

Abstract
Key points
Channelopathies of autoimmune origin are novel and are associated with corrected QT (QTc) prolongation and complex ventricular arrhythmias.
We have recently demonstrated that anti‐SSA/Ro antibodies from patients with autoimmune diseases and with QTc prolongation on the ECG target the human ether‐à‐go‐go‐related gene (HERG) K+ channel by inhibiting the corresponding current, I Kr, at the pore region.
Immunization of guinea‐pigs with a peptide (E‐pore peptide) corresponding to the extracellular loop region connecting the S5 and S6 segments of the HERG channel induces high titres of antibodies that inhibit I Kr, lengthen the action potential and cause QTc prolongation on the surface ECG. In addition, anti‐SSA/Ro‐positive sera from patients with connective tissue diseases showed high reactivity to the E‐pore peptide.
The translational impact is the development of a peptide‐based approach for the diagnosis and treatment of autoimmune‐associated long QT syndrome.
Abstract
We recently demonstrated that anti‐SSA/52 kDa Ro antibodies (Abs) from patients with autoimmune diseases and corrected QT (QTc) prolongation directly target and inhibit the human ether‐à‐go‐go‐related gene (HERG) K+ channel at the extracellular pore (E‐pore) region, where homology with SSA/52 kDa Ro antigen was demonstrated. We tested the hypothesis that immunization of guinea‐pigs with a peptide corresponding to the E‐pore region (E‐pore peptide) will generate pathogenic inhibitory Abs and cause QTc prolongation. Guinea‐pigs were immunized with a 31‐amino‐acid peptide corresponding to the E‐pore region of HERG. On days 10–62 after immunization, ECGs were recorded and blood was sampled for the detection of E‐pore peptide Abs. Serum samples from patients with autoimmune diseases were evaluated for reactivity to E‐pore peptide by enzyme‐linked immunosorbent assay (ELISA), and histology was performed on hearts using Masson's Trichrome. Inhibition of the HERG channel was assessed by electrophysiology and by computational modelling of the human ventricular action potential. The ELISA results revealed the presence of high titres of E‐pore peptide Abs and significant QTc prolongation after immunization. High reactivity to E‐pore peptide was found using anti‐SSA/Ro Ab‐positive sera from patients with QTc prolongation. Histological data showed no evidence of fibrosis in immunized hearts. Simulations of simultaneous inhibition of repolarizing currents by anti‐SSA/Ro Ab‐positive sera showed the predominance of the HERG channel in controlling action potential duration and the QT interval. These results are the first to demonstrate that inhibitory Abs to the HERG E‐pore region induce QTc prolongation in immunized guinea‐pigs by targeting the HERG channel independently from fibrosis. The reactivity of anti‐SSA/Ro Ab‐positive sera from patients with connective tissue diseases with the E‐pore peptide opens novel pharmacotherapeutic avenues in the diagnosis and management of autoimmune‐associated QTc prolongation.
Keywords: autoimmune diseases, HERG channel, Immunization, Long QT syndrome
Key points
Channelopathies of autoimmune origin are novel and are associated with corrected QT (QTc) prolongation and complex ventricular arrhythmias.
We have recently demonstrated that anti‐SSA/Ro antibodies from patients with autoimmune diseases and with QTc prolongation on the ECG target the human ether‐à‐go‐go‐related gene (HERG) K+ channel by inhibiting the corresponding current, I Kr, at the pore region.
Immunization of guinea‐pigs with a peptide (E‐pore peptide) corresponding to the extracellular loop region connecting the S5 and S6 segments of the HERG channel induces high titres of antibodies that inhibit I Kr, lengthen the action potential and cause QTc prolongation on the surface ECG. In addition, anti‐SSA/Ro‐positive sera from patients with connective tissue diseases showed high reactivity to the E‐pore peptide.
The translational impact is the development of a peptide‐based approach for the diagnosis and treatment of autoimmune‐associated long QT syndrome.
Abbreviations
- Abs
antibodies
- AP
action potential
- APD
action potential duration
- APD90
action potential at 90% duration
- ELISA
enzyme‐linked immunosorbent assay
- E‐pore
extracellular pore
- HEK cells
human embryonic kidney cells
- HERG
human ether‐à‐go‐go‐related gene
- IgG
immunoglobulin G
- IKr
rapidly activating delayed K+ current
- IKs
slow delayed rectifier K+ current
- ICaL
L‐type Ca2 + current
- MEM
minimal essential medium
- QTc
corrected QT
- QT1
long QT1
- VA
veterans affairs
Introduction
Autoantibody‐associated cardiac channelopathies are gaining significant interest in the area of heart rhythm disturbances (Qu & Boutjdir 2012; Xiao et al. 2012; Li et al. 2013, 2014; El‐Sherif & Boutjdir, 2015). In a series of papers, we identified L‐type Ca2+ channel‐inhibiting (antagonist‐like) antibodies (Abs) directed against the extracellular loop domain as crucial in the pathophysiology of congenital heart block (Boutjdir et al. 1997, 1998; Qu et al. 2001, 2005; Karnabi et al. 2010, 2011). Activating (agonist‐like) Abs against Ca2+ and K+ channels have also been reported in dilated cardiomyopathy (Xiao et al. 2012) and the short QT syndrome (Li et al. 2013, 2014), respectively. In particular, immunization of rabbits with KCNQ1‐encoded Kv7.1 K+ channel peptide induced high circulating anti‐KCNQ1 antibody titres and accelerated cardiac repolarization as a result of an activating effect on the related I Ks current (Li et al. 2014). Moreover, an autoimmune‐mediated inhibition or activation of Ca2+ and K+ channels can also be the indirect result of Abs that recognize cardiac antigens, such as the β1‐adrenergic receptor, as observed in patients with dilated cardiomyopathy (Christ et al. 2001; Fukuda et al. 2004; Lazzerini et al. 2015). Notably, only rabbit immunization with the second extracellular loop of the human β1‐adrenergic receptor induced functional Abs, whereas Abs resulting from the immunization with the extracellular N‐terminus had no functional effects (Magnusson et al. 1994). Thus, depending on the specific cardiac antigen considered and the fine epitope specificity of the peptide employed, immunization can unpredictably induce activating (agonist‐like), inhibiting (antagonist‐like) or non‐functional Abs.
A novel acquired autoimmune‐associated long QT syndrome has recently been reported in adult patients carrying anti‐SSA/52 kDa Ro (anti‐Ro52) Abs (Lazzerini et al. 2004, 2007, 2008; Bourre‐Tessier et al. 2011). Clinically, patients with connective tissue diseases who tested positive for anti‐Ro52 Abs had a prolonged corrected QT (QTc) and complex ventricular arrhythmias independent of any other QTc‐prolonging factors, such as medication, electrolytes or genetic factors (Lazzerini et al. 2004). The molecular and functional basis for this QTc prolongation has been attributed to a functional inhibition of the human ether‐a‐go‐go‐related gene (HERG) channel conducting the rapidly activating delayed K+ current, I Kr (Nakamura et al. 2007), by direct interaction of the antibodies with the extracellular loop between segments S5 and S6 of the pore‐forming α1 subunit of the HERG channel (Yue et al. 2015; Lazzerini et al. 2016). In the present study, we tested the hypothesis that immunization of guinea‐pigs with a peptide corresponding to the extracellular pore region (E‐pore peptide) of the α1 subunit of HERG will be sufficient to inhibit but not activate I Kr, thus resulting in action potential duration (APD) lengthening and in QTc prolongation on the surface ECG.
Methods
Ethical approval
The experiments used heart tissue from isoflurane‐anaesthetized Dunkin–Hartley adult guinea‐pigs. All animal procedures were performed in conformity with the US National Institutes of Health guidelines for care and use of laboratory animals and the local institutional animal care and use committee at the VA New York Harbor Healthcare System, New York. The investigators understand the ethical principles under which the journal operates, and this work complies with animal ethics checklist.
Synthesis of E‐pore peptides and immunization
Given that the Ro52 antigen is not accessible to circulating Abs, we hypothesized that circulating anti‐Ro Abs must recognize an epitope mimic on the extracellular side of the HERG channel homologous to Ro52 antigen. Homology analysis showed the presence of homology between the Ro52 antigen at the extracellular pore region of the HERG channel (Fig. 1). We thus designed the E‐pore peptide (GNMEQPHMDSRIGWLHNLGDQIGKPYNSSGL) and the scrambled peptide (NEQDRSGYHPMKWMSIILGGSGLNGPQNDLH), which were synthesized at >90% purity by Genscript USA, Inc., (Piscataway, NJ, USA). The E‐pore peptide consisted of a 31‐amino‐acid sequence corresponding to a portion of the 65‐amino‐acid extracellular loop between S5 and S6 of the pore‐forming region of the α1 subunit of the HERG channel where homology with Ro52 antigen exists (Fig. 1). Six‐ to 8‐week‐old Dunkin–Hartley guinea‐pigs (n = 10; four males and six females) were immunized with 75 μg of the peptide in complete Freund's adjuvant subcutaneously in the scapular region (Boutjdir et al. 1997; Miranda‐Carus et al. 1998; Xiao et al. 2001 b; Karnabi et al. 2011; Yue et al. 2015). Subsequent booster injections of 37.3 μg in incomplete Freund's adjuvant were administered 10 and 15 days after the initial immunization. Control experiments were conducted in six guinea‐pigs using the same immunization protocol but with the scrambled peptide. Guinea‐pigs were anaesthetized using a Minivent ventilator attached to an isoflurane vapourizer (Harvard Apparatus, Holliston, MA, USA).
Figure 1. Homology between HERG K+ channel and Ro52 protein .

A, relevant linear homology between Ro52 protein and HERG channel at the pore region. B, schematic representation of the secondary structure of a single HERG channel α1 subunit. The six segments (S1–S6) are shown, along with the intracellularly located N‐ and C‐termini. The pore‐forming extracellular loop is located between S5 and S6, where black and blue circles illustrate similar and identical amino acids between Ro52 protein and HERG channel, respectively. C, schematic illustration of the top view of the tetrameric HERG channel, bringing together the homologous regions at the pore.
Electrocardiographic recordings
The ECG was recorded in lead II at baseline (before immunization) and on days 10, 21, 32, 42, 48 and 62 after immunization. The ECGs were analysed for QTc, heart rate, PR and QRS intervals. The QTc was calculated using Bazett's formula because it is more suitable for anaesthetized guinea‐pigs (Hamlin et al. 2003).
Blood collection
Blood was obtained via the cranial vena cava while the animals were under general anaesthesia on days 0, 10, 21, 42 and 62. The serum was isolated by centrifugation for 20 min at 5000 g, and an enzyme‐linked immunosorbent assay (ELISA) was used to detect the antibody levels (Yue et al. 2015).
Enzyme‐linked immunosorbent assay
An ELISA was used for the detection of antibody levels (Yue et al. 2015). The peptide (E‐pore or scrambled) was coated overnight, washed in PBS–Tween 20 and probed in diluted serum followed by anti‐human immunoglobulin G (IgG) alkaline phosphatase. The plates were developed with disodium p‐nitrophenyl phosphate. All samples were run in triplicate. Results were expressed as the optical density at 405 nm minus that of the reagent blank.
Electrophysiological studies
Expression systems
HEK293 cells stably expressing the HERG channel were a generous gift from Dr Craig January (University of Wisconsin, Madison, WI, USA) and Dr Zhengfeng Zhou (Oregon Health & Science University, Portland, OR, USA). Details of DNA constructs and the methods of stable transfection of HEK293 cells with HERG channel have been reported elsewhere (Zhou et al. 1998). These HEK293 cells were cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum and 400 μg ml−1 geneticin (G418). Cells were washed twice with standard MEM, and stored in this medium at room temperature for later use. Cells were superfused with Hepes‐buffered Tyrode's solution containing (mm): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes (pH adjusted to 7.4 with NaOH). The internal pipette solution contained (mm): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP and 10 Hepes (pH adjusted to 7.2 with KOH). The I Kr was recorded using a standard protocol (shown in Fig. 3 F) as previously reported (Yue et al. 2015; Lazzerini et al. 2016). Experiments were performed at room temperature.
Figure 3. Serum from E‐pore peptide immunized guinea‐pigs inhibits IKr .
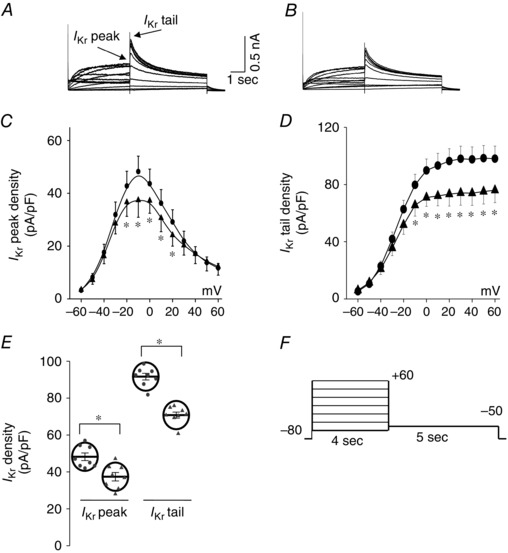
A and B, representative current traces for I Kr recorded from HEK293 cells stably expressing HERG channels at baseline (A) and in the presence of serum (100 μl) from an E‐pore peptide‐immunized guinea‐pig (B). C and D, I–V relationships of I Kr peak (C) and tail current densities (D) at baseline (circles) and after application of serum from E‐pore peptide‐immunized guinea‐pigs (triangles). E, dot‐plot at 0 mV for I Kr peak and tail densities before and after serum application from E‐pore peptide‐immunized guinea‐pigs. F shows the protocol used to record I Kr from HEK293 cells stably expressing HERG channels. *Statistical significance at P < 0.05 (n = 8 each).
Guinea‐pig ventricular myocytes
Guinea‐pigs were anaesthetized with isoflurane using a Harvard apparatus vapourizer, and animals were monitored until recumbent. A thoracotomy was performed and the heart rapidly excised and, in this way, the animal was killed. Hearts were then Langendorf perfused with Tyrode's solution of the following composition (mm): 140 NaCl, 4.5 KCl, 2 CaCl2, 10 dextrose, 1 MgCl2 and 10 Hepes (pH 7.4) for 2 min. The heart was then perfused with Ca2+‐free Tyrode's solution followed by collagenase B (final concentration, 0.6 mg ml−1; Worthington, Lakewood, NJ, USA). The dispersed cells were resuspended in Kraft‐Brühe (KB) solution and maintained at room temperature before use. The external solution used for I Kr recordings contained (mm): 145 NaCl, 4.5 KCl, 1 MgCl2, 1.8 CaCl2, 10 Hepes and 10 glucose (pH 7.35). Calcium currents were blocked by the addition of 10 μm nifedipine in the bath solution, and the slow delayed rectifier K+ current (I Ks) was blocked with 10 μm chromanol. The pipette solution contained (mm): 140 KCl, 10 Hepes, 11 EGTA, 1 MgCl2, 1 CaCl2, 5 MgATP and 5 K2ATP; the pH was adjusted to 7.2 with KOH. Currents were recorded in the whole‐cell, voltage‐clamp configuration of the patch‐clamp technique using an Axopatch‐200B amplifier (Axon Instruments, Inc., Burlingame, CA, USA). The I Kr was recorded using a short (200 ms) depolarizing pulse from a holding potential of −40 mV, and test pulses were applied at various voltages from −40 to +40 mV in 10 mV increments before returning to −40 mV for tail current recording (Yue et al. 2015; Lazzerini et al. 2016). Measurements were repeated every 2 min to allow for complete tail current deactivation. Action potentials were recorded from single ventricular myocytes in current‐clamp mode by passing depolarizing currents at subthreshold (1.4 times) intensity.
Histology and quantification of myocardial fibrosis
Hearts were washed in PBS, fixed in 10% neutral buffered formalin, and embedded in paraffin as described previously (Wolf et al. 2005). Transverse serial sections 5 μm thick were cut every 50 μm from the apex to the base of the hearts and stained with Masson's Trichrome to assess collagen deposition resulting from fibrosis. Fibrotic areas within sections were measured as blue‐stained areas, exclusive of staining that co‐localized with perivascular or intramural vascular structures, the endocardium or left ventricular trabeculae (Wolf et al. 2005). Blue‐stained areas and non‐stained myocyte areas from each section were determined with a colour‐based threshold using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The total fibrotic area was calculated as the summed blue‐stained areas divided by the total ventricular area and expressed as a percentage (Wolf et al. 2005).
Computational modelling
Simulations of the human ventricular action potential (AP) and pseudo‐ECG were performed using the model developed by O'Hara et al. (2011), with the incorporation of a cell cable in the model to produce traces similar to the pseudo‐ECG in humans using MATLAB programming language. The heterogeneity of ionic conductances seen across the epi‐, mid‐ and endocardial cell layers was incorporated according to their specifications; the size of the wall and the relative size of each layer were constructed according to Gima & Rudy (2002). Simulation of alterations in I Kr, I Ks and L‐type Ca current (I CaL) were achieved by changing the conductance values of G Kr, G Ks and G CaL by the corresponding percentage inhibition as appropriate.
Data and statistical analyses
The ECGs were analysed using LabChart Pro software (AD Instruments, Inc., Colorado Springs, CO, USA). Statistical comparisons at baseline and after interventions were evaluated with Student's paired t test and with the non‐parametric Wilcoxon paired test as appropriate using Origin9.1 (OriginLab Corporation, Northampton, MA, USA). Data are presented as means ± SD. A value of P < 0.05 was considered significant.
Results
Immunization with the peptide corresponding to the extracellular pore region of the HERG channel induced QTc prolongation in guinea‐pigs
Electrocardiograms were performed on 16 guinea‐pigs (10 experimental and 6 control) at baseline and at several time points after immunization. In the E‐pore peptide‐immunized guinea‐pigs, the average QTc was 263 ± 4 ms at baseline and 312 ± 5 ms on day 62 (P = 0.0001), an increase of 49 ms. This QTc prolongation was associated with high titres of anti‐E‐pore peptide Abs in immunized guinea‐pigs (optical density increased from 0.02 ± 0.01 at baseline to 2.73 ± 0.73 on day 62, n = 10, P < 0.01), reflecting successful immunization. There were no significant differences between heart rate, PR and QRS intervals at baseline and after immunization. Figure 2 illustrates an example of a guinea‐pig ECG before (Fig. 2 A) and after immunization on day 62 (Fig. 2 B). The QTc values were 254 ms at baseline and 306 ms 62 days after E‐pore peptide immunization, corresponding to an increase in QTc of 54 ms. Control guinea‐pigs immunized with the scrambled E‐pore peptide showed no significant ECG changes compared with baseline except for high antibody titres (optical density increased from 0.08 ± 0.01 at baseline to 1.36 ± 0.13 on day 62, n = 6, P < 0.01). Figure 2 C and D illustrates ECGs at baseline and 62 days after scrambled E‐pore peptide immunization, respectively. Figure 2 E shows that QTc values increased progressively after immunization with E‐pore peptide but not with scrambled E‐pore peptide. The levels of Abs also increased after E‐pore peptide immunization (Fig. 2 F; n = 10 for each time point) and the prolongation in QTc correlated well with the antibody levels (Fig. 2 G) as assessed by the coefficient of determination (R 2 = 0.81, P < 0.01).
Figure 2. E‐pore peptide immunized guinea‐pigs exhibit QTc prolongation .
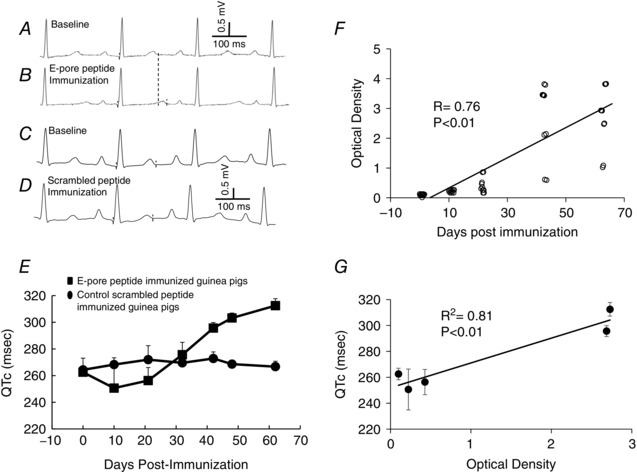
Selected ECG from the same guinea‐pig at baseline (pre‐immunization; A) and after E‐pore peptide immunization (B). C and D represent a control ECG at baseline and an ECG 62 days after scrambled E‐pore peptide‐immunization, respectively. E, QTc values before (n = 10) and after immunization of guinea‐pigs (n = 10) with E‐pore peptide (squares) and scrambled E‐pore peptide (circles) between days 10 and 62. F, enzyme‐linked immunosorbent assay values for anti‐E‐pore peptide antibodies at days between 0 and 62 (n = 10 at each time point). G, the correlation between QTc prolongation and the anti‐E‐pore peptide antibody levels is illustrated by the coefficient of determination, R 2 (each data point represents the QTc and the corresponding optical density values at the days shown in F).
Serum from E‐pore peptide‐immunized guinea‐pigs inhibited I Kr recorded from HEK293 cells
Figure 3 shows the effect of serum from E‐pore peptide‐immunized guinea‐pigs on I Kr recorded from HEK293 cells stably expressing HERG channels. This serum (100 μl) inhibited I Kr peak densities at 0 mV by 23% (from 48.2 ± 5.8 to 37.4 ± 6.5 pA pF−1, P < 0.05, n = 8) and I Kr tail densities by 23% (from 90.6 ± 5.0 to 70.3 ± 4.6 pA pF−1, P < 0.01, n = 8). These effects on I Kr were not seen with serum samples (100 μl) from non‐immunized guinea‐pigs at baseline before immunization (I Kr peak densities at 0 mV from 45.3 ± 5.2 to 41.2 ± 5.3 pA pF−1, P > 0.05, n = 6; I Kr tail from 92.2 ± 7.4 to 88.4 ± 6.4 pA pF−1, P > 0.05, n = 6).
Serum from E‐pore peptide‐immunized guinea‐pigs inhibited I Kr and prolonged the APD recorded from cardiomyocytes
Figure 4 shows I Kr recorded from guinea‐pig ventricular myocytes using a short‐duration (200 ms) pulse protocol from a holding potential of −40 mV, at which the slow delayed rectifier, I Ks, is inactive (and in the presence of 10 μm chromanol, an I Ks blocker). Serum samples (100 μl) from scrambled peptide‐immunized control guinea‐pigs (n = 6) had no effect on I Kr peak or tail currents (Fig. 4 A and C). However, sera from E‐pore peptide‐immunized guinea‐pigs significantly inhibited I Kr peak by 27.6% (P < 0.01, n = 6) and I Kr tail by 29.0% (P < 0.01, n = 6; Fig. 4 B and D). If the QTc prolongation seen in the immunized guinea‐pigs is attributable to I Kr inhibition, then serum samples from immunized guinea‐pigs should be expected to lengthen the ventricular APD. Thus, action potentials (APs) were recorded from guinea‐pig single ventricular myocytes before and after the application of control and E‐pore peptide‐immunized guinea‐pig serum samples at 0.5 Hz in the current‐clamp mode configuration. Figure 4 shows that control serum samples (100 μl) had no effect on AP parameters (Fig. 4 E; n = 6) and on the action potential at 90% duration (APD90; Fig. 4 G; n = 6). However, serum samples (100 μl) from E‐pore peptide‐immunized guinea‐pigs resulted in a prolongation of APD90 by 30.0% (P < 0.01, n = 6) without any changes in the resting membrane potential or AP amplitude (Fig. 4 F and H).
Figure 4. E‐pore peptide immunized guinea‐pigs serum inhibits native IKr and prolong action potential duration .
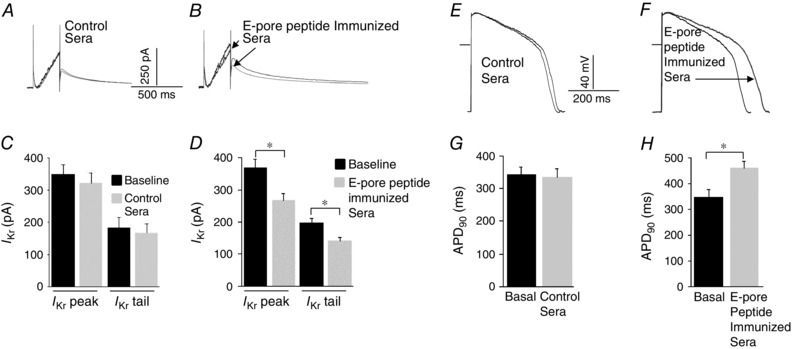
Effect of serum samples from control scrambled peptide‐immunized and serum samples from E‐pore peptide‐immunized guinea‐pigs on I Kr (A and B, respectively) and action potentials (E and F, respectively) recorded from guinea‐pig ventricular myocytes. Averaged data from six experiments each for control and E‐pore‐immunized guinea‐pigs are represented by the histograms for I Kr (C and D) and action potentials (G and H), respectively. *Statistical significance at P < 0.05 (n = 6 each).
Serum from anti‐Ro‐positive patients immunoreacted with the peptide corresponding to the extracellular pore region of the HERG channel
Next, we tested whether serum samples from patients with autoimmune diseases, mainly connective tissue diseases, anti‐Ro Ab positive and with documented QTc prolongation (QTc >460 ms; Lazzerini et al. 2004, 2007), would demonstrate reactivity to the E‐pore peptide. Patients with connective tissue diseases who were anti‐Ro Ab negative and with a normal QTc (<440 ms) were used as a control group. Figure 5 depicts the reactivity status of 13 anti‐Ro Ab‐positive connective tissue disease patients and eight anti‐Ro Ab‐negative connective tissue disease patients. The difference in the average optical density values, indicated by the squares in Fig. 5, was statistically significant (P = 0.00029) between the positive group (1.2 ± 0.1, n = 13) and the negative group (0.2 ± 0.002, n = 8). The optical density values ranged from 0.51 to 2.47 in the positive group and from 0.10 to 0.41 in the negative group.
Figure 5. Reactivity of serum samples from patients to E‐pore peptide assessed by enzyme‐linked immunosorbent assay .
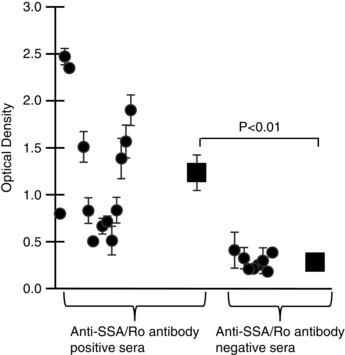
Anti‐Ro antibody‐positive serum samples from patients with connective tissue diseases and QTc prolongation had significant (P < 0.001, n = 13) reactivity to E‐pore peptide (left) compared with anti‐Ro antibody‐negative serum samples from patients with connective tissue diseases but with normal QTc (n = 8, right).
Immunization of guinea‐pigs with the peptide corresponding to the extracellular pore region of the HERG channel did not induce fibrosis
Although previous studies suggested that provocation of an autoimmune response against ion channels can induce electrophysiological changes without eliciting an immune‐mediated tissue inflammatory response (Korkmaz et al. 2013; Li et al. 2014), it has been demonstrated that other cardiac autoantibodies associated with development of arrhythmias, such as anti‐myosin heavy chain autoantibodies, exert their pathological effect by promoting an acute inflammatory response, and resulting in later reparative myocardial fibrosis (Lazzerini et al. 2008; Watanabe et al. 2011). To test whether immunization with the E‐pore peptide might have resulted in myocardial fibrosis, we prepared Masson's Trichrome‐stained sections from immunized and non‐immunized hearts. Figure 6 shows transverse cross‐sections of guinea‐pig hearts across the ventricles from a control non‐immunized (Fig. 6 A) and an E‐pore peptide immunized (Fig. 6 B) guinea‐pig at 62 days after immunization. There was no significant difference in fibrosis (blue staining) between the control and E‐pore peptide‐immunized guinea‐pig ventricles, although the latter showed a slightly greater staining, which was not statistically significant. The average quantification data of fibrosis for the two groups are illustrated in Fig. 6 C (0.21 ± 0.04, n = 5 for immunized group and 0.18 ± 0.03, n = 3 for control non‐immunized; P = 0.129).
Figure 6. Immunization of guinea-pigs with E-pore peptide does not cause fibrosis .

Masson's Trichrome‐stained sections from a non‐immunized control (A) and E‐pore peptide‐immunized (B) hearts from two different guinea‐pigs. No significant difference (P = 0.129) in fibrosis (blue staining) was observed on day 62 after immunization in five hearts compared with three control hearts (C). In Y axis, fibrosis is indicated as percent of the affected area.
Mathematical simulation of the effects of anti‐Ro autoantibodies on repolarization currents and APD
The duration of the QT interval is determined by the APD, which is controlled by three major repolarization currents, I Kr, I Ks and I CaL. Previous experimental data from our laboratory demonstrated that anti‐Ro Abs inhibit both I Kr (Yue et al. 2015; Lazzerini et al. 2016) and I CaL (Boutjdir et al. 1997, 1998; Xiao et al. 2001 a) but not I Ks (Xiao et al. 2001 a) when studied individually in expression systems. Here, we used mathematical modelling of the human ventricular AP developed by O'Hara et al. (2011) to gain insight into changes in APD as a result of both inhibition of individual currents and simultaneous inhibition of I Kr, I Ks and I CaL. Figure 7 shows the results of individual inhibition of I Kr, I Ks or I CaL by 20, 40, 60 and 100%. Only I Kr inhibition resulted in pronounced APD prolongation (Fig. 7 A) that was ‘dose dependent’ and consistent with recent experimental data (Yue et al. 2015). For example, at 60% inhibition of I Kr, APD90 lengthened by 56.3%, whereas 100% inhibition of I Ks resulted in only 6.5% prolongation of APD90, consistent with published reports that I Ks inhibition in the absence of sympathetic stimulation plays little role in increasing normal human AP repolarization (Jost et al. 2005). Although inherited mutations in I Ks channel protein are associated with the occurrence of long QT1 (LQT1), it is known that penetrance is low (Swan et al. 1998) in families with documented LQT1 mutations, histories of seizures and sudden cardiac death; and elevated sympathetic tone may play a role in the phenotype expression. Note that 100% I Kr inhibition resulted in an AP which did not repolarize. In contrast, inhibition of I CaL shortened APD90 in a ‘dose‐dependent’ manner. Inhibition of I CaL by 60% resulted in only 16.3% shortening of APD90. Table 1 summarizes the percentage changes in ADP90 values as a result of inhibition of individual currents.
Figure 7. Computational model of the human ventricular action potential .
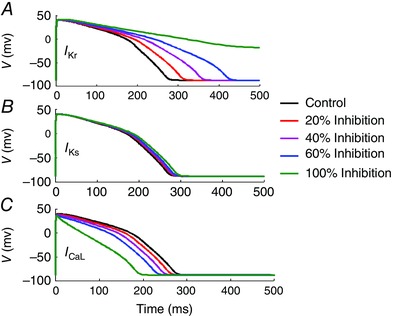
The conductances of I Kr (A), I Ks (B) and I CaL (C) were inhibited individually to mimic autoantibody‐induced inhibition of these currents and to assess their impact on the action potential duration at 90% (APD90). As can be seen, although inhibition of I Ks had little effect on APD, inhibition of I Kr and I CaL resulted in marked prolongation and shortening of APD, respectively.
Table 1.
Mathematical model of the human cardiac action potential: consequences of individual I Kr, I Ks and I CaL block on action potential duration
| Current | 20% block | 40% block | 60% block | 100% block |
|---|---|---|---|---|
| Percentage of APD90 changes for: | ||||
| I Kr | +13.3 | +31.6 | +56.3 | n.a. |
| I Ks | +1 | +2.4 | +3.8 | +6.5 |
| I CaL | −5.1 | −10.4 | −16.3 | −32 |
Key: ‘+’ indicates lengthening and ‘−’ indicates shortening of the action potential at 90% duration (APD90); n.a. indicates that the action potential did not repolarize.
Figure 8 A shows the consequences of simultaneous inhibition of I Kr and I CaL on APD. Given that I CaL inhibition antagonizes I Kr inhibition vis‐à‐vis APD, in this simulation we kept I Kr inhibition constant and evaluated the effects of various degrees of I CaL inhibition (50, 80, 90 and 100%) on APD90. Inhibition of I Kr by 50% resulted in APD90 prolongation by 42.5% (Table 2), and additional incremental inhibition of I CaL by 50, 80, 90 and 100% prolonged APD90 by only 30.9, 18.4, 13.2 and 0.4%, respectively (Table 2), suggesting that dual inhibition of I Kr and I CaL will dictate the net APD and thus QT interval depending on the degree to which I Kr and I CaL conductances are inhibited. This is clinically relevant because anti‐Ro Abs inhibit both I Kr (Yue et al. 2015; Lazzerini et al. 2016) and I CaL (Boutjdir et al. 1997). Finally, we explored the consequences of different degrees of I Kr inhibition and APD prolongation on QT interval. This was achieved by simultaneous simulation of APD and pseudo‐ECG. Figure 8 B and C demonstrates that ‘dose‐dependent’ inhibition of I Kr results in prolongation not only of APD but also of QT interval, thereby highlighting the direct relationship between I Kr inhibition, APD and QT prolongation on the surface ECG.
Figure 8. Computational model of the human ventricular action potential and pseudo‐ECG .

A, simultaneous inhibition of I Kr conductance at 50% and I CaL conductance at 50, 80, 90 and 100% was simulated to assess the overall changes in the action potential duration at 90% (APD90). Inhibition of I Kr (50%) resulted in a significant prolongation in APD90 (red action potential). Incremental inhibition of I CaL, in addition to I Kr, resulted in progressive shortening of APD90 but did not exceed (blue action potential) the control APD90 (black action potential). The consequences of I Kr conductance inhibition in a dose‐dependent manner resulted not only in action potential prolongation (B) but also in QT interval prolongation on the pseudo‐ECG (C).
Table 2.
Mathematical model of the human cardiac action potential: consequences of simultaneous inhibition of I Kr and I CaL on action potential duration
| Percentage of I Kr | Percentage of I CaL | Percentage APD90 | ||
|---|---|---|---|---|
| inhibition | inhibition | APD90 | changes | |
| Control | 0 | 0 | 264.35 | 0 |
| Simulation 1 | 50 | 0 | 376.74 | 42.5 |
| Simulation 2 | 50 | 50 | 345.96 | 30.5 |
| Simulation 3 | 50 | 80 | 313.11 | 18.4 |
| Simulation 4 | 50 | 90 | 299.24 | 13.2 |
| Simulation 5 | 50 | 100 | 265.36 | 0.4 |
Abbreviations: APD90, action potential duration at 90%.
Discussion
The data from this study establish that immunization of guinea‐pigs with a peptide corresponding to the extracellular loop at the pore region between S5 and S6 (E‐pore peptide) of the HERG channel generated high titres of anti‐E‐pore peptide‐inhibitory Abs, which inhibited I Kr from both the expressed and the native HERG channel, and induced QTc prolongation on the surface ECG in the absence of significant fibrosis. These electrophysiological effects were not seen with a control scrambled form of the E‐pore peptide. In addition, anti‐Ro‐positive serum samples from patients with autoimmune diseases and QTc prolongation showed significant reactivity to the E‐pore peptide compared with serum samples from control patients with autoimmune diseases who were anti‐Ro Ab negative and had normal QTc. Mathematical simulation of major repolarizing current inhibition by anti‐Ro Abs suggests that simultaneous inhibition of I Kr and I CaL could account for the variability of QTc prolongation in anti‐Ro‐positive patients with autoimmune diseases. Altogether, these data identify the extracellular loop pore region of the HERG channel as an antigenic target for anti‐Ro Abs, consistent with the inhibition of I Kr and the resulting prolongation of AP and QTc in patients with autoimmune diseases.
Ro antigen is an intracellular ribonucleoprotein that is not accessible to circulating anti‐Ro Abs found in multiple autoimmune diseases, including connective tissue diseases, and is associated with QTc prolongation in these patients (Lazzerini et al. 2004, 2007). Recently, we established an animal model for anti‐Ro Ab‐associated QTc prolongation (Yue et al. 2015) reminiscent of the clinical phenotype and provided biochemical and functional evidence that the HERG channel is a target for these Abs. Given that Ro antigen is not normally accessible to circulating antibodies, we postulated that a potential epitope mimic is present on the HERG channel. Indeed, homology between Ro52 antigen and the pore region of the HERG channel was recently identified (Yue et al. 2015), thereby accounting for the interaction between the Abs and the HERG channel.
Linear homology analysis showed that there is 44% homology between Ro52 protein (amino acids 302–321) and the HERG α1 subunit (amino acids 574–598) at the pore region, of which 25% are identical (Yue et al. 2015). The presence of this homology at the pore region is potentially sufficient for anti‐Ro52 Abs to bind to the HERG channel at this epitope mimic, especially in the tetrameric conformation of the channel, where the four pore extracellular loops come together and are accessible to the Abs (Fig. 1).
The present data demonstrating the induction of QTc prolongation by immunization of guinea‐pigs with the E‐pore peptide, but not with the scrambled control peptide, establishes this HERG channel region as a potential pathogenic epitope that serves as an antigenic target for the circulating anti‐Ro Abs. The translational application of this finding is the potential for the development of short therapeutic decoy biologics that can distract pathogenic anti‐Ro Abs from the HERG channel, thereby preventing the interaction and thus QTc prolongation. To date, it is not clear what specific sequence on the Ro52 antigen is responsible for immunogenicity in patients with autoimmune diseases. In fact, although some groups identified the central part of the antigen (i.e. the coiled‐coil domain), particularly the p200 peptide (amino acids 200–239), as the main immunogenic region (Bozic et al. 1993; Salomonsson et al. 2002; Strandberg et al. 2008; Burbelo et al. 2010; Infantino et al. 2015), others did not confirm these findings, demonstrating a prevalent reactivity towards sequences included in the N‐terminal (RING‐B‐box domain) and C‐terminal parts (B30.2. domain) of the antigen (Fritsch et al. 2006). Hennig et al. (2008) found that in patients with Sjogren's syndrome, a native autoantigenic epitope is located in the zinc‐binding region of the RING‐B‐box domain. Other studies reported a significant immunogenicity also in the B30.2 domain able to generate high antibody titres in patients with autoimmune diseases, particularly when affected by Sjögren's syndrome (Bozic et al. 1993; Burbelo et al. 2010; Infantino et al. 2015). Notably, the C‐terminal B30.2 domain contains the amino acid sequence (amino acids 302–321) that overlaps with HERG. Future in vivo studies are needed to develop non‐pathological peptides able to serve as effective decoys and neutralize the pathogenic Abs.
The QTc prolongation observed in the E‐pore peptide‐immunized guinea‐pigs correlated with the development of the antibody titre. This is consistent with the in vitro and clinical data demonstrating the dose‐dependent inhibition of HERG channels (Yue et al. 2015) and the titre‐dependent association between anti‐Ro52 Abs and QTc prolongation (Lazzerini et al. 2011). These observations may account, in part, for individual variability in QTc prolongation observed in patients with autoimmune diseases (Lazzerini et al. 2004, 2007, 2011). Indeed, among those patients presenting with QTc prolongation, QTc can vary from 460 to >560 ms (Lazzerini et al. 2004, 2007, 2011). In this regard, only patients with a moderate to high concentration (≥50 U ml−1) of circulating anti‐Ro52 Abs exhibit QTc prolongation (Lazzerini et al. 2011).
The titre‐dependent association with QTc prolongation cannot alone account for the variability in QTc prolongation, because dual inhibition of HERG and L‐type Ca2+ channels could be another contributory factor. The current generated through the HERG channel, I Kr, repolarizes the AP, and its inhibition lengthens the APD and QT interval. However, the current generated through L‐type Ca2+ channels, I CaL, delays repolarization, and its inhibition shortens the APD. As such, simultaneous inhibition of both I Kr and I CaL has opposite effects on APD, and the net outcome will depend on the relative degree of I Kr and I CaL inhibition by the Abs. To this end, the mathematical simulation data suggest that although inhibition of I CaL shortens APD, thereby antagonizing ADP prolongation due to I Kr inhibition, which per se seems predominant and leads to more dramatic prolongation of APD. As shown in Fig. 8, the net APD and QT interval durations are determined by the degree of I Kr and I CaL inhibition by the Abs titre level in a given patient; however, the scenario of 100% I CaL inhibition by the Abs is unlikely, because we have previously shown that anti‐Ro Abs inhibit I CaL by only 40–60% maximum (Boutjdir et al. 1997, 1998). Altogether, the Ab titre combined with the balance between the degree of I Kr and I CaL inhibition by Abs will determine the APD and QTc durations and thus could also contribute to the variability of QTc values in patients with autoimmune diseases.
Nakamura and colleagues, in their elegant study (Nakamura et al. 2007), also showed that anti‐Ro Ab‐positive serum and purified IgG, from a female patient (n = 1) with marked QTc and episodes of torsades de pointes, functionally inhibited and biochemically interacted with the HERG channel proteins in a dose‐dependent manner. These effects were observed only when HEK293 cells in culture were incubated with anti‐Ro‐positive serum and purified IgG for 1–5 days, but no acute effects (5 min) were observed. In contrast, the data from the present study and other studies from our group (Yue et al. 2015; Lazzerini et al. 2016) demonstrated a consistent inhibitory effect on I Kr within minutes. Specifically, anti‐Ro Ab‐positive IgG from a total of 10 patients from two different studies (Yue et al. 2015; Lazzerini et al. 2016) each tested in several HEK293 cells (n = 6 each) consistently showed a time‐dependent effect, with a steady state reached within 8–12 min. It is therefore possible that longer periods of time than the 5 min exposure in the study by Nakamura et al. (2007) may have been necessary to see an acute effect similar to the one reported in our experimental conditions. The fact that the study by Nakamura et al. (2007) used only one patient may also have contributed to their findings. Differences in the experimental conditions between the two studies may also, at least in part, have contributed to the differences seen, because no detailed methodology was provided in the the study by Nakamura et al. (2007). Nevertheless, both studies showed that anti‐Ro Abs inhibit I Kr.
Collectively, the findings from the present study are the first to demonstrate that inhibitory antibodies to the HERG E‐pore region peptide but not to HERG E‐pore region scrambled peptide induce QTc prolongation in immunized guinea‐pigs by targeting the HERG channel independently from fibrosis. Furthermore, the reactivity of anti‐Ro Ab‐positive sera from patients with connective tissue diseases with the E‐pore peptide opens novel pharmacotherapeutic avenues in the diagnosis and management of autoimmune‐associated QTc prolongation.
Additional information
Competing interests
None declared.
Author contributions
All experiments were performed at the VA New York Harbor Healthcare System in New York. Y.Y. and F.F. performed the experiments. The rest of the authors contributed to the conception, design and interpretation of the work and review of the manuscript. All authors approved the final version of this manuscript agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The work was supported by Award Number I01BX007080 from the Biomedical Laboratory Research & Development Service of the Veterans Affairs Office of Research and Development.
References
- Bourre‐Tessier J, Clarke AE, Huynh T, Bernatsky S, Joseph L, Belisle P & Pineau CA (2011). Prolonged corrected QT interval in anti‐Ro/SSA‐positive adults with systemic lupus erythematosus. Arthritis Care Res (Hoboken) 63, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Boutjdir M, Chen L, Zhang ZH, Tseng CE, DiDonato F, Rashbaum W, Morris A, el‐Sherif N & Buyon JP (1997). Arrhythmogenicity of IgG and anti‐52‐kD SSA/Ro affinity‐purified antibodies from mothers of children with congenital heart block. Circ Res 80, 354–362. [DOI] [PubMed] [Google Scholar]
- Boutjdir M, Chen L, Zhang ZH, Tseng CE, El‐Sherif N & Buyon JP (1998). Serum and immunoglobulin G from the mother of a child with congenital heart block induce conduction abnormalities and inhibit L‐type calcium channels in a rat heart model. Pediatr Res 44, 11–19. [DOI] [PubMed] [Google Scholar]
- Bozic B, Pruijn GJ, Rozman B & van Venrooij WJ (1993). Sera from patients with rheumatic diseases recognize different epitope regions on the 52‐kD Ro/SS‐A protein. Clin Exp Immunol 94, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Ching KH, Han BL, Bush ER, Reeves WH & Iadarola MJ (2010). Extraordinary antigenicity of the human Ro52 autoantigen. Am J Transl Res 2, 145–155. [PMC free article] [PubMed] [Google Scholar]
- Christ T, Wettwer E, Dobrev D, Adolph E, Knaut M, Wallukat G & Ravens U (2001). Autoantibodies against the β1 adrenoceptor from patients with dilated cardiomyopathy prolong action potential duration and enhance contractility in isolated cardiomyocytes. J Mol Cell Cardiol 33, 1515–1525. [DOI] [PubMed] [Google Scholar]
- El‐Sherif N & Boutjdir M (2015). Role of pharmacotherapy in cardiac ion channelopathies. Pharmacol Ther 155, 132–142. [DOI] [PubMed] [Google Scholar]
- Fritsch C, Hoebeke J, Dali H, Ricchiuti V, Isenberg DA, Meyer O & Muller S (2006). 52‐kDa Ro/SSA epitopes preferentially recognized by antibodies from mothers of children with neonatal lupus and congenital heart block. Arthritis Res Ther 8, R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Miyoshi S, Tanimoto K, Oota K, Fujikura K, Iwata M, Baba A, Hagiwara Y, Yoshikawa T, Mitamura H & Ogawa S (2004). Autoimmunity against the second extracellular loop of β1‐adrenergic receptors induces early afterdepolarization and decreases in K‐channel density in rabbits. J Am Coll Cardiol 43, 1090–1100. [DOI] [PubMed] [Google Scholar]
- Gima K & Rudy Y (2002). Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 90, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin RL, Kijtawornrat A, Keene BW & Hamlin DM (2003). QT and RR intervals in conscious and anesthetized guinea pigs with highly varying RR intervals and given QTc‐lengthening test articles. Toxicol Sci 76, 437–442. [DOI] [PubMed] [Google Scholar]
- Hennig J, Bresell A, Sandberg M, Hennig KD, Wahren‐Herlenius M, Persson B & Sunnerhagen M (2008). The fellowship of the RING: the RING‐B‐box linker region interacts with the RING in TRIM21/Ro52, contains a native autoantigenic epitope in Sjögren syndrome, and is an integral and conserved region in TRIM proteins. J Mol Biol 377, 431–449. [DOI] [PubMed] [Google Scholar]
- Infantino M, Meacci F, Grossi V, Benucci M, Morozzi G, Tonutti E, Tampoia M, Ott A, Meyer W, Atzeni F, Sarzi‐Puttini P, Manfredi M & Bizzaro N (2015). Serological epitope profile of anti‐Ro52‐positive patients with systemic autoimmune rheumatic diseases. Arthritis Res Ther 5, 17–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, Nagy Z, Bogáts G, Lathrop DA, Papp JG & Varró A (2005). Restricting excessive cardiac action potential and QT prolongation: a vital role for I Ks in human ventricular muscle. Circulation 112, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Karnabi E, Qu Y, Mancarella S & Boutjdir M (2011). Rescue and worsening of congenital heart block‐associated electrocardiographic abnormalities in two transgenic mice. J Cardiovasc Electrophysiol 22, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnabi E, Qu Y, Wadgaonkar R, Mancarella S, Yue Y, Chahine M, Clancy RM, Buyon JP & Boutjdir M (2010). Congenital heart block: identification of autoantibody binding site on the extracellular loop (domain I, S5–S6) of α1D L‐type Ca channel. J Autoimmun 34, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz S, Zitron E, Bangert A, Seyler C, Li S, Hegedüs P, Scherer D, Li J, Fink T, Schweizer PA, Giannitsis E, Karck M, Szabó G, Katus HA & Kaya Z (2013). Provocation of an autoimmune response to cardiac voltage‐gated sodium channel NaV1.5 induces cardiac conduction defects in rats. J Am Coll Cardiol 62, 340–349. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Acampa M, Guideri F, Capecchi PL, Campanella V, Morozzi G, Galeazzi M, Marcolongo R & Laghi‐Pasini F (2004). Prolongation of the corrected QT interval in adult patients with anti‐Ro/SSA‐positive connective tissue diseases. Arthritis Rheum 50, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL, Acampa M, Morozzi G, Bellisai F, Bacarelli MR, Dragoni S, Fineschi I, Simpatico A, Galeazzi M & Laghi‐Pasini F (2011). Anti‐Ro/SSA‐associated corrected QT interval prolongation in adults: the role of antibody level and specificity. Arthritis Care Res (Hoboken) 63, 1463–1470. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL, Guideri F, Acampa M, Selvi E, Bisogno S, Galeazzi M & Laghi‐Pasini F (2008). Autoantibody‐mediated cardiac arrhythmias: mechanisms and clinical implications. Basic Res Cardiol 103, 1–11. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL, Guideri F, Bellisai F, Selvi E, Acampa M, Costa A, Maggio R, Garcia‐Gonzalez E, Bisogno S, Morozzi G, Galeazzi M & Laghi‐Pasini F (2007). Comparison of frequency of complex ventricular arrhythmias in patients with positive versus negative anti‐Ro/SSA and connective tissue disease. Am J Cardiol 100, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL, Laghi‐Pasini F (2015). Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med 2, 26. doi:10.3389/fcvm.2015.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzerini PE, Yue Y, Srivastava U, Fabris F, Capecchi PL, Bertolozzi I, Bacarelli MR, Morozzi G, Acampa M, Natale M, El‐Sherif N, Galeazzi M, Laghi‐Pasini F & Boutjdir M (2016). Arrhythmogenicity of anti‐Ro/SSA antibodies in patients with torsades de pointes. Circ Arrhythm Electrophysiol 9, e003419. [DOI] [PubMed] [Google Scholar]
- Li J, Maguy A, Duverger JE, Vigneault P, Comtois P, Shi Y, Tardif JC, Thomas D & Nattel S (2014). Induced KCNQ1 autoimmunity accelerates cardiac repolarization in rabbits: potential significance in arrhythmogenesis and antiarrhythmic therapy. Heart Rhythm 11, 2092–2100. [DOI] [PubMed] [Google Scholar]
- Li J, Seyler C, Wiedmann F, Schmidt C, Schweizer PA, Becker R, Katus HA & Thomas D (2013). Anti‐KCNQ1 K+ channel autoantibodies increase I Ks current and are associated with QT interval shortening in dilated cardiomyopathy. Cardiovasc Res 98, 496–503. [DOI] [PubMed] [Google Scholar]
- Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A & Hoebeke J (1994). Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the β1‐adrenoceptor with positive chronotropic effect. Circulation 89, 2760–2767. [DOI] [PubMed] [Google Scholar]
- Miranda‐Carus ME, Boutjdir M, Tseng CE, DiDonato F, Chan EK & Buyon JP (1998). Induction of antibodies reactive with SSA/Ro‐SSB/La and development of congenital heart block in a murine model. J Immunol 161, 5886–5892. [PubMed] [Google Scholar]
- Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, Morita H, Miura D, Fujimoto Y, Furukawa T, Ueda K, Aizawa Y, Kimura A, Kurachi Y & Ohe T (2007). Anti‐KCNH2 antibody‐induced long QT syndrome: novel acquired form of long QT syndrome. J Am Coll Cardiol 50, 1808–1809. [DOI] [PubMed] [Google Scholar]
- O'Hara T, Virag L, Varro A & Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Baroudi G, Yue Y & Boutjdir M (2005). Novel molecular mechanism involving α1D (Cav1.3) L‐type calcium channel in autoimmune‐associated sinus bradycardia. Circulation 111, 3034–3041. [DOI] [PubMed] [Google Scholar]
- Qu Y & Boutjdir M (2012). Pathophysiology of autoimmune associated congenital heart block In From Prediction to Prevention of Autoimmune Diseases, ed. Conrad K, Chan KL, Fritzler MJ, Humbel RL. & Shoenfeld Y, 16, 96–112. [Google Scholar]
- Qu Y, Xiao GQ, Chen L & Boutjdir M (2001). Autoantibodies from mothers of children with congenital heart block downregulate cardiac L‐type Ca channels. J Mol Cell Cardiol 33, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Salomonsson S, Dörner T, Theander E, Bremme K, Larsson P & Wahren‐Herlenius M (2002). A serologic marker for fetal risk of congenital heart block. Arthritis Rheum 46, 1233–1241. [DOI] [PubMed] [Google Scholar]
- Strandberg L, Winqvist O, Sonesson S‐E, Mohseni S, Salomonsson S, Bremme K, Buyon JP, Julkunen H & Wahren‐Herlenius M (2008). Antibodies to amino acid 200–239 (p200) of Ro52 as serological markers for the risk of developing congenital heart block. Clin Exp Immunol 154, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan H, Saarinen K, Kontula K, Toivonen L & Viitasalo M (1998). Evaluation of QT interval duration and dispersion and proposed clinical criteria in diagnosis of long QT syndrome in patients with a genetically uniform type of LQT1. J Am Coll Cardiol 32, 486–491. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Sukumaran V, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, Arozal W, Sari FR, Lakshmanan AP, Arumugam S, Soetikno V, Rajavel V & Suzuki K (2011). Regulation of inflammation and myocardial fibrosis in experimental autoimmune myocarditis. Inflamm Allergy Drug Targets 10, 218–225. [DOI] [PubMed] [Google Scholar]
- Wolf CM, Moskowitz IP, Arno S, Branco DM, Semsarian C, Bernstein SA, Peterson M, Maida M, Morley GE, Fishman G, Berul CI, Seidman CE & Seidman JG (2005). Somatic events modify hypertrophic cardiomyopathy pathology and link hypertrophy to arrhythmia. Proc Natl Acad Sci USA 102, 18123–18128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao GQ, Hu K & Boutjdir M (2001. a). Direct inhibition of expressed cardiac L‐ and T‐type calcium channels by IgG from mothers whose children have congenital heart block. Circulation 103, 1599–1604. [DOI] [PubMed] [Google Scholar]
- Xiao GQ, Qu Y, Hu K & Boutjdir M (2001. b). Down‐regulation of L‐type calcium channel in pups born to 52 kDa SSA/Ro immunized rabbits. FASEB J 15, 1539–1545. [DOI] [PubMed] [Google Scholar]
- Xiao H, Wang M, Du Y, Yuan J, Zhao G, Tu D & Liao YH (2012). Agonist‐like autoantibodies against calcium channel in patients with dilated cardiomyopathy. Heart Vessels 27, 486–492. [DOI] [PubMed] [Google Scholar]
- Yue Y, Castrichini M, Srivastava U, Fabris F, Shah K, Li Z, Qu Y, El‐Sherif N, Zhou Z, January C, Hussain MM, Jiang XC, Sobie EA, Wahren‐Herlenius M, Chahine M, Capecchi PL, Laghi‐Pasini F, Lazzerini PE & Boutjdir M (2015). Pathogenesis of the novel autoimmune‐associated long‐QT syndrome. Circulation 132, 230–240. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA & January CT (1998). Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74, 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]