic attacks, which appear to be unique to this case and remain unexplained despite extensive neurological study of the patient. It is possible that they are related to the extent of basal ganglion calcification, although other cases of pRTA exhibit similar calcifications apparently without neurological signs. Plasma [HCO3 −] in the patient (11 mm) is in accordance with values reported from pRTA cases involving homozygous SLC4A4 mutations (10 ± 1 mm, n = 15), although the acidosis in the patient (pH 7.27) is not as severe as that so far reported for any homozygous cases (pH range 7.07–7.23) indicating some compensatory ability. Migraines have been reported as a feature in some cases of pRTA (including an R510H homozygote) and even in some heterozygous carriers of pRTA‐causing mutations (including the mother of the aforementioned R510H homozygote) (Suzuki et al. 2010). Our finding that neither the R510H/Q913R patient, nor his parents report a history of migraine does not constitute proof of absence but may suggest variable penetrance or expressivity of this phenotype.
The SLC4 family of transporters includes 10 members in mammals: three anion exchangers (AE1–3), five Na+‐coupled HCO3 − transporters and two unusual members (SLC4A9 and bicarbonate‐transporter related protein 1) (Parker & Boron, 2013). The importance of Arg510 and Gln913 in NBCe1‐A is indicated by the conservation of arginine residues at the 510‐equivalent position in nine of the 10 human SLC4 proteins (bicarbonate‐transporter related protein 1 has a tyrosine at this location) and by the conservation of glutamine at the 913‐equivalent position in all 10 human SLC4 proteins (Parker & Boron, 2013) The recent determination of SLC4‐related structures, most notably that of the transmembrane domain of the human anion exchanger AE1 (Arakawa et al. 2015), permits us to predict the location of R510H and Q913R in three dimensions. Although this approach does explain how or why R510H and Q913R are disruptive, it does validate their assignment in the topology model that places them on opposite sides of the membrane (Fig. 1 K); thus, they probably do not interact with each other. WT NBCe1 is a dimer (Sergeev et al. 2012) and so we expect that the patient's NBCe1‐A protein complement is a mixture of R510H homodimers, Q913R homodimers and R510H/Q913R heterodimers. It is these subpopulations that we have studied in our model expression systems.
Features of R510H
Other studies have described homozygous inheritance of R510H in pRTA (Igarashi et al. 1999; Shiohara et al. 2000; Suzuki et al. 2010) and attributed pRTA to intracellular retention of NBCe1 protein, away from its normal site of action in the plasma membrane, as indicated by a cytoplasmic distribution of R510H‐mutant NBCe1‐constructs in C6 glioma cells (Suzuki et al. 2010) and polarized MDCK cells (Li et al. 2005 a). The present study supports these observations by reporting a cytoplasmic distribution of R510H‐EGFP in polarized MDCK‐II cells (Fig. 3 B and Fig. 4 B), as well as in live HEK293 cells (Fig. 2 B). It is clear from one previous study that R510H‐mutant molecules retain some NBCe1 activity; when expressed in an endothelial cell line, the R510H‐equivalent mutant R554H (in the longer NBCe1‐B variant) mediates a pH recovery as a result of HCO3 − influx that is 50% of that mediated by wild‐type NBCe1 (Satoh et al. 2003). However, extrapolation to per molecule activity data is complicated by not knowing the driving forces acting on the transport process in each case. Although some R510H mutant NBCe1 is expressed into the plasma membrane of oocytes, detailed electrophysiological studies in those cells have been hindered by low expression levels (Horita et al. 2005; Li et al. 2005 a). We also find that R510H‐EGFP does not express robustly in oocytes, although we have been able to detect sufficient activity to perform function studies. We show, for the first time, that R510H‐EGFP action is capable of hyperpolarizing oocytes to the same extent as WT‐EGFP (Table 1) and that R510H‐EGFP mediates a Na+‐ and HCO3 −‐dependent conductance reminiscent of WT‐EGFP activity (Fig. 5 C). The HCO3 −‐dependent conductance and the plasma membrane expression of R510H‐EGFP in oocytes were both <5% of WT‐EGFP and thus the reduced action of R510H‐EGFP vs. WT‐EGFP is probably explained by the reduction in expression. Thus, although the R510H mutation results in aberrant processing of NBCe1‐A, if it were possible to correct the NBCe1‐A trafficking defect in R510H homozygotes, we would expect the rescued NBCe1‐A to be capable of contributing to HCO3 − reabsorption.
Features of Q913R
No individual with homozygous inheritance of Q913R has been described, although the compound heterozygote R510H/Q913R patient's cells probably express some Q913R homodimers. We find the distribution of Q913R‐EGFP in mammalian cell lines to be similar to that of R510H‐EGFP, exhibiting a cytoplasmic distribution in HEK293 and in polarized MDCK‐II cells that is consistent with substantial intracellular retention (Figs 2 C, 3 C and 4 C). Furthermore, our colocalization analysis between the Na,K‐ATPase and the Q913R‐EGFP describes a reduction of transporter accumulation in the basolateral membranes of polarized MDCK‐II cells (Fig. 4 D). This finding can be compared with the result of a two previous cysteine‐scanning mutagenesis studies: one describes an inability to detect the presence of a Q913C mutant in the plasma membrane of HEK293 cells using an antibody directed to an extracellular epitope in NBCe1 (Zhu et al. 2010), whereas the other described a lack of Cl‐HCO3 exchange activity in the plasma membrane of HEK293 cells expressing the Q913C‐homologous AE1 mutant Q840C (Zhu & Casey, 2004). Together, these data suggest that the machinery for processing NBCe1 protein in mammalian cells has little tolerance for the substitution of Q913. We also find that Q913R‐EGFP exhibits a substantial expression defect in oocytes but, as with R510H‐EGFP, the molecules of mutant transporter expressed into the oocyte plasma membrane appear to be capable of close‐to‐normal per molecule NBCe1‐A activity once corrected for abundance (Figs 5 and 7). Co‐expression of R510H‐EGFP does not appear to exert any synergistic influence upon Q913R‐EGFP or vice versa (Figs 5 and 6).
Four pieces of evidence suggest that oocytes expressing Q913R‐EGFP (or Q913R‐EGFP together with R510H‐EGFP) exhibit an unusual, HCO3 − independent ion‐leak even in the presence of HCO3 −. Compared to oocytes expressing WT‐EGFP, oocytes expressing Q913R‐EGFP are: (1) more depolarized at rest (Table 1); (2) do not hyperpolarize to the same extent in response to HCO3 − application despite an apparently normal E rev for Na/HCO3 cotransport and apparently sufficient G m to dominate oocyte V m (Figs 5 and 7 and Table 1); (3) exhibit a membrane conductance with unusual inward rectification (Fig. 5); and (4) exhibit unusually large HCO3 − independent currents compared to WT‐EGFP (Figs 8 and 9), considering their relative abundance in the plasma membrane (Fig. 6). The evidence comprising points (1) and (2) indicates that a depolarizing influence is contributing to V m in Q913R‐EGFP‐expressing cells. Our ion substitution experiments (Figs 8 and 9) indicate that the leak currents are predominantly carried by Cl−, and our isotopic flux experiments (Fig. 10) show that Q913R‐EGFP‐expressing cells are substantially permeable to 36Cl at rest.
Oocytes co‐expressing R510H‐EGFP with Q913R‐EGFP exhibit features consistent with the presence of the depolarizing Cl−‐leak (Fig. 5 E and Table 1). However, the lower plasma membrane abundance of NBCe1‐A‐EGFP protein in these cells (Fig. 6) means that the leak conductance expressed in their membranes is too small to allow us to reliably determine whether the presence of R510H‐EGFP within a heterodimer alters the properties of the Q913R‐EGFP‐dependent leak. It is interesting to note that the combined plasma membrane abundance of NBCe1‐A‐EGFP protein in R510H‐EGFP/Q913R‐EGFP‐expressing cells (Fig. 6) is less than that which would be expected if the two mutant proteins were not interacting, indicating that the R510H‐EGFP is exerting a dominant effect with respect to Q913R‐EGFP plasma membrane accumulation.
As an aside, it is interesting to note that even WT‐EGFP enhances the Cl−‐permeability of oocytes (Fig. 10). NBCe1 is not known to be Cl‐permeable but WT‐EGFP does express a small bicarbonate independent conductance, as seen in Fig. 5 F. Although not greatly Cl−‐conductive under voltage clamped conditions (Fig. 9), this pathway could be somewhat Cl−‐permeable and could therefore be responsible for the observed 36Cl accumulation over the 2 h assay period. It is also important to consider that WT‐EGFP is five times more abundant in the oocyte plasma membrane than Q913R‐EGFP (Fig. 6) and so the per molecule accumulation of 36Cl for WT‐EGFP is not as robust as it might appear in Fig. 10.
A key question is whether the Cl− leak that we observe in Q913R‐EGFP‐expressing oocytes would also manifest in the patients cells, or whether it merely represents an endogenous oocyte Cl−‐channel that has been upregulated by overexpression of a mutant NBCe1 protein. The leak was not specifically blocked by NBCe1‐interacting molecules such as DIDS, tenidap or niflumic acid (Fig. 11 D–F). Although this does not rule out the possibility that the leak pathway is intrinsic to NBCe1, it does preclude definitive attribution of the leak to the Q913R molecule. However (1) expression of a Cl− leak is not a general feature of oocytes overexpressing mutant NBCe1 molecules as indicated by the behaviour of R510H in the present study and in comparable studies of the mutants A799V and R881C (Toye et al. 2006; Parker et al. 2012); (2) the Cl− leak is not blocked by 100 μm niflumic acid (Fig. 11), a dose that should block contribution from the endogenous oocyte Ca2+‐activated Cl−‐channel to membrane currents (White & Aylwin, 1990); and (3) no other endogenous anion channel is predicted to be active under the conditions of the present study (i.e. in non‐hyperpolarized oocytes bathed in isosmotic, Ca2+‐containing solution: Sobczak et al. 2010; Terhag et al. 2010). There are several precedents in oocytes for the introduction of non‐HCO3 − ion‐leak pathways into SLC4 molecules by mutation (Bruce et al. 2005; Parker et al. 2007, 2012; Yang et al. 2009), whereas some other wild‐type SLC4 and Slc4‐like transporters include HCO3 − independent leak pathways when expressed in oocytes that have been demonstrated to persist in vertebrate cells (Fiévet et al. 1995; Choi et al. 2000; Koomoa et al. 2002; Cooper et al. 2005). The case of a Cl− leak constitutively associated with the expression of the trout Cl‐HCO3 exchanger AE1 in oocytes is especially interesting because it represents a swelling‐activated osmolyte pathway that is required for erythrocyte osmoregulation (Fiévet et al. 1995); the constitutive nature of the pathway in oocytes perhaps reflecting the different geometry of oocytes vs. red cells. Taken together, it does not appear to be probable that the leak observed in Q913R‐expressing oocytes represents an endogenous anion channel upregulated by Q913R expression in oocytes, although, if the leak is intrinsic to Q913R, we do not know whether the leak would be a feature, or an inducible feature, of Q913R expressed in mammalian cells.
Physiological implications
The molecular basis of many of the other signs of pRTA is still a matter of active investigation (Seki et al. 2013). Beyond the effects of whole‐body acidosis caused by loss of NBCe1 from renal PT cells, disturbed NBCe1 expression in the enamel organ (Lacruz et al. 2010; Jalali et al. 2014), skeletal muscle,(Kristensen et al. 2004), brain (Schmitt et al. 2000; Majumdar et al. 2008) and eye (Bok et al. 2001) probably results in defective pH regulation in those cells and directly contributes to non‐renal signs of pRTA such as blindness (Parker & Boron, 2013; Seki et al. 2013). In the case of the R510H/Q913R heterozygous individual, if the behaviour of NBCe1‐A mutants in HEK293 cells, MDCK‐II cells and oocytes is predictive of their behaviour in vivo, the retention of NBCe1 protein from its normal site of action in the basolateral membrane of PT epithelia and other cells underlies metabolic acidosis and other signs of pRTA in the patient.
An ion‐leak expressed together with NBCe1 would be undesirable at many sites of NBCe1 expression; expression of a leaky NBCe1 in PT cells could cause a local depolarization of the basolateral membrane potential that would tend to reduce the electrical driving force for HCO3 − reabsorption (Parker et al. 2012); a leaky NBCe1 in secretory epithelia could short circuit anion secretion across the apical membranes of those cells; and a leaky NBCe1 in excitable cells such as neurons or myocytes could result in complex effects with respect to excitability and contractility. By way of example, pathological ion leaks in skeletal muscle cells are associated with myotonia (Cannon, 2000), hypokalaemic paralysis (Sokolov et al. 2007) and malignant hyperthermia (Tong et al. 1997). Further work will be required to determine how much (if any) Q913R‐mutant protein is expressed in the plasma membrane of diverse cell types in the patient, or whether Q913R would exhibit a substantial anion leak in those cells. Either way, it is premature to assume that the leak, even if intrinsic to Q913R, is contributing to the pathology of pRTA in this case.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
EJM, LY, W‐BX and MDP were responsible for the conception and design of the experiments. LY, Y‐YL, YJ, YP, OW, ML, X‐PX, W‐BX (clinical observations), EJM (electrophysiology), EJM, MAF and MDP (cell culture and fluorescence microscopy) and EJM, AM and MDP (molecular biology) were responsible for performing the experiments and data collection. EJM, LY, W‐BX and MDP drafted the work and revised it for intellectual content. All authors contributed to the data analysis and interpretation. All authors approved the final version of the manuscript, agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved, and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by start‐up funding from the Dean of the School of Medicine and Biomedical Sciences and the Department of Physiology and Biophysics at UB:SUNY to MDP; a Carl W. Gottschalk Research Scholar Grant from the ASN Foundation for Kidney Research to MDP; the National Natural Science Foundation of China (No. 81070687 and 81170805); the National Science and Technology Major Projects for ‘Major New Drugs Innovation and Development’ (Grant2008ZX09312‐016); the Beijing Natural Science Foundation (No. 7121012), and the National Key Program of Clinical Science (WBYZ2011‐873). MAF thanks the Dean of the School of Pharmacy and Pharmaceutical Sciences and the Department of Pharmaceutical Sciences at UB:SUNY for providing start‐up funding.
Translational perspectives
Whole‐body pH in proximal renal tubular acidosis patients can be controlled by alkali therapy, relieving growth defects if administered from an early age (Shiohara et al. 2000) but this treatment does not address those pathologies that are related to defects in control of intracellular pH caused by NBCe1 loss from non‐renal cells. Importantly in this regard, both R510H and Q913R‐mutant proteins exhibit a per molecule Na/HCO3 cotransport activity that similar to wild‐type, suggesting that some signs could be corrected by increasing the plasma membrane abundance of these mutants (Chu et al. 2013; Chiu et al. 2015). This may be a valuable therapy for R510H homozygotes. However, a similar intervention for individuals expressing Q913R‐mutant protein could have undesirable consequences if the anion‐leak is intrinsic to the mutant transporter and the therapy does not also block the leak.
Acknowledgements
The authors thank the patient and his family for their involvement in this study. EJM and MDP are grateful for the assistance of Dr Wade Sigurdson in the Confocal Microscope and Flow Cytometry Facility in the School of Medicine and Biomedical Sciences, University at Buffalo. MAF thanks Scott Ferguson (UB) for helpful discussions.
Contributor Information
Wei‐Bo Xia, Email: xiaweibo8301@163.com.
Mark D. Parker, Email: parker28@buffalo.edu.
References
- Alper SL (2010). Familial renal tubular acidosis. J Nephrol 23 (Suppl 16), S57–S76. [PubMed] [Google Scholar]
- Arakawa T, Kobayashi‐Yurugi T, Alguel Y, Iwanari H, Hatae H, Iwata M, Abe Y, Hino T, Ikeda‐Suno C, Kuma H, Kang D, Murata T, Hamakubo T, Cameron AD, Kobayashi T, Hamasaki N & Iwata S (2015). Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 350, 680–684. [DOI] [PubMed] [Google Scholar]
- Bevensee MO, Schmitt BM, Choi I, Romero MF & Boron WF (2000). An electrogenic Na+‐HCO3 – cotransporter (NBC) with a novel COOH‐terminus, cloned from rat brain. Am J Physiol Cell Physiol 278, C1200–C1211. [DOI] [PubMed] [Google Scholar]
- Bok D, Schibler MJ, Pushkin A, Sassani P, Abuladze N, Naser Z & Kurtz I (2001). Immunolocalization of electrogenic sodium‐bicarbonate cotransporters pNBC1 and kNBC1 in the rat eye. Am J Physiol Renal Physiol 281, F920–F935. [DOI] [PubMed] [Google Scholar]
- Boron WF (2012). Acid Base Physiology In Medical Physiology. A Cellular and Molecular Approach, 2nd updated edn, ed. Boron WF. & Boulpaep EL, pp. 652–671. Elsevier Saunders, Philadelphia, PA. [Google Scholar]
- Boron WF & Boulpaep EL (1983). Intracellular pH regulation in the renal proximal tubule of the salamander. Basolateral HCO3 – transport. J Gen Physiol 81, 53–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce LJ, Robinson HC, Guizouarn H, Borgese F, Harrison P, King M‐J, Goede JS, Coles SE, Gore DM, Lutz HU, Ficarella R, Layton DM, Iolascon A, Ellory JC & Stewart GW (2005). Monovalent cation leaks in human red cells caused by single amino‐acid substitutions in the transport domain of the band 3 chloride‐bicarbonate exchanger, AE1. Nat Genet 37, 1258–1263. [DOI] [PubMed] [Google Scholar]
- Cannon SC (2000). Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses. Kidney Int 57, 772–779. [DOI] [PubMed] [Google Scholar]
- Chen A‐P, Holmes HL, Chang M & Romero MF (2014). Targeted NBCe1A Deletion Causes Proximal RTA: Whole nbce1 (sh_nbce1) KO versus nbce1A KO Mice. J Am Soc Nephrol 25, 71A. [Google Scholar]
- Chiu AM, Mandziuk JJ, Loganathan SK, Alka K & Casey JR (2015). High throughput assay identifies glafenine as a corrector for the folding defect in corneal dystrophy‐causing mutants of SLC4A11. Invest Ophthalmol Vis Sci 56, 7739–7753. [DOI] [PubMed] [Google Scholar]
- Choi I, Aalkjaer C, Boulpaep EL & Boron WF (2000). An electroneutral sodium/bicarbonate cotransporter NBCn1 and associated sodium channel. Nature 405, 571–575. [DOI] [PubMed] [Google Scholar]
- Choi I, Hu L, Rojas JD, Schmitt BM & Boron WF (2003). Role of glycosylation in the renal electrogenic Na+‐HCO3 – cotransporter (NBCe1). Am J Physiol Renal Physiol 284, F1199–F1206. [DOI] [PubMed] [Google Scholar]
- Choi I, Romero MF, Khandoudi N, Bril A & Boron WF (1999). Cloning and characterization of a human electrogenic Na+‐HCO3 – cotransporter isoform (hhNBC). Am J Physiol 276, C576–C584. [DOI] [PubMed] [Google Scholar]
- Chu CYS, King JC, Berrini M, Alexander RT & Cordat E (2013). Functional rescue of a kidney anion exchanger 1 trafficking mutant in renal epithelial cells. PloS ONE 8, e57062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DS, Saxena NC, Yang HS, Lee HJ, Moring AG, Lee A & Choi I (2005). Molecular and functional characterization of the electroneutral Na/HCO3 cotransporter NBCn1 in rat hippocampal neurons. J Biol Chem 280, 17823–17830. [DOI] [PubMed] [Google Scholar]
- Demirci FYK, Chang M‐H, Mah TS, Romero MF & Gorin MB (2006). Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol Vis 12, 324–330. [PubMed] [Google Scholar]
- Dinour D, Chang M‐H, Satoh J, Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ & Romero MF (2004). A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem 279, 52238–52246. [DOI] [PubMed] [Google Scholar]
- Dukes JD, Whitley P & Chalmers AD (2011). The MDCK variety pack: choosing the right strain. BMC Cell Biol 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y‐W, Yang S‐S, Chau T, Nakamura M, Yamazaki O, Seki G, Yamada H, Hsu H‐M, Cheng C‐J & Lin S‐H (2015). Therapeutic effect of prenatal alkalization and PTC124 in Na+/HCO3 – cotransporter 1 p.W516* knock‐in mice. Gene Ther 22, 374–381. [DOI] [PubMed] [Google Scholar]
- Fiévet B, Gabillat N, Borgese F & Motais R (1995). Expression of band 3 anion exchanger induces chloride current and taurine transport: structure‐function analysis. EMBO J 14, 5158–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML & Shull GE (2007). Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3 – cotransporter. J Biol Chem 282, 9042–9052. [DOI] [PubMed] [Google Scholar]
- Giebisch G & Windhager E (2012). Transport of acids and bases In Medical Physiology. A Cellular and Molecular Approach, 2nd updated edition, ed. Boron WF. & Boulpaep EL, pp. 851–865. Elsevier Saunders, Philadelphia, PA. [Google Scholar]
- Grichtchenko II & Boron WF (2002). Surface‐pH gradient measurements in Xenopus oocytes co‐expressing the Na+‐driven Cl‐HCO3 exchanger (NDCBE1) and CAIV: evidence for CO3 = transport. FASEB J 16, A797. [Google Scholar]
- Handlogten ME, Osis G, Lee H‐W, Romero MF, Verlander JW & Weiner ID (2015). NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309, F658–F666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer M, Müller‐Berger S, Romero MF, Boron WF & Frömter E (1999). Stoichiometry of the rat kidney Na+‐HCO3 – cotransporter expressed in Xenopus laevis oocytes. Pflüg Arch Eur J Physiol 438, 322–329. [DOI] [PubMed] [Google Scholar]
- Horita S, Yamada H, Inatomi J, Moriyama N, Sekine T, Igarashi T, Endo Y, Dasouki M, Ekim M, Al‐Gazali L, Shimadzu M, Seki G & Fujita T (2005). Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J Am Soc Nephrol 16, 2270–2278. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, Mori T, Shiobara M, Seki G & Endou H (1999). Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 23, 264–266. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Inatomi J, Sekine T, Seki G, Shimadzu M, Tozawa F, Takeshima Y, Takumi T, Takahashi T, Yoshikawa N, Nakamura H & Endou H (2001). Novel nonsense mutation in the Na+/HCO3 – cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J Am Soc Nephrol 12, 713–718. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Sekine T, Inatomi J & Seki G (2002). Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol 13, 2171–2177. [DOI] [PubMed] [Google Scholar]
- Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama N, Kudo A, Kawakami H, Shimadzu M, Endou H, Fujita T, Seki G & Igarashi T (2004). Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflüg Arch Eur J Physiol 448, 438–444. [DOI] [PubMed] [Google Scholar]
- Jalali R, Guo J, Zandieh‐Doulabi B, Bervoets TJM, Paine ML, Boron WF, Parker MD, Bijvelds MJC, Medina JF, DenBesten PK & Bronckers ALJJ (2014). NBCe1 (SLC4A4) a potential pH regulator in enamel organ cells during enamel development in the mouse. Cell Tissue Res 358, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari JA, El Desoky SM, Singh AK, Gari MA, Kleta R & Bockenhauer D (2014). The case | Renal tubular acidosis and eye findings. Kidney Int 86, 217–218. [DOI] [PubMed] [Google Scholar]
- Koomoa D‐LT, Musch MW & Goldstein L (2002). Comparison of the osmolyte transport properties induced by trAE1 versus IClswell in Xenopus oocytes. J Membr Biol 185, 57–63. [DOI] [PubMed] [Google Scholar]
- Kristensen JM, Kristensen M & Juel C (2004). Expression of Na+/HCO3 – co‐transporter proteins (NBCs) in rat and human skeletal muscle. Acta Physiol Scand 182, 69–76. [DOI] [PubMed] [Google Scholar]
- Lacruz RS, Nanci A, White SN, Wen X, Wang H, Zalzal SF, Luong VQ, Schuetter VL, Conti PS, Kurtz I & Paine ML (2010). The sodium bicarbonate cotransporter (NBCe1) is essential for normal development of mouse dentition. J Biol Chem 285, 24432–24438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S‐K, Grichtchenko II & Boron WF (2011). Distinguishing HCO3 – from CO3 2– transport by NBCe1‐A. FASEB J 25, 656.9. [Google Scholar]
- Li HC, Szigligeti P, Worrell RT, Matthews JB, Conforti L & Soleimani M (2005. a). Missense mutations in Na+:HCO3 – cotransporter NBC1 show abnormal trafficking in polarized kidney cells: a basis of proximal renal tubular acidosis. Am J Physiol Renal Physiol 289, F61–F71. [DOI] [PubMed] [Google Scholar]
- Li J, Sun XC & Bonanno JA (2005. b). Role of NBC1 in apical and basolateral HCO3 – permeabilities and transendothelial HCO3 – fluxes in bovine corneal endothelium. Am J Physiol Cell Physiol 288, C739–C746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Williams JB, Sumpter BR & Bevensee MO (2007). Inhibition of the Na/bicarbonate cotransporter NBCe1‐A by diBAC oxonol dyes relative to niflumic acid and a stilbene. J Membr Biol 215, 195–204. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xu J‐Y, Wang D‐K, Wang L & Chen L‐M (2011). Cloning and identification of two novel NBCe1 splice variants from mouse reproductive tract tissues: a comparative study of NCBT genes. Genomics 98, 112–119. [DOI] [PubMed] [Google Scholar]
- Lo Y‐F, Yang S‐S, Seki G, Yamada H, Horita S, Yamazaki O, Fujita T, Usui T, Tsai J‐D, Yu I‐S, Lin S‐W & Lin S‐H (2011). Severe metabolic acidosis causes early lethality in NBC1 W516X knock‐in mice as a model of human isolated proximal renal tubular acidosis. Kidney Int 79, 730–741. [DOI] [PubMed] [Google Scholar]
- Lu J & Boron WF (2007). Reversible and irreversible interactions of DIDS with the human electrogenic Na/HCO3 cotransporter NBCe1‐A: role of lysines in the KKMIK motif of TM5. Am J Physiol Cell Physiol 292, C1787–C1798. [DOI] [PubMed] [Google Scholar]
- Majumdar D, Maunsbach AB, Shacka JJ, Williams JB, Berger UV, Schultz KP, Harkins LE, Boron WF, Roth KA & Bevensee MO (2008). Localization of electrogenic Na/bicarbonate cotransporter NBCe1 variants in rat brain. Neuroscience 155, 818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa‐Aziz R, Boron WF & Parker MD (2010). Using fluorometry and ion‐sensitive microelectrodes to study the functional expression of heterologously‐expressed ion channels and transporters in Xenopus oocytes. Methods 51, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MD & Boron WF (2013). The divergence, actions, roles, and relatives of sodium‐coupled bicarbonate transporters. Physiol Rev 93, 803–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MD, Qin X, Williamson RC, Toye AM & Boron WF (2012). HCO3 – ‐independent conductance with a mutant Na+/HCO3 – cotransporter (SLC4A4) in a case of proximal renal tubular acidosis with hypokalaemic paralysis. J Physiol 590, 2009–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MD, Young MT, Daly CM, Meech RW, Boron WF & Tanner MJA (2007). A conductive pathway generated from fragments of the human red cell anion exchanger AE1. J Physiol 581, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MF, Hediger MA, Boulpaep EL & Boron WF (1997). Expression cloning and characterization of a renal electrogenic Na+/HCO3 – cotransporter. Nature 387, 409–413. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shiigai T, Yoshiyama N & Takeuchi J (1987). Mechanism of bicarbonate exit across basolateral membrane of rabbit proximal straight tubule. Am J Physiol 252, F11–F18. [DOI] [PubMed] [Google Scholar]
- Satoh H, Moriyama N, Hara C, Yamada H, Horita S, Kunimi M, Tsukamoto K, Iso‐O N, Inatomi J, Kawakami H, Kudo A, Endou H, Igarashi T, Goto A, Fujita T & Seki G (2003). Localization of Na+‐HCO3 – cotransporter (NBC‐1) variants in rat and human pancreas. Am J Physiol Cell Physiol 284, C729–C737. [DOI] [PubMed] [Google Scholar]
- Schmitt BM, Berger UV, Douglas RM, Bevensee MO, Hediger MA, Haddad GG & Boron WF (2000). Na/HCO3 cotransporters in rat brain: expression in glia, neurons, and choroid plexus. J Neurosci Off J Soc Neurosci 20, 6839–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciortino CM & Romero MF (1999). Cation and voltage dependence of rat kidney electrogenic Na+‐HCO3 – cotransporter, rkNBC, expressed in oocytes. Am J Physiol 277, F611–F623. [DOI] [PubMed] [Google Scholar]
- Seki G, Coppola S & Frömter E (1993). The Na+‐HCO3 – cotransporter operates with a coupling ratio of 2 HCO3 – to 1 Na+ in isolated rabbit renal proximal tubule. Pflüg Arch Eur J Physiol 425, 409–416. [DOI] [PubMed] [Google Scholar]
- Seki G, Horita S, Suzuki M, Yamazaki O, Usui T, Nakamura M & Yamada H (2013). Molecular mechanisms of renal and extrarenal manifestations caused by inactivation of the electrogenic Na+‐HCO3 – cotransporter NBCe1. Front Physiol 4, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeev M, Godin AG, Kao L, Abuladze N, Wiseman PW & Kurtz I (2012). Determination of membrane protein transporter oligomerization in native tissue using spatial fluorescence intensity fluctuation analysis. PloS ONE 7, e36215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiohara M, Igarashi T, Mori T & Komiyama A (2000). Genetic and long‐term data on a patient with permanent isolated proximal renal tubular acidosis. Eur J Pediatr 159, 892–894. [DOI] [PubMed] [Google Scholar]
- Sobczak K, Bangel‐Ruland N, Leier G & Weber W‐M (2010). Endogenous transport systems in the Xenopus laevis oocyte plasma membrane. Methods 51, 183–189. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T & Catterall WA (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78. [DOI] [PubMed] [Google Scholar]
- Suzuki M et al (2010). Defective membrane expression of the Na+‐HCO3 − cotransporter NBCe1 is associated with familial migraine. Proc Natl Acad Sci USA 107, 15963–15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Vaisbich MH, Yamada H, Horita S, Li Y, Sekine T, Moriyama N, Igarashi T, Endo Y, Cardoso TP, de Sá LCF, Koch VH, Seki G & Fujita T (2008). Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflüg Arch Eur J Physiol 455, 583–593. [DOI] [PubMed] [Google Scholar]
- Terhag J, Cavara NA & Hollmann M (2010). Cave Canalem: how endogenous ion channels may interfere with heterologous expression in Xenopus oocytes. Methods 51, 66–74. [DOI] [PubMed] [Google Scholar]
- Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV & MacLennan DH (1997). Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem 272, 26332–26339. [DOI] [PubMed] [Google Scholar]
- Toye AM, Parker MD, Daly CM, Lu J, Virkki LV, Pelletier MF & Boron WF (2006). The human NBCe1‐A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am J Physiol Cell Physiol 291, C788–C801. [DOI] [PubMed] [Google Scholar]
- Weber W (1999). Ion currents of Xenopus laevis oocytes: state of the art. Biochim Biophys Acta 1421, 213–233. [DOI] [PubMed] [Google Scholar]
- White MM & Aylwin M (1990). Niflumic and flufenamic acids are potent reversible blockers of Ca2+‐activated Cl– channels in Xenopus oocytes. Mol Pharmacol 37, 720–724. [PubMed] [Google Scholar]
- Yang HS, Kim E, Lee S, Park HJ, Cooper DS, Rajbhandari I & Choi I (2009). Mutation of Aspartate 555 of the Sodium/Bicarbonate Transporter SLC4A4/NBCe1 Induces Chloride Transport. J Biol Chem 284, 15970–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitomi K, Burckhardt BC & Frömter E (1985). Rheogenic sodium‐bicarbonate cotransport in the peritubular cell membrane of rat renal proximal tubule. Pflüg Arch Eur J Physiol 405, 360–366. [DOI] [PubMed] [Google Scholar]
- Zhu Q & Casey JR (2004). The substrate anion selectivity filter in the human erythrocyte Cl‐/HCO3‐ exchange protein, AE1. J Biol Chem 279, 23565–23573. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Kao L, Azimov R, Abuladze N, Newman D, Pushkin A, Liu W, Chang C & Kurtz I (2010). Structural and functional characterization of the C‐terminal transmembrane region of NBCe1‐A. J Biol Chem 285, 37178–37187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Shao XM, Kao L, Azimov R, Weinstein AM, Newman D, Liu W & Kurtz I (2013). Missense mutation T485S alters NBCe1‐A electrogenicity causing proximal renal tubular acidosis. Am J Physiol Cell Physiol 305, C392–C405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhorn IS, Kalicharan D, Robillard GT & Hoekstra D (2007). Adhesion receptors mediate efficient non‐viral gene delivery. Mol Ther J Am Soc Gene Ther 15, 946–953. [DOI] [PubMed] [Google Scholar]