

It has been known for more than a century that bile acids and gallstones may represent an aetiological factor in acute pancreatitis (AP). Importantly, while bile is responsible for around 40% of AP, its aetiological role in the chronic form of the disease (CP) is close to zero. Only 4% of patients suffering from CP have gallstones, and it is still not clear whether this is only an association or whether bile or gallstones play any pathophysiological role in the development of the chronic inflammation. In this issue of The Journal of Physiology, Ferdek et al. have offered the first explanation for this phenomenon at the cellular level (Ferdek et al. 2016). They have shown for the first time that bile acids elicit dramatic necrosis in pancreatic stellate cells (PSCs) but not in pancreatic acinar cells (PACs). In the presence of calcium, sodium cholate induces 73% necrosis in PSCs but only around 10% in PACs, suggesting that the PSC, the key player in the extracellular matrix in the pancreas, is the cell type most endangered by bile (Ferdek et al. 2016). Since the effects of bile acids on pancreatic ductal cells (PDCs) have also been characterized, the chronological events in how bile acids affect the exocrine pancreas and induce acute but not chronic pancreatitis can be followed (Fig. 1).
-
(1)
Obstruction. Gallstones impact the ampulla of Vater, and a common channel is formed by the bile and pancreatic ducts (Opie et al. 1901).
-
(2)
Reflux. Although the pressure in the pancreatic duct is higher than in the bile duct when the common channel is formed, it quickly equalizes and bile can reflux into the pancreas due to the chemical gradient (Opie et al. 1901).
-
(3)
Defence. Bile acid reaching PDCs from the luminal side in low concentration stimulates fluid and bicarbonate secretion. While the stimulated fluid attempts to flush the toxic bile out of the pancreas, the high concentration of bicarbonate will elevate the pH in the lumen, inhibiting the autoactivation of trypsinogen. These effects not only attempt to defend the other cell types, such as PACs and PSCs, but may aid in removing the impacted stone from the pancreas due to the elevated pressure (Hegyi et al. 2013).
-
(4)
Ductal cell damage. If the PDC defence mechanism is not sufficient, bile will reach the PDC in high concentration, inducing a toxic calcium signal, mitochondrial damage, ATP depletion and almost total inhibition of fluid and bicarbonate secretion. Moreover, bile acids enhance the permeability of the epithelial barrier, making the pancreas more vulnerable. These alterations will create the opportunity for bile acids to reach acinar cells (Venglovecz et al. 2008).
-
(5)
Acinar cell damage. Bile acids can reach acinar cells from both the basolateral and the luminal sides. They will trigger a toxic calcium signal, mitochondrial damage, ATP depletion (as we can see in the PDCs), zymogen activation, liberation of kallikreins and cell necrosis (Hegyi et al. 2013).
-
(6)
Acute inflammatory response. Bile acids activate the innate immune system via toll‐like receptor 4 (TLR4) and the necrotized cell. Activation of TLR4 leads to downstream release of inflammatory modulators, including TNF‐α and interleukins (Hegyi et al. 2013). Moreover, it has been shown that bile acids also induce the production of the inflammatory mediator nitric oxide (NO) in pancreatic stellate cells (Jakubowska et al. 2016).
-
(7)
Stellate cell necrosis. Bile acids induce a severe toxic calcium signal and a large amount of stellate cell necrosis, which can even be enhanced by the inflammatory mediator bradykinin. The stellate cell damage will inhibit fibrogenesis and extracellular matrix protein synthesis in the pancreas preventing the development of chronic inflammation (Ferdek et al. 2016).
Figure 1. Chronological events in how bile acids affect the exocrine pancreas .
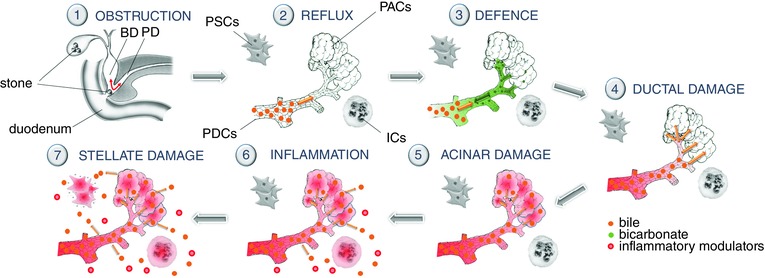
BD, bile duct; PAC, pancreatic acinar cell; PD, pancreatic duct; PDC, pancreatic ductal cell; PSC, pancreatic stellate cell; IC, inflammatory cell.
It is almost unnecessary to point out that besides the cellular explanation, other mechanisms can also decrease the potential for the development of bile‐induced chronic pancreatitis. It is well known that recurrent acute inflammation and continuous or frequent activation of PSCs can transform into an activated, myofibroblast‐like PSCs form that secretes large amounts of extracellular matrix protein that lead to fibrosis and chronic pancreatitis. Since in most cases the papilla of Vater is opened by sphincterectomy and the gallbladder is removed in bile‐induced AP, no additional injury can be caused by bile.
The fascinating observations by Ferdek et al. not only provide the first cellular explanation for the lack of chronic inflammation in bile‐induced pancreatic damage, but also highlight bile acids as a new therapeutic window in chronic pancreatitis. The best candidate seems to be the ursodeoxycholic acids, which have been shown (1) to have protective effects on pancreatic ductal cells against chenodeoxycholic acid‐induced injury (Katona et al. 2016) and (2) to have beneficial effects in autoimmune pancreatitis and to have a protective effect in a number of liver diseases, where hepatic fibrogenesis occurs. I hope that the recent observation by Ferdek et al. will initiate both preclinical and clinical investigations.
Additional information
Competing interests
None declared.
Linked articles This Perspective highlights an article by Ferdek et al. To read this paper, visit http://dx.doi.org/10.1113/JP272774.
References
- Ferdek PE, Jakubowska MA, Gerasimenko JV, Gerasimenko OV & Petersen OH (2016). Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium‐driven bile uptake. J Physiol 594, 6147–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi P & Petersen OH (2013). Rev Physiol Biochem Pharmacol 165, 1–30. [DOI] [PubMed] [Google Scholar]
- Jakubowska MA, Ferdek PE, Gerasimenko OV, Gerasimenko JV & Petersen OH (2016). Nitric oxide signals are interlinked with calcium signals in normal pancreatic stellate cells upon oxidative stress and inflammation. Open Biol 6, 160149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona M, Hegyi P, Kui B, Balla Z, Rakonczay Z Jr, Rázga Z, Tiszlavicz L, Maléth J & Venglovecz V (2016). A novel, protective role of ursodeoxycholate in bile‐induced pancreatic ductal injury. Am J Physiol Gastrointest Liver Physiol 310, G193–204. [DOI] [PubMed] [Google Scholar]
- Opie EL (1901). The etiology of acute haemmorhagic pancreatitis. Bull Johns Hopkins Hosp 12, 182–188 [Google Scholar]
- Venglovecz V, Rakonczay Z Jr, Ozsvári B, Takács T, Lonovics J, Varró A, Gray MA, Argent BE & Hegyi P (2008). Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 57, 1102–1112. [DOI] [PubMed] [Google Scholar]