

Abstract
Key points
Heart Failure (HF) is accompanied by reduced ventricular function, activation of compensatory neurohormonal mechanisms and marked autonomic dysfunction characterized by exaggerated sympathoexcitation and reduced parasympathetic activity.
With 6 weeks of exercise training, HF‐related loss of choline acetyltransferase (ChAT)‐positive vagal preganglionic neurones is avoided, restoring the parasympathetic tonus to the heart, and the immunoreactivity of dopamine β‐hydroxylase‐positive premotor neurones that drive sympathetic outflow to the heart is reduced. Training‐induced correction of autonomic dysfunction occurs even with the persistence of abnormal ventricular function.
Strong positive correlation between improved parasympathetic tonus to the heart and increased ChAT immunoreactivity in vagal preganglionic neurones after training indicates this is a crucial mechanism to restore autonomic function in heart failure.
Abstract
Exercise training is an efficient tool to attenuate sympathoexcitation, a hallmark of heart failure (HF). Although sympathetic modulation in HF is widely studied, information regarding parasympathetic control is lacking. We examined the combined effects of sympathetic and vagal tonus to the heart in sedentary (Sed) and exercise trained (ET) HF rats and the contribution of respective premotor and preganglionic neurones. Wistar rats submitted to coronary artery ligation or sham surgery were assigned to training or sedentary protocols for 6 weeks. After haemodynamic, autonomic tonus (atropine and atenolol i.v.) and ventricular function determinations, brains were collected for immunoreactivity assays (choline acetyltransferase, ChATir; dopamine β‐hydroxylase, DBHir) and neuronal counting in the dorsal motor nucleus of vagus (DMV), nucleus ambiguus (NA) and rostroventrolateral medulla (RVLM). HF‐Sed vs. SHAM‐Sed exhibited decreased exercise capacity, reduced ejection fraction, increased left ventricle end diastolic pressure, smaller positive and negative dP/dt, decreased intrinsic heart rate (IHR), lower parasympathetic and higher sympathetic tonus, reduced preganglionic vagal neurones and ChATir in the DMV/NA, and increased RVLM DBHir. Training increased treadmill performance, normalized autonomic tonus and IHR, restored the number of DMV and NA neurones and corrected ChATir without affecting ventricular function. There were strong positive correlations between parasympathetic tonus and ChATir in NA and DMV. RVLM DBHir was also normalized by training, but there was no change in neurone number and no correlation with sympathetic tonus. Training‐induced preservation of preganglionic vagal neurones is crucial to normalize parasympathetic activity and restore autonomic balance to the heart even in the persistence of cardiac dysfunction.
Keywords: autonomic dysfunction, aerobic training, ChAT immunoreactivity, DBH immunoreactivity, heart failure
Key points
Heart Failure (HF) is accompanied by reduced ventricular function, activation of compensatory neurohormonal mechanisms and marked autonomic dysfunction characterized by exaggerated sympathoexcitation and reduced parasympathetic activity.
With 6 weeks of exercise training, HF‐related loss of choline acetyltransferase (ChAT)‐positive vagal preganglionic neurones is avoided, restoring the parasympathetic tonus to the heart, and the immunoreactivity of dopamine β‐hydroxylase‐positive premotor neurones that drive sympathetic outflow to the heart is reduced. Training‐induced correction of autonomic dysfunction occurs even with the persistence of abnormal ventricular function.
Strong positive correlation between improved parasympathetic tonus to the heart and increased ChAT immunoreactivity in vagal preganglionic neurones after training indicates this is a crucial mechanism to restore autonomic function in heart failure.
Abbreviations
- AOI
area of interest
- AP
arterial pressure
- ChAT
choline acetyltransferase
- DΒH
dopamine β‐hydroxylase
- DMV
dorsal motor nucleus of the vagus
- ET
exercise trained
- FS
fractional shortening
- HF
heart failure
- HR
heart rate
- IHR
intrinsic heart rate
- ir
immunoreactivity
- LVEDD
left ventricle end diastolic diameter
- LVEDV
left ventricle end diastolic volume
- LVEDP
left ventricle end diastolic pressure
- LVEF
left ventricle ejection fraction
- LVESD
left ventricle end systolic diameter
- LVESV
left ventricle end systolic volume
- MAP
mean arterial pressure
- NA
nucleus ambiguus
- NTS
nucleus of the solitary tract
- PVN
paraventricular nucleus of the hypothalamus
- RVLM
rostroventrolateral medulla
- Sed
sedentary
Introduction
Heart failure (HF), resulting from structural or functional impairment of left ventricular filling or ejection, is a leading cause of death in developed countries (Braunwald, 2013). As a response to this dysfunction, compensatory neurohumoral mechanisms, such as the sympathetic nervous system and the renin–angiotensin–aldosterone system, are activated in an attempt to maintain cardiovascular homeostasis (Braunwald, 2013). In spite of their importance in the acute phase of the disease, in the long term they are harmful, leading to the progression of HF and to a higher mortality (Braunwald, 2013). Sympathoexcitation, the hallmark of chronic HF, is caused by an increased activation of specific neuronal networks responsible for the sympathetic output to the cardiovascular system. Indeed, several studies in myocardial infarcted rats have shown increased glutamatergic and angiotensinergic signalling in the both paraventricular nucleus of the hypothalamus (PVN; Li et al. 2003; Carillo et al. 2012) and rostroventrolateral medulla (RVLM; Gao et al. 2008; Wang et al. 2009), decreased GABAergic activation in the PVN (Zheng et al. 2012) and reduced nitrergic signalling in the nucleus of the solitary tract (NTS; Hirooka et al. 2003; Sakai et al. 2005). It is well established that exaggerated sympathetic activity causes baroreflex dysfunction, alters the autonomic balance to the heart and increases peripheral vasoconstriction, being responsible for the worsening of the disease's prognosis (Barreto et al. 2009; Negrao et al. 2015).
Clinical trials and experimental studies have indicated that exercise training is an efficient adjuvant therapy to reduce the autonomic dysfunction in HF in both animals and humans (Zucker et al. 2009; Negrao et al. 2015) and to decrease mortality in HF patients (Piepoli et al. 2004; Bellardinelli et al. 2012). In patients it was shown that aerobic training decreased neurohumoral activation, reduced sympathetic activity (Coats et al. 1992; Roveda et al. 2003; Fraga et al. 2007; Haack and Zucker, 2015), increased vascular conductance and augmented the perfusion of peripheral organs, thus enhancing the cardiac function (Erbs et al. 2010). It was also shown that exercise training exerts its beneficial effects in animals by attenuating HF‐induced sympathoexcitation within the PVN (Kleiber et al. 2008; Zheng et al. 2012), NTS (Kar et al. 2010) and RVLM (Haack et al. 2012).
Although the involvement of the sympathetic axis in the maintenance/progression and/or regression of HF has been extensively investigated, there is scarce information on the role played by the parasympathetic axis, the counter‐regulatory autonomic axis. The observations that HF was accompanied by abnormal vagal control (Bibevski & Dunlap, 2011) and that stimulation of the vagus nerve improved the prognosis of HF in both men (Schwartz et al. 2008; De Ferrari et al. 2011) and animals (Li et al. 2004; Zhang et al. 2009) indicate that depressed parasympathetic control participates in the genesis of HF. However, there is little information on which brain areas are driving parasympathetic dysfunction in HF and whether an improved central parasympathetic drive participates in the amelioration of cardiovascular control in trained HF rats.
Knowing that aerobic training not only decreases sympathetic outflow, but improves vagal activity in spontaneously hypertensive rats (Ceroni et al. 2009; Cavalleri et al. 2011; Masson et al. 2014), we hypothesized that exercise training is also able to augment the parasympathetic outflow in HF rats, thus contributing to a more complete correction of autonomic dysfunction. Therefore, we evaluated in sedentary and trained HF rats the haemodynamic variables, ventricular function and sympathetic and parasympathetic tonus to the heart. We also quantified the level of expression of enzymatic markers and the number of both parasympathetic preganglionic and sympathetic premotor neurones in brainstem autonomic areas. Intact (SHAM) sedentary and trained rats were included in the study as controls.
Methods
Ethical approval
All the procedures and experimental protocols were in accordance with the Ethical Principles in Animal Research of the Brazilian Society of Science in Laboratory Animals and conformed to The Journal of Physiology’s principles and regulations. They were approved by the Institutional Animal Care and Use Committee of the University of Sao Paulo.
Animals and induction of myocardial infarction
Eight‐week‐old male Wistar rats, weighing 250–300 g, were housed (3–4 animals per cage) in the Animal Facility of the Department of Physiology and Biophysics, Institute of Biomedical Sciences, University of Sao Paulo and kept under controlled temperature (22–24°C) and a 12:12 h light–dark cycle. Food and tap water were provided ad libitum. Forty‐seven rats were used in this study: 10 died within 24 h after the myocardial infarction surgery, seven were excluded for not meeting the echocardiographic criteria for inclusion and were killed with a bolus of ketamine + xylazine (300 mg kg−1 + 60 mg kg−1), and two died within the 6 weeks of training or sedentary protocols. Twenty‐eight rats completed the experiments and were allocated to the four groups (6–8 rats per group).
For the HF induction, rats were deeply anaesthetized (100 mg kg−1 of ketamine hydrochloride (Cetamin 10%, Syntec, Cotia, Brazil) and 10 mg kg−1 of xylazine hydrochloride (Xilasin 2%, Syntec) i.p.) Once the pedal reflex was lost, rats were submitted to orotracheal intubation plus artificial ventilation (Small Animal Ventilator, Model 683, 55–65 insufflations min–1, 2.5 ml per insufflation, Harvard Apparatus, South Natick, MA, USA) with room air. Left thoracotomy was performed and the anterior descendent coronary artery was ligated. The thorax was closed and the pneumothorax drained. For the SHAM group the same procedures were performed, except for the coronary ligation. Whenever needed (i.e. return of whiskers twitching or pedal reflex before the end of the surgical procedure) supplemental doses of ketamine + xylazine i.p. were administered.
After the surgery, rats were treated with ketoprofen (Biofen, 2 mg kg−1 day−1 s.c.; Biofarm, Jaboticabal, Brazil) and enrofloxacin (Baytril, 5 mg kg−1 day−1 s.c.; Bayer, Sao Paulo, Brazil) and placed in individual cages under an electric air heater. Animals were constantly monitored for signs of excessive distress and turned from side to side every 15 min until total recovery from anaesthesia. Rats that failed to recover from anaesthesia or developed respiratory arrest were killed with a bolus of ketamine + xylazine (300 mg kg−1 + 60 mg kg−1). After anaesthestic recovery, rats were provided with food and water ad libitum and placed overnight in a quiet ambient environment, isolated from the surgical room, under controlled temperature (24°C) before returning to the collective cage in the Animal Facility. The antibiotic and analgesic treatment was extended for 3 days.
Echocardiography
Four weeks after the coronary artery ligation, HF and SHAM rats were submitted to echocardiography in order to guarantee that induction of myocardial infarction was effective, leading to an important loss of ventricular function, and that both ET and sedentary (Sed) animals started the training or sedentary protocols in similar conditions. Rats were anaesthetized with ketamine + xylazine (80 mg kg−1 + 8 mg kg−1) for echocardiographic measurements with a 4–12 MHz transducer (Vivid 9, GE Healthcare, Fairfield, CT, USA). HF rats that showed an ejection fraction higher than 40% were excluded from the study.
Training and sedentary protocols
One week before the echocardiography, rats started their adaptation to the treadmill (KT‐300, Inbramed, Porto Alegre, Brazil) with daily sessions of 10 min, the speed ranging from 0.3 to 0.8 km h−1, 0% inclination. One day after the echocardiography, all rats were submitted to exercise testing (starting with 0.3 km h−1, with 0.3 km h−1 increments every 3 min until exhaustion) to determine maximal exercise capacity and calculate the intensity of the training protocol (Ceroni et al. 2009). Slight manual touches in the tail were used in order to stimulate the animals to run until exhaustion (defined as the moment the rat stopped running, lying on the treadmill floor). Maximal exercise tests were repeated at the end of the third and sixth weeks to adjust exercise intensity in exercise trained (ET) and sedentary (Sed) groups and to determine the efficacy of training protocols, respectively. The training protocol consisted of running sessions of 1 h day−1, 5 days week−1, 0% inclination, for a total of 6 weeks. Training intensity ranged from 50% (in the beginning of the 1st week) to 60% (at the end of the 3rd week) of the average maximal speed attained by rats of each group during maximal exercise tests (Ceroni et al. 2009; Cavalleri et al. 2011). Sed rats were handled every day and submitted to a short session of exercise once a week (10 min week−1, 0.3–0.8 km h−1, 0% inclination, which has no training effect) in order to guarantee similar conditions in both groups.
Haemodynamic variables and sympathetic and parasympathetic tonus determinations
At the end of experimental protocols, rats were anaesthetized (ketamine + xylazine i.p.) for the implantation of arterial and venous catheters into the left femoral artery and vein. The catheters were exteriorized through the subcutaneous tissue in the back of the neck and fixed with sutures. Resting values of arterial pressure (AP) and heart rate (HR) were recorded in conscious rats on the next day (PowerLab, ADInstruments, Bella Vista, NSW, Australia, 40 min recording with a sampling frequency of 2000 Hz) at least 24–30 h after the last session of training, as previously described (Ceroni et al. 2009; Masson et al. 2014). Rats were then injected with the muscarinic receptor antagonist atropine sulfate (2 mg kg−1) and 15 min later with the β‐blocker atenolol (4 mg kg−1). On the next day, this procedure was repeated alternating the order of the drugs. Drug choice and dosage were determined according to previous animal and human studies using similar techniques (reviewed by Boyett et al. 2013). Animals showed an agitated behaviour right after atropine injection but returned to resting state in a few minutes. Post‐drug HR values were obtained only after haemodynamic variables were relatively stable and the animals returned to the resting condition. Results are reported separately for each day and intrinsic heart rate (IHR) was calculated as the average HR value obtained after atropine + atenolol and atenolol + atropine. Sympathetic tonus was calculated as the HR after atropine minus the IHR and the parasympathetic tonus as the HR after atenolol minus the IHR.
Ventricular function
On the following day, rats were anaesthetized (ketamine + xylazine i.p.) for left ventricle cannulation in order to evaluate the effects of training and sedentary protocols on the left ventricle function. The ventricular catheter was connected to the recording system for the measurement of ventricular pressure and the determination of left ventricle end diastolic pressure (LVEDP), and positive and negative peaks of the first derivative of the ventricular pressure, indicative of inotropic (+dP/dt) and lusitropic (–dP/dt) properties of the ventricular myocytes.
Tissue sampling
Anaesthesia was then deepened by a bolus of ketamine + xylazine (300 mg kg−1 + 60 mg kg−1). Immediately after the respiratory arrest rats were subjected to transcardiac perfusion (300 ml of Dubecco's modified Eagle's medium, D‐8900, Sigma‐Aldrich, St Louis, MO, USA; peristaltic pump, model 3386, from Daigger, Vernon Hills, IL, USA) followed by 300 ml of 4% paraformaldehyde in 0.01 m phosphate‐buffered saline (PBS). The brains were removed and post‐fixed for 48 h in 4% paraformaldehyde and cryoprotected with 20% and 30% sucrose in 0.01 m PBS (24 h in each solution).
Immunofluorescence assays
Since preganglionic parasympathetic neurones innervating the heart are mainly located in the nucleus ambiguus (NA) and dorsal motor nucleus of the vagus (DMV; Dampney, 1994) in the brainstem, immunofluorescence analysis was directed to these areas. We also analysed the RVLM area that contains most of the premotor sympathetic neurones controlling sympathetic activity to the heart (Dampney, 1994). Serial coronal sections (30 μm) through the brainstem, including the NA, DMV and RVLM, were cut in a cryostat (Leica CM1850, Nussloch, Germany) and every third slice was collected in 0.1 m PBS and stored in anti‐freezing solution containing 30% ethylene glycol and 30% sucrose in 0.01 m PBS. Slices were washed in a solution containing 0.3% Triton X‐100 and 10% normal donkey serum (Millipore, Temecula, CA, USA) in Tris‐PBS. Slices were then incubated overnight at room temperature with both primary antibodies: goat anti‐choline acetyltransferase (ChAT, 1:400, Millipore, AB144P) and mouse anti‐dopamine β‐hydroxylase (DBH, 1:3000, Millipore, MAB318). Brain sections were washed three times in Tris‐PBS, 10 min each, and then incubated with the secondary antibodies Alexa 488 donkey anti‐goat (1:500, Jackson Immunoreasearch Laboratories, West Grove, PA, USA) and Cy3 donkey anti‐mouse (1:500, Jackson Immunoresearch Laboratories) for 2 h. Sections were washed again with Tris‐PBS, ordered on slides and covered with a coverslip and Slowfade® Gold Antifade reagent (Life Technologies, Carlsbad, CA, USA). Negative controls omitted the primary or the secondary antibodies.
ChAT and DBH expression analysis and neuronal counting
DMV, NA and RVLM were identified according to the atlas of Paxinos and Watson (2007), in images obtained through a fluorescence microscope (DMLB, Leica, Wetzlar, Germany; x200 magnification) connected to a digital camera (Axio‐Cam HRC, Carl Zeiss Vision GmbH, Aalen, Germany). The RVLM was identified as an area containing sparse located DBH‐positive neurones caudal to the facial nucleus and ventral to the NA. Areas of interest (AOIs) were carefully drawn surrounding each of the three nuclei, avoiding the inclusion of neighbouring structures (for example, hypoglossal motoneurones located ventrally to the DMV were excluded from the analysis). Predetermined AOIs from DMV, NA and RVLM (corresponding to 101,352, 75,055 and 203,895 μm2, respectively) were used for all immunofluorescence analyses. ChAT and DBH immunoreactivity (ChATir and DBHir) were calculated as the percentage of the area occupied by thresholded signal within each AOI, and were analysed with ImageJ software (NIH) as previously described (Cavalleri et al. 2011). Only signal intensities 3 times higher than the background were considered for the analysis. ChATir was quantified within the intermediate part of the DMV (corresponding to intermediate NTS, as indicated by the whole extension of the area postrema) and in different NA subnuclei, as the pars compacta, pars sub‐compacta and loose part; DBHir was quantified within the RVLM. In each area, measurements were made bilaterally in four to five slices per rat that were carefully chosen to cover similar levels (usually at the mid‐level of the nucleus). ChATir and DBHir were analysed in four rats per group.
In the DMV, NA subnuclei and RVLM, we also counted the number of neurones. All ChAT‐positive and DBH‐positive neurones in which both the cell body and the nucleus were visible and distinguishable from the background were counted; if necessary, slices were rechecked at ×400 magnification. No threshold filters were applied for neuronal counting that was made bilaterally in the whole DMV, NA and RVLM nuclei. Counting was carefully performed in every third section (that is 90 μm apart, avoiding double counting the neurones) in all sections containing the nucleus of interest in five rats per group. Since the objective of our study was to compare the number of neurones, rather than find the absolute number of neurones, no correction factor was applied.
Statistical analysis
Data are presented as means ± SEM. Treadmill performance and comparison of mean arterial pressure before and after drug injection in HF and SHAM, Sed and ET rats were analysed by three‐way ANOVA with repeated measurements (group, condition and time or drug). For the other measurements, comparisons between groups (HF vs. SHAM) and conditions (ET vs. Sed) were made by two‐way ANOVA. Tukey's test was used as the post hoc test. Correlation analyses were performed using Pearson's statistics. Differences were considered significant when P < 0.05.
Results
Effects of myocardial infarction and training on haemodynamic variables and ventricular function
The echocardiographic data were used as the exclusion criteria and only animals showing ejection fraction < 40% were included in the experimental groups. Table 1 shows that ligation of the coronary artery augmented the ventricular size in HF groups, as evidenced by the higher values of left ventricle end systolic volume (LVESV), left ventricle end diastolic volume (LVEDV), left ventricle end systolic diameter (LVESD) and left ventricle end diastolic diameter (LVEDD) compared with SHAM controls. In addition, HF rats showed lower left ventricle ejection fraction (LVEF) and fractional shortening (FS) values (–46% and –69%, respectively) when compared with SHAM controls, indicating an important systolic dysfunction. The similarity between trained and sedentary rats indicated that both groups started the experimental protocols in comparable physiological conditions.
Table 1.
Effects of myocardial infarction on echocardiographic variables, treadmill performance during maximal exercise tests, and haemodynamic and ventricular variables
| Parameter | SHAM‐Sed | SHAM‐ET | HF‐Sed | HF‐ET |
|---|---|---|---|---|
| Echocardiographic variables before training | ||||
| LVESV (cm³) | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.44 ± 0.02* | 0.48 ± 0.02* |
| LVEDV (cm³) | 0.32 ± 0.03 | 0.35 ± 0.02 | 0.71 ± 0.03* | 0.78 ± 0.03* |
| LVEF (%) | 68.18 ± 0.72 | 73.15 ± 1.67 | 38.08 ± 1.09* | 38.60 ± 1.60* |
| LVESD (mm) | 3.92 ± 0.15 | 4.12 ± 0.15 | 8.65 ± 0.30* | 9.11 ± 0.33* |
| LVEDD (mm) | 7.16 ± 0.31 | 6.92 ± 0.18 | 10.11 ± 0.29* | 10.35 ± 0.25* |
| FS (%) | 45.06 ± 2.29 | 40.37 ± 1.76 | 14.44 ± 1.32* | 12.10 ± 1.16* |
| Treadmill performance (km h–1) | ||||
| Week 0 | 1.63 ± 0.06 | 1.50 ± 0.01 | 1.16 ± 0.07* | 1.16 ± 0.07* |
| Week 3 | 1.41 ± 0.10 | 2.07 ± 0.06†, ‡ | 1.05 ± 0.06 | 1.54 ± 0.07*, †‡ |
| Week 6 | 1.50 ± 0.16 | 2.37 ± 0.06†‡ | 0.95 ± 0.08* | 1.84 ± 0.10*, †‡ |
| Gain | −0.13 ± 0.16 | 0.87 ± 0.06† | −0.19 ± 0.05 | 0.68 ± 0.11† |
| Haemodynamic variables after training | ||||
| SAP (mmHg) | 130 ± 2 | 122 ± 3 | 109 ± 4* | 114 ± 4 |
| DAP (mmHg) | 105 ± 4 | 99 ± 3 | 85 ± 3* | 96 ± 4 |
| MAP (mmHg) | 118 ± 3 | 110 ± 2 | 96 ± 4* | 104 ± 4 |
| HR (beats min–1) | 334 ± 6 | 314 ± 10 | 343 ± 17 | 332 ± 10 |
| Ventricular variables after training | ||||
| LVEDP (mmHg) | 1.00 ± 0.04 | 2.12 ± 0.06 | 12.83 ± 3.65* | 8.60 ± 1.59* |
| +dP/dt (mmHg s–1) | 4638 ± 245 | 4419 ± 207 | 3442 ± 266* | 3104 ± 263* |
| –dP/dt (mmHg s–1) | −3593 ± 169 | −3672 ± 134 | −2242 ± 110* | −2236 ± 187* |
Values are means ± SEM. n = 6–8 rats in each group. +dP/dt, positive peak of the first derivative of ventricular pressure; –dP/dt, negative peak of the first derivative of ventricular pressure; DAP, diastolic arterial pressure; FS, fractional shortening; HR, heart rate; LVEDP, left ventricle end diastolic pressure; LVEDV, left ventricle end diastolic volume; LVEF, left ventricle ejection fraction; LVESD, left ventricle end systolic diameter; LVESV, left ventricle end systolic volume; LVEDD, left ventricle end diastolic diameter; MAP, mean arterial pressure; SAP, systolic arterial pressure. Significant difference (P < 0.05): * vs. SHAM, † vs. Sed, ‡ vs. week 0.
At the beginning of protocols (week 0), HF rats showed a reduced exercise capacity when compared to SHAM operated groups (Table 1). Treadmill performance was not changed in HF and SHAM groups submitted to the sedentary protocol, but training significantly improved it in both groups. Observe that the attained velocity in HF‐ET rats was lower than in SHAM‐ET rats during the entire protocol. However, the performance gain from the beginning to the end of the training protocol was similar in both groups (Table 1). There were no significant differences in body mass (–10% and –7% for SHAM‐ET and HF‐ET vs. 430 ± 10 and 430 ± 15 g in SHAM‐Sed and HF‐Sed, respectively).
Haemodynamic recordings in conscious rats showed that resting systolic, diastolic and mean AP values were lower in HF‐Sed when compared to SHAM‐Sed rats (Table 1). AP was not significantly changed by training in HF and SHAM groups (ET vs. respective Sed controls, Table 1), but at the end of protocols AP values exhibited by HF‐ET rats did not differ from those of the SHAM‐ET group. There were no significant differences in resting HR between the groups (Table 1). Table 2 shows detailed information regarding resting mean AP and HR, the effects of atenolol, atropine and double blockade on these variables, as well as the calculation of sympathetic and parasympathetic tonus to the heart. There were no carryover effects of the drugs on resting HR and mean arterial pressure (MAP) from the first to the second day (Table 2).
Table 2.
Effects of atropine and/or atenolol administration on heart rate (HR) and mean arterial pressure (MAP) and the calculation of sympathetic and parasympathetic tonus
| Parameter | SHAM‐Sed | SHAM‐ET | HF‐Sed | HF‐ET |
|---|---|---|---|---|
| Day 1 | ||||
| Resting | ||||
| HR (beats min–1) | 334 ± 6 | 314 ± 10 | 343 ± 17 | 332 ± 10 |
| MAP (mmHg) | 118 ± 3 | 110 ± 2 | 96 ± 4* | 104 ± 4 |
| After Atropine | ||||
| HR (beats min–1) | 437 ± 12‡ | 427 ± 13‡ | 434 ± 9‡ | 428 ± 8‡ |
| MAP (mmHg) | 119 ± 3 | 115 ± 3 | 98 ± 4* | 109 ± 4 |
| After atropine + atenolol | ||||
| HR (beats min–1) | 361 ± 5§ | 354 ± 4§ | 323 ± 11§, * | 358 ± 6§† |
| MAP (mmHg) | 115 ± 2 | 116 ± 2 | 91 ± 3* | 109 ± 4 |
| Day 2 | ||||
| Resting | ||||
| HR (beats min–1) | 333 ± 9 | 313 ± 8 | 342 ± 17 | 333 ± 9 |
| MAP (mmHg) | 118 ± 1 | 113 ± 3 | 99 ± 1* | 104 ± 2 |
| After Atenolol | ||||
| HR (beats min–1) | 279 ± 5‡ | 270 ± 3‡ | 274 ± 7‡ | 274 ± 4‡ |
| MAP (mmHg) | 109 ± 3 | 104 ± 1 | 102 ± 3 | 102 ± 6 |
| After atenolol + atropine | ||||
| HR (beats min–1) | 337 ± 6§ | 333 ± 6§ | 307 ± 6* | 327 ± 4§† |
| MAP (mmHg) | 112 ± 2 | 110 ± 3 | 95 ± 2* | 105 ± 5 |
| Autonomic tonus | ||||
| IHR | 349 ± 5 | 344 ± 5 | 316 ± 8* | 342 ± 4† |
| Sympathetic tonus (beats min–1) | 88 ± 8 | 83 ± 9 | 118 ± 4* | 86 ± 6† |
| Parasympathetic tonus (beats min–1) | −70 ± 4 | −74 ± 6 | −42 ± 4* | −68 ± 3† |
Values are means ± SEM. n = 6–8 rats in each group. Results for each experimental day are reported separately. Intrinsic heart rate (IHR) was calculated as the mean of ‘after atropine + atenolol’ and ‘after atenolol + atropine’ values. Sympathetic and parasympathetic tonus were calculated as ‘after atropine’ HR minus IHR and ‘after atenolol’ HR minus IHR, respectively. Significant difference (P < 0.05): ‡ vs. respective resting value; § vs. atropine or atenolol; * vs. SHAM; † vs. Sed.
Myocardial infarction was accompanied by a significant reduction of IHR (316 ± 8 vs. 349 ± 5 beats min−1 in SHAM‐Sed), higher sympathetic tonus (HF‐Sed = +118 ± 4 beats min−1, a 36% increase over the SHAM‐Sed value, P < 0,05) and lower parasympathetic tonus (–42 ± 4 vs. –70 ± 4 beats min−1 in SHAM‐Sed, P < 0.05). In contrast, training restored these deficits: HF‐ET exhibited higher IHR, reduced sympathetic and increased parasympathetic tonus (all values significant when compared to HF‐Sed and similar to those observed in SHAM groups; Table 2). Compared to resting pressure there were no significant changes in mean AP following the drugs used. Notice that except for the values after atenolol, BP maintain the same pattern observed in the resting condition, that is, significantly reduced in HF‐Sed vs. SHAM‐Sed and normalized by training in HF‐ET rats (Table 2). The relationship between sympathetic and parasympathetic tonus in the determination of resting HR is depicted in Fig. 1. Observe that opposite to SHAM groups, the resting HR (represented by the arrow) in HF‐Sed rats is driven by the sympathetic tonus. In contrast, baseline HR of HF‐ET rats is maintained by the parasympathetic outflow (arrow in the HF‐ET group, a pattern similar to that observed in SHAM groups), demonstrating that cardiac vagal tonus is mostly important for cardiac function in infarcted hearts after training.
Figure 1. Comparison of cardiac sympathetic (ST, open bars) and parasympathetic tonus (PT, filled bars), intrinsic heart rate (intersection between ST and PT) and resting heart rate (indicated by arrows) in infarcted (HF) and SHAM groups submitted to sedentary (Sed) and training (ET) protocols .
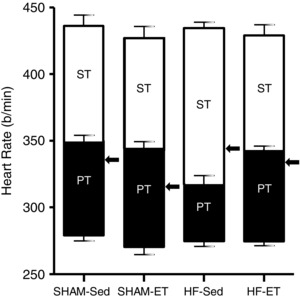
n = 6–8 rats in each group. Significant differences are shown in Table 2.
After the completion of haemodynamic recordings, rats were anaesthetized for left ventricle catheterization. LVEDP was markedly increased in HF rats (12.8 ± 3.7 vs. 1.0 ± 0.4 mmHg in SHAM‐Sed, Table 1), while both positive and negative dP/dt were significantly reduced (3442 ± 266 and –2242 ± 110 mmHg s−1, respectively, corresponding to 26% reduction of LV inotropism and 38% decrease of LV lusitropism when compared to SHAM‐Sed, Table 1). Six weeks of exercise training did not change these variables.
Effects of myocardial infarction and training on ChAT‐ and DBH‐positive neurones in autonomic areas controlling sympathetic and parasympathetic outflow
ChATir was are shown in Table 2 analysed in both brainstem areas containing the cell bodies of parasympathetic preganglionic neurones. In the whole extension of NA, the establishment of HF was accompanied by a large reduction of ChATir (Fig. 2 A–C). In contrast, training avoided the loss of ChATir and abrogated HF‐induced changes, but had no effect in SHAM rats. Quantitative analysis revealed that HF was accompanied by a robust reduction in ChATir in all NA subnuclei: –35% in pars compacta, –54% in pars sub‐compacta and –53% in the loose part (Fig. 2 D, E and F, respectively). Notice that 6 weeks of exercise training was able to completely reverse HF effects on NA ChAT expression: in the three subnuclei ChATir of HF‐ET was significantly higher that of HF‐Sed and did not differ from those exhibited by SHAM groups (Fig. 2 D–F). These changes were confirmed by neuronal counting. When compared to SHAM‐Sed, the number of ChAT‐positive neurones in HF‐Sed was reduced in all NA subnuclei (pars compacta: from 616 ± 33 to 409 ± 11; pars subcompacta: from 312 ± 22 to 163 ± 18; loose part: from 247 ± 15 to 181 ± 20; Fig. 2 G, H and I, respectively). Exercise training significantly reversed these alterations, since HF‐ET rats showed a significant increase when compared to HF‐Sed and no difference when compared to SHAM groups.
Figure 2. Effects of heart failure (HF) and exercise training on the expression of ChAT‐positive neurones within the nucleus ambiguus .
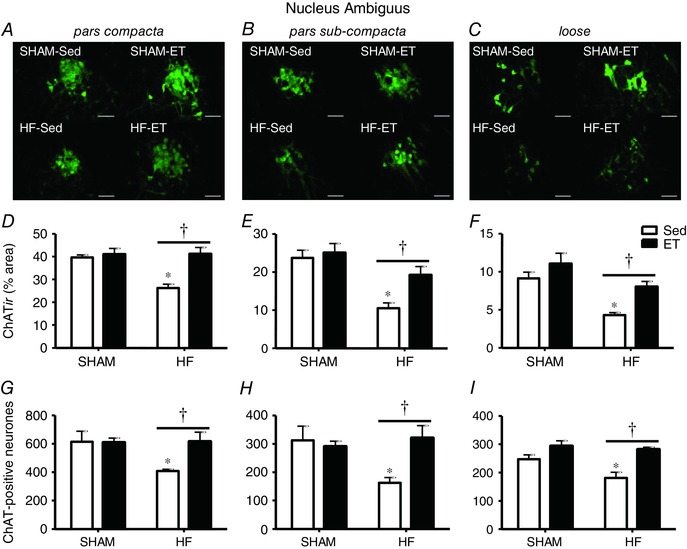
A–C, photomicrographs comparing the effects of infarction and training on ChATir in different parts of the nucleus ambiguus of SHAM and HF rats submitted to sedentary (Sed) or training (ET) protocols. Scale bars: 100 μm. D–F, percentage area occupied by ChAT‐positive neurones in pars compacta (D), pars sub‐compacta (E) and loose part (F) of the nucleus ambiguus in each experimental group. n = 4–5 slices/rat, 4 rats/group. G–I, number of ChAT‐positive neurones in pars compacta (G), pars sub‐compacta (H) and loose part (I) of the nucleus ambiguus in each experimental group. n = 5 rats/group. Significant difference (P < 0.05): ∗ vs. SHAM; † vs. respective Sed controls.
Comparison of ChATir in the central part of the DMV showed similar effects (photomicrographs and quantitative data in Fig. 3 A and C, respectively). HF‐Sed vs. SHAM‐Sed exhibited a 50% reduction in ChATir, which is significantly increased by training. Despite a training‐induced augmentation of 46% in the DMV of HF‐ET vs. HF‐Sed rats, ChATir was still reduced compared to that of the SHAM groups (Fig. 3 C). Neuronal counting within the DMV also confirmed these changes: HF‐Sed vs. SHAM‐Sed showed a significant reduction in the number of ChAT‐positive neurones (from 2832 ± 84 to 1927 ± 193; Fig. 3 E), which was completely normalized following exercise training. Notice that within both DMV and NA of SHAM rats, training had no effect on ChATir and neuronal counting.
Figure 3. Effects of heart failure (HF) and exercise training on the expression of ChAT‐positive neurones in the dorsal motor nucleus of the vagus and DBH‐positive neurones in the rostroventrolateral medulla .
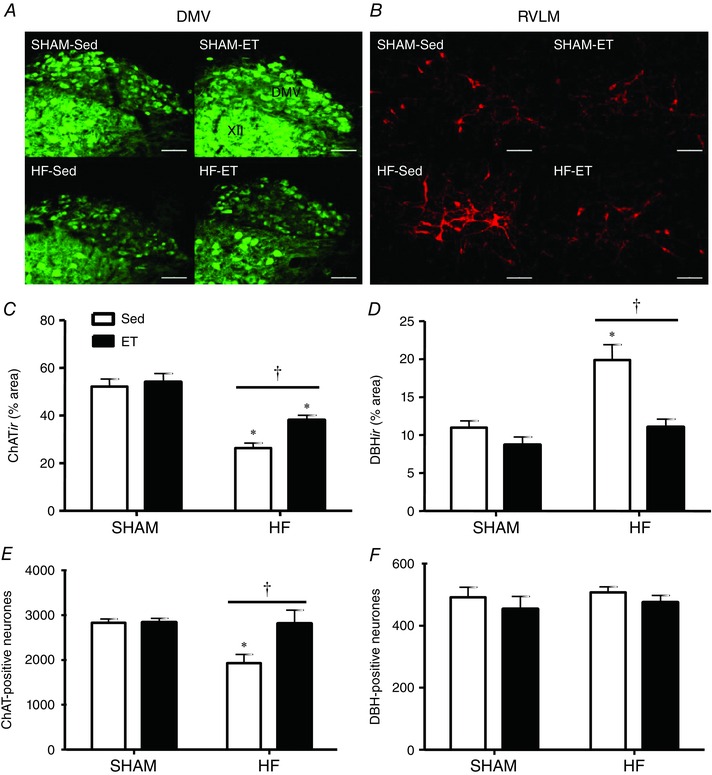
A and B, photomicrographs comparing the effects of infarction and training on ChATir in the intermediate part of the dorsal motor nucleus of the vagus (DMV) and on dopamine β‐hydroxylase immunoreactivity (DBHir) in the rostroventrolateral medulla (RVLM) of SHAM and HF rats submitted to sedentary (Sed) or training (ET) protocols. Scale bars: 100 μm. C and D, percentage area occupied by ChAT‐positive neurones in the DMV (C) and DBH‐positive neurones in the RVLM (D) in each experimental group. n = 4–5 slices/rat, 4 rats/group. E and F, number of DMV ChATir‐positive neurones (E) and RVLM DBHir‐positive neurones (F) in each experimental group. n = 5 rats/group. Significant difference (P < 0.05): ∗ vs. SHAM; † vs. respective Sed controls.
In the same rats, counting of DBH‐positive neurones revealed that HF and training were unable to change neuronal counting within the RVLM (Fig. 3 F). Opposite to that observed for parasympathetic preganglionic neurones, HF was accompanied by a large increase in RVLM DBHir, which was reversed by training (photomicrographs in Fig. 3 B). Observe that DBH‐positive neurones in a HF‐Sed rat occupied a larger percentage area (increased dendritic branching?) when compared to a SHAM‐Sed, a pattern not observed in the HF‐ET. Group analysis confirmed these observations: DBHir was increased by 82% in HF‐Sed vs. SHAM‐Sed, but was significantly reduced by training (11 ± 1% in the HF‐ET, a value similar to that observed in SHAM groups; Fig. 3 D). Again, training did not change DBHir in SHAM‐ET rats when compared to respective Sed controls.
Interestingly, we found significant strong correlations between the parasympathetic tonus and ChATir within both NA pars sub‐compacta and DMV (r = –0.739 and –0.820, respectively; Fig. 4 and Table 3), indicating that the higher the immunofluorescence area of parasympathetic preganglionic neurones in these areas, the higher the parasympathetic outflow to the heart. Observe that parasympathetic tonus is represented by reductions in HR below the IHR yielding negative values; therefore, in this case negative r indicates a positive correlation. We also obtained significant relationships between changes in parasympathetic tonus and ChATir within the other NA subnuclei, but the correlations were moderate (r = –0.583 and –0.652 for pars compacta and loose part, respectively; Table 3). Notice that in trained rats, sympathetic tonus to the heart decreased proportionally to the reduction in RVLM DBHir, but the correlation was weaker (r = 0.475) and did not attain significance (Table 3).
Figure 4. Correlations between parasympathetic tonus and ChATir content within the nucleus ambiguus pars subcompacta (NA; A) and the dorsal motor nucleus of the vagus (DMV; B) .
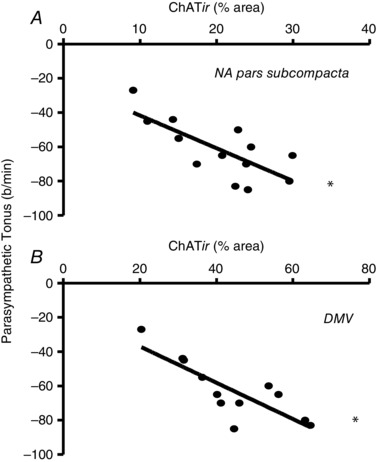
Linear regression equations and regression coefficients are shown in Table 3. *Significant correlation (P < 0.05).
Table 3.
Regression equations correlating ChATir with parasympathetic tonus and DΒHir with sympathetic tonus in SHAM and HF groups submitted to sedentary (Sed) or training (ET) protocols
| Correlations | Regression equation | r | P |
|---|---|---|---|
| Parasympathetic tonus × ChATir | |||
| NA pars compacta | y = −1.17x – 16 | −0.583 | 0.037* |
| NA pars subcompacta | y = −1.91x – 23 | −0.739 | 0.004* |
| NA loose | y = −3.87x – 31 | −0.652 | 0.016* |
| DMV (intermediate part) | y = −1.06x – 16 | −0.820 | 0.001* |
| Sympathetic tonus × DBHir | |||
| RVLM | y = 2.08x + 68 | 0.475 | 0.086 |
In each brain area, ChATir or DBHir represents the area of immunofluorescence of preganglionic vagal or premotor neurones that is correlated with respective parasympathetic or sympathetic tonus measured in the conscious state. n = 15–16 values in each correlation. ∗Significant correlation.
Discussion
Our results confirm that myocardial ischaemia alters the functionality of the heart, reducing both the inotropism and lusitropism of the left ventricle, and increases the sympathetic and decreases the parasympathetic outflow to the heart, leading to heart failure. In addition, the present set of data revealed several new observations: (i) decreased vagal tonus to the heart after myocardial infarction is associated with reduced number of ChAT‐positive preganglionic parasympathetic neurones in the NA and DMV, the main sources of vagal innervations to the heart; (ii) with exercise training HF‐induced loss of ChAT‐positive preganglionic parasympathetic neurones in both brainstem nuclei is avoided; (iii) training‐induced preservation of ChATir in preganglionic vagal neurones correlates with higher parasympathetic tonus to the heart, being an essential mechanism to reverse cardiac autonomic dysfunction in HF rats; (iv) HF does not change the number of DBH‐positive neurones within the RVLM but increases the DBHir; (v) training abrogates both the increased sympathetic tonus to the heart and DBHir in HF rats; (vi) besides improving autonomic function, exercise training corrects the decreased intrinsic pacemaker rate found in HF rats; and (vii) together these training‐induced changes restore the autonomic balance to the heart even in the persistence of ventricular dysfunction.
Exercise training is a safe and efficient treatment for HF patients since it positively affects , peripheral vasculature and muscle function, corrects the autonomic balance, increases the quality of life and reduces mortality (Piepoli et al. 2004; McKelvie, 2008; Belardinelli et al. 2012). Previous studies on the mechanisms conditioning the beneficial effects of exercise training on autonomic balance were focused almost exclusively on changes in sympathetic activity and neurohormonal profile (Fraga et al. 2007; Gao et al. 2008; Kleiber et al. 2008; Zheng et al. 2012). Indeed, reduced sympathoexcitation (as indicated by downregulation of the renin–angiotensin system, decreased glutamatergic activation within brain areas controlling sympathetic activity, improved baroreflex control and reduced peripheral vasoconstriction) is a common feature observed in trained HF animals (Liu et al. 2000; Rondon et al. 2006; Kleiber et al. 2008; Kar et al. 2010; Haack et al. 2012; Zheng et al. 2012) and humans (Roveda et al. 2003; Fraga et al. 2007). Our study confirms those previous findings, showing in addition that normalized sympathetic outflow to the heart in trained HF rats was accompanied by decreased DBHir in the RVLM, which could be due to lower content of the enzyme within DBH‐positive neurones and/or reduced neuronal branching. Interestingly, HF and training did not change the number of DBH‐positive neurones. A previous study by Mischel et al. (2014) had already shown that C1 presympathetic neurones from sedentary rats exhibited unchanged cell bodies but were more branched than those of physically active rats. The larger surface area, facilitating synapse formation in inactive rats, may enhance the overall sensitivity to excitatory stimuli of RVLM neurones (Mischel et al. 2014). Coherently, a similar structural pattern in DBH‐positive neurones now observed in HF‐Sed and HF‐ET rats could explain the changes in DBHir and sympathetic drive presently observed.
Two other original observations were that (i) autonomic dysfunction to the heart in HF‐Sed rats is accompanied by a significant reduction of ChAT‐positive vagal preganglionic neurones and marked reduction in the area occupied by ChATir in both NA and DMV; and (ii) exercise training restores both neurone number and ChATir and normalizes the reduced parasympathetic tonus to the heart in HF. A significant loss of cardiac vagal preganglionic neurones has been previously reported in 12‐week‐old SHR when compared to both age‐matched normotensive controls and 4‐week‐old SHR (Corbett et al. 2007). Importantly, the present set of data showed that training was able to reverse HF‐induced reduction in neuronal counting and ChATir and that training‐induced preservation of preganglionic vagal neurones was positively correlated with the normalization of the parasympathetic tonus to the heart. These observations highlight the importance of exercise as a trigger of beneficial cardiac adjustments and that of parasympathetic outflow as a major contributor of improved cardiovascular control. La Rovere et al. (1998) have already emphasized that preservation of cardiac vagal activity after myocardial infarction has significant prognostic value independently of the ejection fraction. Indeed, previous studies showed that direct stimulation of the vagus nerve in rats (Li et al. 2004), dogs (Zhang et al. 2009) and humans (Schwartz et al. 2008; De Ferrari et al. 2011) and its activation through cholinergic drugs in rats (Lataro et al. 2013; Sabino et al. 2013) are beneficial for the treatment of HF. Our study is the first demonstration that aerobic training, by restoring a near normal number of vagal preganglionic neurones, is as effective as pharmacological or direct stimulation of the vagus nerve to improve cardiovascular control in HF individuals. This finding is of great importance not only for a better understanding of the physiopathology of HF, but also for directing our attention to an effective therapeutic approach, without side effects. It is not known whether the reduced number of ChAT‐positive neurones in the HF‐Sed rats is due to neurones switching off or neuronal death, and also the cellular mechanism(s) responsible for the great improvement on neuronal plasticity/activity after training remains to be identified. Future studies will address these questions.
The lack of significant improvement in ventricular function, as depicted by our results of LVEDP and positive and negative dP/dt, despite the great improvements in autonomic function, should not lead to the interpretation that exercise training has no beneficial effects. While some studies pointed towards an amelioration of cardiac function in heart failure trained animals (Kleiber et al. 2008; Zheng et al. 2012) and patients (Erbs et al. 2010), others have found no improvement in ventricular function in animals (Liu et al. 2000; Rondon et al. 2006; Kar et al. 2010; Haack et al. 2012) or humans (Roveda et al. 2003). We believe this divergence could be due to differences in intensity and duration of training protocols. Indeed, Wisløff et al. (2007) have shown that high intensity interval training has better effects on left ventricle remodelling when compared to moderate continuous training. Independently of the effects of exercise on ventricular function, correction of autonomic tonus to the heart, notably the parasympathetic tonus, is of great importance in the treatment of heart failure. Several studies using different markers of vagal activity and autonomic balance (La Rovere et al. 1998; Cole et al. 1999; Schmidt et al. 1999) have demonstrated that low vagal tone controlling the heart is a strong predictor of higher mortality, independently of ventricular function.
Similar to the studies by Sanders et al. (2004) and Yanni et al. (2011), we also observed reduced IHR in HF rats, the so‐called sick sinus syndrome. Interestingly, aerobic training was also able to correct the dysfunctional sinus node and normalize the intrinsic pacemaker rate, highlighting another potential mechanism through which exercise can benefit HF individuals. With regard to a recent CrossTalk debate in The Journal of Physiology on the mechanisms conditioning bradycardia in athletes (Coote & White, 2015; D'Souza et al. 2015), our data showed that the beneficial effects of training in HF are driven by both increased cardiac parasympathetic activity and training‐induced increase in IHR. While D'Souza et al. (2015) showed a training‐induced downregulation of the pacemaker channel in athletes, our data in downregulated HF rats suggest a complete reversion after training, showing the potentiality of this mechanism to correct HR dysfunction in whichever direction it occurs.
One limitation of this study is that we did not identify, within the ChAT‐positive cells, those projecting to the heart. However, the strong correlations between parasympathetic tonus × ChAT neuronal profile within the intermediate DMV and NA pars sub‐compacta draw our attention to these areas as the main sources of vagal innervation to the heart. Indeed, our data showed that improvement in the expression/activity of vagal preganglionic neurones is crucial for parasympathetic control of the heart. The huge augmentation of ChATir in HF‐ET rats indicated that training should also affect other neurones such as those responsible for the innervation of the gastrointestinal tract, suggesting a broad effect on body homeostasis. In addition, we cannot exclude the participation of other structures such as the parasympathetic ganglia (Bibevski & Dunlap, 1999), the postganglionic intramural neurones and even the myocardial muscarinic receptors (Dunlap et al. 2003) in the improvement of autonomic control in HF‐ET rats. Future experiments may clarify these points.
The chosen criteria to define HF in our rats (LV ejection fraction < 40%) could be another potential limitation of this study. Indeed, data from studies in humans showed that LV ejection fraction has little or no correlation with exercise capacity, an important variable to determine LV systolic dysfunction in HF patients (Franciosa et al. 1981). On the other hand, studies in humans (Ahnve et al. 1986; Wong et al. 2004) and animals (Sjaastad et al. 2000) showed a good correlation between worsening of the LVEF and increased morbidity and mortality. It should be noted that besides reduced LVEF, our rats also exhibited clear signals of cardiomegaly, reduced fractional shortening, higher LVEDP, depressed ventricular contraction and relaxation and, importantly, worse performance in maximal exercise tests. Therefore, we believe that our HF rats developed a persistent heart failure.
In conclusion, data in the present study show that HF rats exhibit parasympathetic depression in addition to sympathoexcitation, leading to a marked autonomic dysfunction. Importantly, training‐induced preservation of preganglionic vagal neurones (occurring simultaneously with reduced DBH expression in premotor sympathetic neurones) is crucial to normalize parasympathetic activity, restore autonomic balance and protect the heart even in the persistence of depressed cardiac function.
Additional information
Competing interests
The authors declare they have no competing interests
Author contributions
M.H.A.I.: myocardial infarction, exercise training, functional recordings, immunofluorescence assays, assembly of data, manuscript writing. C.R.S.: immunofluorescence assays; C.P.J.: echocardiography; A.C.: functional recordings; C.E.N.: conception and design, discussion of data; L.C.M.: conception and design, data analysis and interpretation, manuscript writing. All persons listed as authors qualify for authorship. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the following research grants: Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP, grants 2014/06772‐8 (fellowship to M.H.A.I.) and 2011/51410‐9 (research grant to L.C.M.)) and Conselho Nacional de Pesquisa (CNPq, grants 304060/2011‐9 and 301867/2010‐0: research fellow to L.C.M. and C.E.N., respectively).
Translational perspective
This study demonstrates that (i) cardiac dysfunction, in addition to sympathoexcitation, progresses with the loss of vagal preganglionic neurones innervating the heart and simultaneous worsening of parasympathetic control, and (ii) exercise training is able to block these alterations in addition to correcting the reduced intrinsic heart rate. This extends our knowledge on the pathophysiology of heart failure and has clear clinical implications. It leads to the possibility of new and more efficient therapeutic strategies to treat patients with heart failure (without the side effects inherent in the usual treatments) and perhaps offers a better prognosis for them. Future experiments investigating the mechanisms by which cardiac vagal preganglionic neurones are lost and/or preserved will refine understanding of the underlying factors and will help to define more precise therapeutic tools.
References
- Ahnve S, Gilpin E, Henning H, Curtis G, Collins D & Ross J Jr (1986). Limitations and advantages of the ejection fraction for defining high risk after acute myocardial infarction. Am J Cardiol 58, 872–878. [DOI] [PubMed] [Google Scholar]
- Barretto AC, Santos AC, Munhoz R, Rondon MU, Franco FG, Trombetta IC, Roveda F, de Matos LN, Braga AM, Middlekauff HR & Negrão CE (2009). Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 135, 302–307. [DOI] [PubMed] [Google Scholar]
- Belardinelli R, Georgiou D, Cianci G & Purcaro A (2012). 10‐year exercise training in chronic heart failure: a randomized controlled trial. J Am Coll Cardiol 60, 1521–1528. [DOI] [PubMed] [Google Scholar]
- Bibevski S & Dunlap ME (1999). Ganglionic mechanisms contribute to diminished vagal control in heart failure. Circulation 99, 2958–2963. [DOI] [PubMed] [Google Scholar]
- Bibevski S & Dunlap ME (2011). Evidence for impaired vagus nerve activity in heart failure. Heart Fail Rev 16, 129–135. [DOI] [PubMed] [Google Scholar]
- Boyett MR, D'Souza A, Zhang H, Morris GM, Dobrzynski H & Monfredi O (2013). Viewpoint: Is the resting bradycardia in athletes the result of remodeling of the sinoatrial node rather than high vagal tone? J Appl Physiol 114, 1351–1355. [DOI] [PubMed] [Google Scholar]
- Braunwald E (2013). Heart failure. JACC Heart Fail 1, 1–20. [DOI] [PubMed] [Google Scholar]
- Carillo BA, Oliveira‐Sales EB, Andersen M, Tufik S, Hipolide D, Santos AA, Tucci PJ, Bergamaschi CT & Campos RR (2012). Changes in GABAergic inputs in the paraventricular nucleus maintain sympathetic vasomotor tone in chronic heart failure. Auton Neurosci 171, 41–48. [DOI] [PubMed] [Google Scholar]
- Cavalleri MT, Burgi K, Cruz JC, Jordão MT, Ceroni A & Michelini LC (2011). Afferent signaling drives oxytocinergic pre‐autonomic neurons and mediates training‐induced plasticity. Am J Physiol Regul Integr Comp Physiol 301, 958–996. [DOI] [PubMed] [Google Scholar]
- Ceroni A, Chaar LJ, Bombein RL & Michelini LC (2009). Chronic absence of baroreceptor inputs prevents training‐induced cardiovascular adjustments in normotensive and spontaneously hypertensive rats. Exp Physiol 94, 630–640. [DOI] [PubMed] [Google Scholar]
- Coats AJ, Adamopoulos S, Radaelli A, McCance A, Meyer TE, Bernardi L, Solda PL, Davey P, Ormerod O & Forfar C (1992). Controlled trial of physical training in chronic heart failure. Exercise performance, hemodynamics, ventilation, and autonomic function. Circulation 85, 2119–2131. [DOI] [PubMed] [Google Scholar]
- Cole CR, Blackstone EH, Pashkow FJ, Snader CE & Lauer MS (1999). Heart rate recovery immediately after exercise as a predictor of mortality. N Engl J Med 341, 1351–1357. [DOI] [PubMed] [Google Scholar]
- Coote JH & White MJ (2015). CrossTalk proposal: Bradycardia in the trained athlete is attributable to high vagal tone. J Physiol 593, 1745–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett EK, Mary DA, McWilliam PN & Batten TF (2007). Age‐related loss of cardiac vagal preganglionic neurones in spontaneously hypertensive rats. Exp Physiol 92, 1005–1013. [DOI] [PubMed] [Google Scholar]
- Dampney RA (1994). Functional organization of central pathways regulating the cardiovascular system. Physiol Rev 74, 323–364. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Crijns HJ, Borggrefe M, Milasinovic G, Smid J, Zabel M, Gavazzi A, Sanzo A, Dennert R, Kuschyk J, Raspopovic S, Klein H, Swedberg K & Schwartz PJ (2011). Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur Heart J 32, 847–855. [DOI] [PubMed] [Google Scholar]
- Dunlap ME, Bibevski S, Rosenberry TL & Ernsberger P (2003). Mechanisms of altered vagal control in heart failure: influence of muscarinic receptors and acetylcholinesterase activity. Am J Physiol Heart Circ Physiol 285, 1632–1640. [DOI] [PubMed] [Google Scholar]
- D'Souza A, Sharma S & Boyett MR (2015). CrossTalk opposing view: Bradycardia in the trained athlete is attributable to a downregulation of a pacemaker channel in the sinus node. J Physiol 593, 1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbs S, Höllriegel R, Linke A, Beck EB, Adams V, Gielen S, Möbius‐Winkler S, Sandri M, Kränkel N, Hambrecht R & Schuler G (2010). Exercise training in patients with advanced chronic heart failure (NYHA IIIb) promotes restoration of peripheral vasomotor function, induction of endogenous regeneration, and improvement of left ventricular function. Circ Heart Fail 3, 486–494. [DOI] [PubMed] [Google Scholar]
- Fraga R, Franco FG, Roveda F, de Matos LN, Braga AM, Rondon MU, Rotta DR, Brum PC, Barretto AC, Middlekauff HR & Negrão CE (2007). Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur J Heart Fail 9, 630–636. [DOI] [PubMed] [Google Scholar]
- Franciosa JA, Park M & Levine TB (1981). Lack of correlation between exercise capacity and indexes of resting left ventricular performance in heart failure. Am J Cardiol 47, 33–39. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang WZ, Wang W & Zucker IH (2008). Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension 52, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Engler CW, Papoutsi E, Pipinos II, Patel KP & Zucker IH (2012). Parallel changes in neuronal AT1R and GRK5 expression following exercise training in heart failure. Hypertension 60, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK & Zucker IH (2015). Central mechanisms for exercise training‐induced reduction in sympatho‐excitation in chronic heart failure. Auton Neurosci 188, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirooka Y, Shigematsu H, Kishi T, Kimura Y, Ueta Y & Takeshita A (2003). Reduced nitric oxide synthase in the brainstem contributes to enhanced sympathetic drive in rats with heart failure. J Cardiovasc Pharmacol 42 Suppl 1, S111–S115. [DOI] [PubMed] [Google Scholar]
- Kar S, Gao L & Zucker IH (2010). Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing‐induced heart failure. J Appl Physiol 108, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiber AC, Zheng H, Schultz HD, Peuler JD & Patel KP (2008). Exercise training normalizes enhanced glutamate‐mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol 294, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT Jr, Marcus FI, Mortara A & Schwartz PJ (1998). Baroreflex sensitivity and heart‐rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet 351, 478–484. [DOI] [PubMed] [Google Scholar]
- Lataro RM, Silva CA, Fazan R Jr, Rossi MA, Prado CM, Godinho RO & Salgado HC (2013). Increase in parasympathetic tone by pyridostigmine prevents ventricular dysfunction during onset of heart failure. Am J Physiol Regul Integr Comp Physiol 304, 908–916. [DOI] [PubMed] [Google Scholar]
- Li M, Zheng C, Sato T, Kawada T, Sugimachi M & Sunagawa K (2004). Vagal nerve stimulation markedly improves long‐term survival after chronic heart failure in rats. Circulation 109, 120–124. [DOI] [PubMed] [Google Scholar]
- Li YF, Cornish KG & Patel KP (2003). Alteration of NMDA NR1 receptors within the paraventricular nucleus of hypothalamus in rats with heart failure. Circ Res 93, 990–997. [DOI] [PubMed] [Google Scholar]
- Liu JL, Irvine S, Reid IA, Patel KP & Zucker IH (2000). Chronic exercise reduces sympathetic nerve activity in rabbits with pacing‐induced heart failure: A role for angiotensin II. Circulation 102, 1854–1862. [DOI] [PubMed] [Google Scholar]
- Masson GS, Costa TSR, Yshii L, Fernandes DC, Soares PP, Laurindo FR, Scavone C & Michelini LC (2014). Time‐dependent effects of training on cardiovascular control in spontaneously hypertensive rats: Role for brain oxidative stress and inflammation and baroreflex sensitivity. PLoS One 9, e94927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKelvie RS (2008). Exercise training in patients with heart failure: clinical outcomes, safety, and indications. Heart Fail Rev 13, 3–11. [DOI] [PubMed] [Google Scholar]
- Mischel NA, Llewellyn‐Smith IJ & Mueller PJ (2014). Physical (in)activity‐dependent structural plasticity in bulbospinal catecholaminergic neurons of rat rostral ventrolateral medulla. J Comp Neurol 522, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrão CE, Middlekauff HR, Gomes‐Santos IL & Antunes‐Correa LM (2015). Effects of exercise training on neurovascular control and skeletal myopathy in systolic heart failure. Am J Physiol Heart Circ Physiol 308, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G & Watson C (2007). The Rat Brain in Stereotaxic Coordinates Academic Press. [DOI] [PubMed] [Google Scholar]
- Piepoli MF, Davos C, Francis DP & Coats AJ; ExTraMATCH Collaborative (2004). Exercise training meta‐analysis of trials in patients with chronic heart failure (ExTraMATCH). BMJ 328, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon E, Brasileiro‐Santos MS, Moreira ED, Rondon ME, Mattos KC, Coelho MA, Silva GJ, Brum PC, Fiorino P, Irigoyen MC, Krieger EM, Middlekauff HR & Negrão CE (2006). Exercise training improves aortic depressor nerve sensitivity in rats with ischemia‐induced heart failure. Am J Physiol Heart Circ Physiol 291, 2801–2806. [DOI] [PubMed] [Google Scholar]
- Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM & Negrão CE (2003). The effects of exercise training on sympathetic neural activation in advanced heart failure: A randomized controlled trial. J Am Coll Cardiol 42, 854–860. [DOI] [PubMed] [Google Scholar]
- Sabino JP, da Silva CA, de Melo RF, Fazan R Jr & Salgado HC (2013). The treatment with pyridostigmine improves the cardiocirculatory function in rats with chronic heart failure. Auton Neurosci 173, 58–64. [DOI] [PubMed] [Google Scholar]
- Sakai K, Hirooka Y, Shigematsu H, Kishi T, Ito K, Shimokawa H, Takeshita A & Sunagawa K (2005). Overexpression of eNOS in brain stem reduces enhanced sympathetic drive in mice with myocardial infarction. Am J Physiol Heart Circ Physiol 289, 2159–2166. [DOI] [PubMed] [Google Scholar]
- Sanders P, Kistler PM, Morton JB, Spence SJ & Kalman JM (2004). Remodeling of sinus node function in patients with congestive heart failure: Reduction in sinus node reserve. Circulation 110, 897–903. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, De Ferrari GM, Sanzo A, Landolina M, Rordorf R, Raineri C, Campana C, Revera M, Ajmone‐Marsan N, Tavazzi L & Odero A (2008). Long term vagal stimulation in patients with advanced heart failure. First experience in man. Eur J Heart Fail 10, 884–891. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Sejersted OM, Ilebekk A & Bjørnerheim R (2000). Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol 89, 1445–1454. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Malik M, Barthel P, Schneider R, Ulm K, Rolnitzky L, Camm AJ, Bigger JT Jr & Schomig A (1999). Heart‐rate turbulence after ventricular premature beats as a predictor of mortality after acute myocardial infarction. Lancet 353, 1390–1396. [DOI] [PubMed] [Google Scholar]
- Wang WZ, Gao L, Wang HJ, Zucker IH & Wang W (2009). Tonic glutamatergic input in the rostral ventrolateral medulla is increased in rats with chronic heart failure. Hypertension 53, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisløff U, Støylen A, Loennechen JP, Bruvold M, Rognmo Ø, Haram PM, Tjønna AE, Helgerud J, Slørdahl SA, Lee SJ, Videm V, Bye A, Smith GL, Najjar SM, Ellingsen Ø & Skjærpe T (2007). Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients. Circulation 115, 3086–3094. [DOI] [PubMed] [Google Scholar]
- Wong M, Staszewsky L, Latini R, Barlera S, Glazer R, Aknay N, Hester A, Anand I & Cohn JN (2004). Severity of left ventricular remodeling defines outcomes and response to therapy in heart failure: Valsartan heart failure trial (Val‐HeFT) echocardiographic data. J Am Coll Cardiol 43, 2022–2027. [DOI] [PubMed] [Google Scholar]
- Yanni J, Tellez JO, Maczewski M, Mackiewicz U, Beresewicz A, Billeter R, Dobrzynski H & Boyett MR (2011). Changes in ion channel gene expression underlying heart failure‐induced sinoatrial node dysfunction. Circ Heart Fail 4, 496–508. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR & Mazgalev TN (2009). Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high‐rate pacing model. Circ Heart Fail 2, 692–699. [DOI] [PubMed] [Google Scholar]
- Zheng H, Sharma NM, Liu X & Patel KP (2012). Exercise training normalizes enhanced sympathetic activation from the paraventricular nucleus in chronic heart failure: role of angiotensin II. Am J Physiol Regul Integr Comp Physiol 303, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Patel KP, Wang W & Gao L (2009). Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol 297, 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]