

Abstract
Remarkable clinical responses have been seen in patients with metastatic melanoma with targeted therapy (BRAFi vemurafenib, MEKi) and with modern immune cell-based approaches such as TCR engineered adoptive cell transfer (ACT) and earlier experiences with high-dose IL-2. The proximal mediators of these immune therapies are tumor-reactive CTL. Various mechanisms of resistance to immune-mediated apoptotic signals have been described, including phenotypic changes, effector cell exhaustion, functional tolerance, deficiencies in Ag processing and presentation, and mutation or down-regulation of antigenic epitopes. The immune system and drugs eradicate tumors via apoptosis. Therefore, tumors’ resistance to apoptosis may be a determining factor that limits the efficacy of immunotherapies. It is predicted that these therapies have limited efficacy in patients whose melanomas have developed resistance to targeted therapy such as vemurafenib. Upregulation of the immune checkpoint molecule CTLA-4 on activated T cells and its interaction with CD80/86 blocks T cell activation. The fully humanized mAb ipilimumab blocks this interaction, resulting in sustained T cell stimulation. Likewise, the programmed death receptor 1 (PD-1) is another member of the B7:CD28 family of costimulatory molecules that regulates T cell activation, whose ligand (PD-L1) is expressed on melanomas. The human anti-PD-1 mAb, Pembrolizumab, overcomes tolerance, has a favorable pharmacokinetics profile with minimal undesired toxic side effects and has shown remarkable improvement in melanoma therapy. This review focuses on recent advances in the development of various anti-PD-1 checkpoint blockade antibodies and will summarize recent clinical data using immune checkpoint blocking antibodies.
Keywords: Pembrolizumab, nivolumab, PD-1, PD-L1, melanoma, apoptosis, TCR, CTLA-4, immunotherapy, tremelimumab, ipilimumab, MED14736
Introduction to immunotherapy and PD-1 inhibition in the treatment of melanoma
Metastatic melanoma is an aggressive disease with only 16% five-year survival rate. The incidence of melanoma is increasing due to its unresponsiveness to currently available chemotherapeutic drugs such as dacarbazine and carmustine [1]. An exciting and alternative approach involves the utilization of various immune-based modalities, including high-dose interleukin (IL)-2 therapy and TCR-engineered immunotherapy. However, recent introduction of immune checkpoint blockade, such as antagonistic monoclonal antibodies (mAbs) directed against cytotoxic T-lymphocyte antigen 4 (CTLA-4) and PD-1, has revolutionized the treatment of metastatic melanoma patients [1]. In this review we will briefly discuss various immune-based approaches in the treatment of advanced metastatic melanoma. We will further discuss recent advances in melanoma therapy including review of the clinical data of using antagonistic mAbs directed against immune checkpoint molecules CTLA-4 and PD-1, as well as comparison of the efficacy of various mAbs disrupting the PD-1/PD-L1 interaction (Figure 1).
Figure 1.
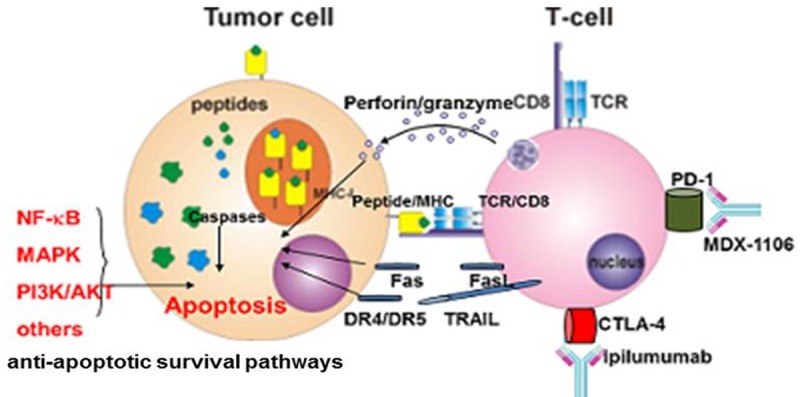
Current immunotherapies in the treatment of melanomas. Upon recognition, through interaction of peptide/MHC with TCR/CD8, cytotoxic T cells induce apoptosis in sensitive melanomas via perforin/granzyme or death receptor-mediated (Fas/FasL, TRAIL/DR4, DR5) pathways. Checkpoints blockade (using anti-CTLA-4 and anti-PD-1 mAbs) and ACT of antigen-specific cytotoxic T cells have remarkably improved melanoma treatment. Constitutively hyper-activated anti-apoptotic cell survival pathways (BRAFV600E, NF-kB, AKT, MAPK, etc.) confer apoptosis-resistance by regulating the expression pattern of pro- and anti-apoptotic gene products, thus, limiting the efficacy of treatment.
High dose interleukin-2 (IL-2)
After being approved by the US Food and Drug Administration (FDA) in 1992, the type I cytokine interleukin (IL)-2, an immune stimulatory cytokine, was clinically used in patients with advanced melanoma. IL-2 has no direct effect on tumor cells. Its antitumor responses are mediated by activating immune effector (cytotoxic T lymphocytes, CTL) cells [2]. This cytokine-based therapy has resulted in long-lasting responses in very few patients. In a clinical study only 22% of those given treatment experienced successful antitumor responses [3]. In a pooled analysis of 270 patients, there was a 7% complete response (CR) rate [3]. However, clinical utilization of high dose IL-2 is hampered by severe multiorgan toxicity. Adverse effects (AEs) include hypertension, pulmonary edema, fever, and bacterial infection. In another clinical trial, 283 patients with metastatic melanoma or metastatic renal cell carcinoma (RCC) who had failed standard treatment were given IL-2 at a dose of 720,000 IU/kg intravenously every 8 hours for a maximum of 15 doses per cycle [2]. IL-2 administration was stopped when grade 3 or 4 toxicity that could not be reversed by standard supportive measures appeared. Out of 134 melanoma patients, nine (7%) had complete regression and 14 (10%) had partial regression with an overall response rate (ORR) of 17%. Out of 149 patients with RCC, 10 patients (7%) had complete regression and 20 patients (13%) had partial regression with an ORR of 20%. Overall, 19% of patients achieved objective regression with this therapy. Collectively, clinical data suggest that immunotherapy using single agent IL-2 does not prove to be very effective.
Adoptive cell transfer (ACT) with genetically modified tumor infiltrating lymphocytes (TILs) carrying IL-12
Adoptive cell transfer (ACT) therapy is an effective immunotherapy treatment for patients with metastatic melanoma. ACT involves ex vivo identification and isolation of tumor reactive CTLs that are then expanded to higher numbers in vitro and transferred back into the patients [4]. With ACT, the exact populations of T cells capable of in vitro tumor killing are identified; these T cells are then selected for expansion. There have been several studies that show promising results of ACT therapy. Conditioning regimen by non-myeloablative lymphodepleting drugs (fludarabine and cyclophosphamide) followed by adoptively transferring autologous tumor-infiltrating lymphocytes (TILs) in conjunction with high-dose IL-2 elicits objective tumor regression in 50% to 70% of melanoma patients based on RECIST criteria [2]. Lymphodepleting drugs help create a lymphopenic environment, which has reduced numbers of immunosuppressive regulatory T cells and myeloid derived suppressor cells [5], allowing rapid proliferation and enhanced activity of adoptively transferred TILs. Moreover, the lymphopenic environment decreases the competition between native lymphocytes and adoptively transferred TILs for cytokines IL-7 and IL-15, thus providing a favorable environment for TILs to proliferate and survive [6].
Interleukin-12 (IL-12), a member of a family of heterodimeric cytokines, has powerful proinflammatory activities. IL-12 has potent antitumor effects when administered in murine tumor model [7]; however, it is toxic when administered directly to humans. There is ongoing research on engineering TILs to carry IL-12 gene. Clinical utilization of TILs containing IL-12 gene has been promising [8]. In this trial, patients who were 18 years of age or older with evaluable metastatic melanoma and a melanoma lesion suitable for resection to generate TIL cultures were given a bolus intravenous (i.v.) infusion of TILs genetically modified by a retroviral vector encoding Nuclear factor of activated T-cells (NFAT). IL-12. After the infusion, patients received a lymphodepleting chemotherapy regimen. The trial was designed as cell dose-escalation, starting with 1×106 cells and then with increasing numbers of cells by half-log increments. Out of 33 patients, 11 achieved an objective response according to RECIST criteria. A single objective response was seen in 17 patients treated with 0.1×109 or fewer cells (5.9%). 10 out of the 16 patients treated with higher dose, 0.3 to 3×109 NFAT. IL-12 cell cultures, exhibited objective responses (62.5%). Tumor regression was seen at multiple sites, including brain, lung, lymph nodes, and subcutaneous tissues. There was a wide range of AEs, including persistent fever and liver abnormalities. The highest levels of serum IL-12 could be lethal and required intensive care unit management in some patients. High level of circulating IL-12 in the body is alarming as it can inhibit the proliferation of lymphocytes. Although there are still problems with treatment using engineered TILs to carry IL-12 genes, the observed response rate was 63% in patients treated with 0.3×109 or greater NFAT. IL-12-engineered T cells compares favorably with previous response rates in patients treated with 10 to 100 higher numbers of T cells along with high-dose IL-2. With more research on ways to control the expression of IL-12 to modulate circulating serum levels, genetically modified TILs can increase the efficacy of ACT therapy.
BRAF inhibitors: the first targeted therapy for advanced melanoma
In 2011, the FDA approved vemurafenib, a BRAFV600E kinase inhibitor (BRAFi). Vemurafenib is used in the treatment of patients having advanced melanoma that cannot be surgically resected and a mutation in the BRAF gene. In one trial, 337 of 675 patients were randomly assigned to vemurafenib, receiving 960 mg tablets twice daily while 338 patients were randomly assigned to dacarbazine treatments intravenously with 1000 mg/m2 every three weeks [9]. Overall survival was significantly improved in patients receiving vemurafenib compared to patients receiving dacarbazine at a median of 6.2 months versus 4.5 months. Progression-free survival was also significantly improved for vemurafenib patients at 5.3 months. The ORR was 48.4% for patients receiving vemurafenib while only 5.5% for patients treated with dacarbazine. However, common AEs occurred in 38% of vemurafenib patients including cutaneous squamous cell carcinoma (SCC) and fatigue. Another BRAFi, dabrafenib was approved by the FDA in 2013. In a clinical study, the complete response (CR) rate was only in 3% of patients receiving 150 mg dabrafenib twice daily. The multikinase inhibitor, sorafenib was studied as well. It acts to inhibit the mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3 kinase (PI3K/Akt) pathways that are constitutively activated in melanoma cells. This drug targets C-Raf, MEK, and ERK growth factors. However, in phase I and phase II trials, the results of treatment with sorafenib were also unsuccessful with responses in less than 10% of patients [10].
T-cell receptor (TCR)-engineered immunotherapy
One of the current most effective immunotherapy regimens for metastatic melanoma involves the generation of T lymphocytes with high affinity TCR for specific melanoma peptides presented in the context of MHC complexes. TCR receptors on the surfaces of T lymphocytes recognize tumor associated antigens [11]. A single TCR can recognize a variety of MHC peptide complexes [12]. Upon T cell recognition of an antigenic epitope via interaction of TCR with peptide/MHC complex and subsequent signaling through accessory molecules, CTLs are activated and induced to proliferate and differentiate. Effector CD8+CTLs kill tumor targets by directly inducing apoptosis or indirectly through the release of cytokines. These cytokines include interferon (IFN)-γ and tumor necrosis factor (TNF)-α that trigger the caspase cascade, causing apoptosis of melanoma targets. Selected patients have been found to have highly reactive T cells that recognize and lyse target tumor cells. Autologous TILs have been found to mediate objective cancer regression. These preexisting tumor-reactive cells can be isolated and expanded ex vivo. The genes encoding the TCR specific for a variety of tumor associated antigens (TAA) can be cloned. However it is often difficult to identify these tumor reactive lymphocytes. Genes encoding for TCR α/β are cloned from these existing T cells, inserted into retroviruses, and transduced into another patient’s own T cells. The transduction with TCR-encoding retroviral vectors has resulted in TCR-engineered cells that are capable of recognizing and destroying specific cancer cells in clinical settings [11]. Transduction of peripheral blood lymphocytes (PBLs) from melanoma patients with retroviral vector encoding TCRα/β genes specific for MART-1, in vitro expansion, and subsequent ACT of these TCR-engineered PBLs has clinical benefit. Peptide-specific TCR genes were cloned from a patient with near complete tumor regression. Gene transfer (transduction) resulted in expression of TCR in 30% of transduced CD8+ cells. Out of the 17 patients that received treatment, 14 patients exhibited persistence of the transduced cells at 1 and 4 weeks after treatment. Two patients experienced a sustained objective regression of their metastatic melanoma after the transfer of genetically engineered PBLs, thus resulting in a response rate of 13%. No toxicities in any patients were attributed to the gene marked cells. More research is still being conducted to improve TCR chain pairing and antigen affinity [13]. Researchers are investigating the use of lentiviral vectors and higher affinity TCRs. The development and use of engineered TCRs is documented to have high clinical efficacy.
Adoptive immunotherapy with TCR-gene transduced T-cells is a novel and promising treatment option for advanced melanoma since it enhances the activity and functionality of patient’s immune system to become reactive against tumor cells. This approach has fewer undesired toxicity than chemotherapy. CD4+ T-cells can recognize peptide epitopes in the context of MHC class II molecules, but MHC class II molecules are either down-regulated or absent on the majority of tumor cells. Therefore, it is difficult to incorporate CD4+ T-cells into therapeutic protocols. TCR-engineered CD4+ T-cells are developed to overcome this limitation. TCR-engineered CD4+ T-cells recognize the antigenic peptide presented by MHC class I molecules with or without a co-receptor, and exhibit effector functions including cytokine secretion and tumor reactivity [14]. A study conducted in 2010 shows that MHC class I-restricted MART-127-35 epitope-specific TCR transduced CD4+CD25- T cells undergo several rounds of division and exhibit different effector functions, such as synthesizing IFN-γ, TNF-α, and IL-2, mobilizing lytic granules and exhibiting cytolytic effector function against melanoma targets [15]. These T-cells do not require a co-receptor, and they also amplify the expansion of the MART-127-35 epitope- specific CD8+ T cells in an epitope-specific CTL generation assay (in vitro).
Another example of TCR-engineered immunotherapy for melanoma involves tumor specific TCRs directed against antigens such as NYESO1 and MART1. Studies involving engineered TCR resulted in objective clinical responses in 12-45% of patients with metastatic melanoma [16]. In one study, researchers generated NYESO1-specific TCR and transduced cytokine-induced killer (CIK) cells using lentiviral particles at an MOI of 50 [17]. Transduction efficiency was 38% and the TD-CIK cells were tested against three melanoma cell lines. TD-CIK cells killed NY-ESO1+/HLA-A2+ melanoma cells with an increase of 4.3 fold efficiency compared to non-transduced CIK (NT-CIK). Tumor antigen-specific TCRs on CIK cells were responsible for improved anti-tumor activity of the engineered cells. However, treatment related AEs such as severe skin and ear inflammation, neurological toxicities, and lethal cardiac toxicities ensued from studies with these TCRs [16]. Furthermore, antitumor responses were not sustained and were often incomplete.
Additional clinical trials are required to determine the efficacy of TCR-engineered immunotherapy, but the results obtained so far are promising. The NY-ESO-1 antigen was expressed in 10% to 50% of metastatic melanomas. Out of all patients who received NY-ESO-1 specific T-cells, 45% melanoma patients showed measurable response rate [18]. Moreover, compared to patients receiving on-target/off-tumor toxicity seen in the melanoma antigen TCR, none of the patients who received NY-ESO-1-specific T cells experienced such toxicity [19].
Cytotoxic T lymphocyte antigen-4 (CTLA-4)
Inhibition of the Cytotoxic T Lymphocyte Antigen-4 (CTLA-4) pathway is yet another type of immunotherapy that induces or enhances antitumor immune responses. Upon binding, CTLA-4 (CD152) can inhibit T-cell maturation and proliferation by competing with CD28 receptor in binding to B7.1 (CD80) and B7.2 (CD86) ligands on antigen presenting cells (APCs) [20]. Therapeutic monoclonal antibodies (mAbs) are developed which can block CTLA-4 checkpoint (Figure 2).
Figure 2.
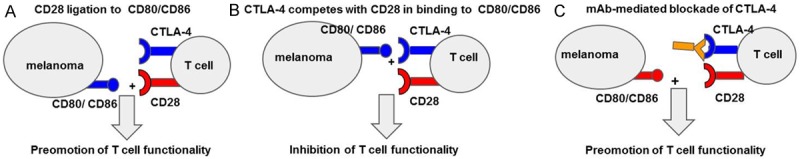
CTLA-4 check point blockade in the treatment of melanomas. A. Under normal conditions ligation of Cd28 with B7.1, B7.2 (CD80, CD86) provides signal II for T cell activation. The negative regulatory signaling molecule CTLA-4 competes with CD28 in binding to B7 family ligands. B. CTLA-4 ligation to B7 family ligands inhibits T cell functionality (proliferation, activation, cytotoxic potential). C. mAbs directed against CTL-4 check point regulator inhibit its binding to B7 family ligands, thus, promoting T cell functionality.
There are two types of signals essential for optimal T-cell activations: signal 1 and signal 2 [21]. The interaction of T-cells receptors (TCRs) with MHC/peptide complex on the surface of tumor cells provides signal 1. This determines the antigen-specificity of the response [22]. The interaction between costimulatory molecules on APCs and counter-receptors on T-cells, such as B7-CD28 interaction, provides the second signal required for T-cell activation. T-cells are rendered inactive in the presence of signal 1 alone. The absence of costimulatory signal 2 causes T-cells to enter a state of unresponsiveness referred to as anergy, which eventually leads to tolerance. Simultaneous presence of both signal 1 and 2 are required for naive T-cells to become active and transition to effector cells. The inhibitory molecule CTLA-4 competes with CD28 receptor in binding to B7.1 (CD80) and B7.2 (CD86) ligands, which can inhibit T-cell maturation and proliferation, a process that can be prevented using recently developed antagonistic CTLA-4 mAbs. Below is a brief description of various anti-CTLA-4 Abs and their clinical implication in melanoma therapy.
Ipilimumab (yervoy)
Ipilimumab is the first FDA approved (2011) anti-CTLA-4 checkpoint inhibitor [23]. By targeting the CTLA-4 checkpoint, Ipilimumab activates CTLs to recognize and destroy cancer cells. Ipilimumab has undergone extensive phase II and phase III clinical trials. In a phase III MDX010-20 study, ipilimumab was shown to improve the overall survival of the patients [24]. 676 patients were randomized to take ipilimumab 3 mg/kg and gp100 vaccine, ipilimumab 3 mg/kg and placebo, or gp100 vaccine alone. One-fifth of the patients survived at least two years. 474 were randomized 2 years before the study cut-off date, 284 of whom were treated with ipilimumab plus gp100, 95 of whom were treated with ipilimumab alone, and 95 of whom were treated with vaccine alone. Out of these 474 patients, 94 patients survived at least 2 years, comprising of 54 (19%) patients treated with ipilimumab plus gp100, 24 patients (25%) treated with ipilimumab alone, and 16 patients (17%) treated with vaccine alone. Of all patients surviving at least 2 years, 42 patients (45%) survived at least 3 years. In another study, Ipilimumab was given to patients with metastatic melanoma every 3 weeks for four cycles. Following the initial 4 doses, ipilimumab was administered every 12 weeks. This study supported the idea that ipilimumab was dose-dependent and that increasing doses from 0.3 to 10 mg/kg increased the percentage of response in patients. CTLA-4 inhibitors have been proved to be effective checkpoint inhibitors, but their side effects are severe. Ipilimumab has been proven as an effective treatment for melanoma, resulting in a higher overall survival (OS) rate than dacarbazine or chemotherapy [24].
However, ipilimumab has resulted in many immune-related (IR) AEs [25]. Although 3 mg/kg doses produce weaker responses than 10 mg/kg doses, higher doses result in higher toxicity levels [26]. The most common IRAEs are rash, colitis, hepatitis, and hypophysitis. More than 50% of patients had severe rash that was associated with deep dermal and perivascular infiltrates of lymphocytes. Patients also experienced enterocolitis of grade 3-4 in 16% of cases. IRAEs are unavoidable, but they may correlate with tumor regression in patients with metastatic melanoma. Ipilimumab-treated patients experiencing grade 3/4 IRAEs were reported to have a significantly higher rate of tumor regression than those without IRAEs (36% versus 5% of patients). The objective response rate was reported to be significantly higher in ipilimumab-treated melanoma patients who developed enterocolitis compared with those who did not (36% versus 11%). CTLA-4 pathway acts on early T-cell activation, while PD-1 pathway affects T-cell at the effector stage [20].
Tremelimumab (ticilimumab, CP-675,206)
Tremelimumab is a human monoclonal non-complement fixing IgG2 isotype that has been approved for phase II clinical trials [27]. This drug is a human monoclonal antibody directed against CTLA-4. It binds to CTLA-4 thus inhibiting its binding to APC ligands CD-80 and CD-86 and resulting in activation of T-lymphocytes. This leads to CTL-mediated T-cell activation. In a phase I study, 39 patients underwent single dose escalation regimens to determine the maximum tolerated dose (MTD). Tremelimumab was administered at 0.01, 1, 3, 6, 10 or 15 mg/kg doses. 3 out of the 5 patients who received 15 mg/kg experienced dose limiting toxicities such as dermatitis or diarrhea. Researchers determined the MTD was 10 mg/kg. In another phase I study, researchers investigated administering 10 mg/kg once a month or 15 mg/kg doses every 3 months [28]. No dose limiting toxicity was observed. ORR was 4 of 41 for the 10 mg/kg compared to ORR of 4 of 43 for the 15 mg/kg every three months. Median time to response was 21 weeks. In phase II trial, 8 of 84 (10%) of patients attained objective antitumor responses. Only partial responses were seen in patients receiving the 10 mg/kg doses. Median overall survival was 9.97 months for 10 mg/kg and 11.53 months for 15 mg/kg. Both treatments were associated with durable tumor responses. Most frequent AEs included diarrhea, rash, and pruritus. AEs were lower among patients treated with 15 mg/kg every 3 months. Results from treatment with Tremelimumab have not been significant but research continues to be conducted to evaluate the dosing regimen and relationship between tumor response and survival.
Programmed Cell Death Ligand 1 (PD-L1) pathway and role in immunotherapy
Programmed Cell Death Ligand (PD-L) 1 and 2 are ligands of programmed cell death 1 (PD-1) [29]. PD-1 is an immunoinhibitory receptor a member of the CD28 family. It is expressed on T and B cells and, upon ligation to PD-L1 and PD-L2, inhibits immune cell activation. PD-L1 is expressed on T cells, B cells, macrophages, dendritic cells, and other nonimmune cells. PD-L2 is expressed mainly on activated macrophages and dendritic cells. The expression of PD-L1 and PD-L2 and subsequent binding to PD-1 induces T cells to undergo apoptosis. By blocking the PD-L1 inhibitory pathway using anti-PD-L1 Abs, T cells can be activated and become more efficient in tumor surveillance (Figure 3).
Figure 3.
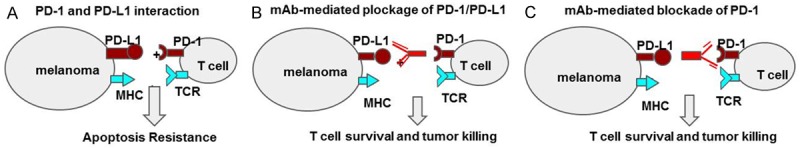
PD-1 check point blockade in the treatment of melanomas. (A) Under normal conditions ligation of PD-1 with PDL-1, PDL-2 provides a negative stimulatory signal to T cells, thus, even in the presence of TCR recognition of peptide/MHC complex, T cells are rendered inactive to kill tumor cells. The negative regulatory signaling provided by PD-1 PD-L engagement can be easily blocked by mAbs directed against check point molecules. (B) PDL-1 or (C) PD-1, promoting the functionality of T cell and tumor eradication.
The PD-L1 checkpoint inhibitor MEDI4736 was administered intravenously every 2 or 3 weeks in a 3+3 dose escalation in 26 patients with various tumor malignancies [30]. 34% of all patients experienced AEs; however, these AEs were only grade 1-2. These AEs were mainly diarrhea, fatigue, rash and vomiting. No IRAEs had been recorded. MEDI4736 could induce 4 partial remissions and 5 additional minor responses. These clinical responses did not occur in melanoma alone, but also in patients with non-small cell lung cancer (NSCLC), indicating that MEDI4736 is a potentially effective checkpoint inhibitor.
Ongoing and future considerations of the clinical efficacy of various immune-checkpoint inhibitors are underway. Currently, there are 5 anti-PD-1 and 4 anti-PD-L1 agents used in clinical trials [1]. Despite the current safety and tolerability of single agent blockade using anti-PD-1 monoclonal antibodies such as pembrolizumab and nivolumab, trials with combination therapies are being studied with the potential to minimize toxicity and reduce the incidence of IRAEs while increasing response rates. Combination therapy trials are currently underway to examine advanced melanoma patient response rates relative to anti-PD-1 and anti-CTLA-4 monotherapies. A phase I trial involving nivolumab and ipilimumab in advanced melanoma showed 20% response. However, those with high dose of 3 mg/kg of nivolumab with 3 mg/kg of ipilimumab every two weeks exceeded the tolerate toxicity [1]. 53% of patients with smaller dose regimens experienced grade 3 and grade 4 AEs. With follow-up periods, development of new immunotherapy agents, and more trials, the use of checkpoint inhibitors in the treatment of advanced melanoma is expected to benefit more patients with metastatic melanoma.
Programmed Cell Death (PD-1) pathway and role in immunotherapy
The programmed cell death (PD)-1 checkpoints function as a control over immune response hyperactivity. However, these immune checkpoints are also means by which tumors can inhibit T cells and block antitumor immune responses [1]. Interaction between PD-1 and PD-L1 and PD-L2 normally inhibits immune response by reducing T lymphocyte function. Signaling inhibits T cell activation, proliferation, and cytokine production [1]. As mentioned above, regulation of T cell activation involves two complementary signals: TCR recognition of peptide/MHC and costimulatory signal provided by CD28 ligation to B7.1 or B7.2 on APCs. Many tumors express PD-L1 in order to induce negative regulation of T cells by the PD-1 checkpoint. The anti-PD-1 mAb nivolumab has recently been approved by the FDA. Studies with nivolumab have shown improved overall survival. This drug inhibits checkpoint binding of PD-1 and PD-L1 between T cells and the tumor cells, thus inducing immune response. Compared to existing treatments involving ipilimumab and nivolumab, the use of pembrolizumab may be the new effective treatment for patients with advanced melanoma.
Below is a brief description of various anti-PD-1 mAbs and their clinical implication in melanoma therapy (Table 1).
Table 1.
Summary of clinical data of targeted therapy and immune check-point blockade in melanoma treatment
| Treatment | Phase | Overall Response Rate | Overall Survival | References |
|---|---|---|---|---|
| Vemurafenib | III | 48.40% | 84% (at 6 months) | [9] |
| Decarbazine | III | 5.50% | 64% (at 6 months) | [9] |
| Ipilimumab + gp100 vaccine | III | 20.10% | 19% (at 2 years) | [24] |
| Ipilimumab | III | 28.50% | 25% (at 2 years) | [24] |
| gp100 vaccine | III | 11.00% | 17% (at 2 years) | [24] |
| Tremelimumab | II | 10% | 32% (at 12 months) | [26] |
| Nivolumab | III | 40% | 72.9% (at 12 months) | [32] |
| Pembrolizumab | I | 38% | 81% (at 12 months) | [33] |
Nivolumab (opdivo, ONO-4538)
Nivolumab is an FDA approved PD-1 checkpoint inhibitor. In one open-label, phase II trial in Japan, almost one-quarter of the patients with previously treated stage III/IV melanoma achieved a partial tumour response when given intravenous nivolumab 2 mg/kg every 3 weeks [31]. These patients had undergone other cancer treatments before. Nivolumab was administered until disease progressed, AEs became intolerable, or a complete response occurred. The median progression free survival lasted for 172 days. In a phase I trial, 94 patients were treated with up to 10 mg/kg of nivolumab every 2 weeks for at most 96 weeks [32]. 5% of patients stopped treatments due to IRAEs such as pneumonitis, colitis, and hepatitis. Some AEs such as diarrhea were managed with the use of corticosteroids. However, 31% of patients responded to treatment and of these responders, 45% responded within the first eight weeks of treatment. A phase III trial of nivolumab had a response rate of 40% with a 72.9% one-year survival rate.
Pembrolizumab (keytruda, lambrolizumab, MK-3475)
With promising safety, tolerability, and efficacy, a new drug, pembrolizumab has been the center of many clinical studies and trials. This anti-PD-1 monoclonal antibody has been reported to increase survival rates and lower incidence of AEs [12]. Pembrolizumab inhibits the PD-1 immune checkpoint and has significant antitumor activity in patients with advanced melanoma. Compared to other drugs and therapies pembrolizumab has prolonged progression-free survival and greater overall survival. Pembrolizumab has resulted in durable antitumor activity. Furthermore this treatment is associated with significantly less high-grade toxicity. Based on various clinical trials it is concluded that pembrolizumab is safe and tolerable in multiple tumor types.
Review of pharmacology, mode of action, pharmacokinetics of pembrolizumab
Unlike chemotherapy which relies on cytotoxic effects of powerful chemicals to induce DNA damage and cause tumor regression or inhibition of tumor progression, programmed death (PD)-1 checkpoint inhibition enhances the antitumor responses of the immune system to tumor cells harboring multiple mutations. Under normal physiological conditions, T-cells can induce apoptosis in tumor cells by two main mechanisms [32]. The first mechanism is through granule-exocytosis pathway. In this pathway, T-cells release perforin or granzyme CD8, which are highly toxic proteins, into the defined intercellular space between CTL and the target cell membranes. Once these proteins are inside the target tumor cells, they induce apoptosis. The second mechanism is death receptor signaling pathway. In this pathway, caspases activation patterns in Type 1 and Type 2 cells can both induce apoptosis. However, tumors have the ability to develop mechanisms to evade antitumor immune responses. One of such mechanisms is PD-1 ligands expression. After a prolonged period of activation, T cells upregulate surface PD-1 expression. Tumor cells express PD-L1 and PD-L2 to bind to PD-1 and send a negative signal that inhibits apoptosis. The humanized IgG4 mAb Pembrolizumab is a checkpoint inhibitor acting selectively on the PD-1 pathway. The drug is designed to bind to PD-1 receptors, thus preventing the interaction between PD-1 and PD-L1 or PD-L2. By interfering with this interaction, pembrolizumab disrupts PD-1 pathway and restores antitumor immune response. Pembrolizumab is administered for patients with unresectable or metastatic melanoma or with a mutated BRAFV600E/K gene, which are unresponsive to treatments involving BRAF inhibitors [33]. Pembrolizumab is also administered for patients that show no tumor regression after treatment with CTLA-4 checkpoint inhibitor ipilimumab. The FDA approved dose for patients is 2 mg intravenous (IV) infusion, administered over 30 minutes and every 3 weeks until patients are no longer responsive to the drug or the AEs are lethal. Pembrolizumab had a mean clearance and elimination half-life (t1/2) of 0.22 L/day and 26 days, respectively. Doses of 2-10 mg/kg of pembrolizumab every 3 weeks showed a linear pharmacokinetics, with dose-proportional increases in peak concentration (Cmax), trough concentration (Cmin) and steady state area under the plasma concentration-time curve (AUCss).
Efficacy studies
After patients no longer show tumor regression upon treatment with ipilimumab, they are treated with PD-1 checkpoint inhibitors [33]. PD-1 checkpoint inhibitors display a wide range of response patterns, depending on the characteristics of each individual’s immune system and its tumors. A patient with extensive PD-1 expressing T cells could have a rapid response with pembrolizumab, while a patient with low numbers of pre-existing PD-1 expressing T cells might show a delayed or no response to pembrolizumab [20]. Even though more data is needed to give a definitive efficacy of pembrolizumab, clinical trials conducted so far show promising results. Treatment with checkpoint inhibition has been shown to correlate with durable, long-lasting responses, even in patients that have discontinued therapy. Except for cases in which the tumor progresses, many patients who discontinued either anti-PD-1 or anti-CTLA-4 therapy had persistent responses, indicating a sustained antitumor immune response. In phase I KEYNOTE-001 trial, patients showed a high rate of tumor regression when treated with pembrolizumab [34]. A total of 1137 patients with melanoma, NSCLC or other carcinomas received IV pembrolizumab 10 mg/kg every 2 or 3 weeks, or 2 mg/kg every 3 weeks and were assessed for tumor response every 12 weeks. The median time to response in the 2 mg/kg and 10 mg/kg groups was 12 weeks. The ORR according to RECIST was 38%. At the time of analysis, response durations ranged from 1.9 to 10.8 months. The ORR was highest among patients receiving pembrolizumab 10 mg/kg every 2 weeks, but they also had the highest IRAEs. In a follow-up after ≥ 13 months of 135 nonrandomized patients, pembrolizumab was shown to have durable effects [33]. The ORR by RECIST was 41%. Responses could be delayed as long as 36 weeks of treatment. Among 71 patients with evaluable tumor PD-L1 expression, the expression of PD-L1 was associated with improved RECIST ORR (51 vs. 6%; P = 0.0012) compared to non-PD-L1-expression (Table 2 summarizes the clinical efficacy of various immune check point blocking mAbs currently in the treatment of metastatic melanoma).
Table 2.
Immune check-point blocking mAbs used in immunotherapy of metastatic melanoma
| Name | Type of Ab | Mechanism of Action | References |
|---|---|---|---|
| Ipilimumab | anti-CTLA-4 (IgG1к) | Inhibits CTLA-4 binding to CD80 and CD86. Activates T cell responses | [23] |
| Tremelimumab | anti-CTLA-4 (IgG2) | Inhibits CTLA-4 binding to CD80 and CD86. Activates T cell responses | [27] |
| Nivolumab | anti-PD-1 (IgG4) | Inhibits binding of PD-1 to PD-L1. Activates T cells-mediated immune responses | [31] |
| Pembrolizumab | anti-PD-1 (IgG4) | Inhibits CTLA-4 binding to CD80 and CD86. Activates T cells responses | [12] |
| MEDI4736 | anti-PD-L1 (IgG1к) | Inhibits binding of PD-1 to PD-L1 or PD-L2. Activates T cells responses | [30] |
Unlike other anti-PD-1 monoclonal antibodies, pembrolizumab has the highest affinity for PD-1. This increased efficacy has been seen in many Phase I trials. A phase I trial involved 135 patients with metastatic melanoma. Patients were either given regimens involving 10 mg/kg doses every 2 weeks or 10 mg/kg doses every 3 weeks. Only 13% of patients exhibited low grade IRAEs. High dose patients had 54% response rates compared to only 35% response undergoing doses every 3 weeks. Of the 52 responders, a majority responded within the first 12 weeks of treatment [1]. In a pooled study of 411 phase I patients, those given pembrolizumab who were previously untreated exhibited higher response rates at 40%, compared to ipilimumab treated patients with 28% response rates [1].
Safety and tolerability of pembrolizumab
Studies are underway to explore the safety of pembrolizumab for patients with advanced melanoma. Recent results show promising tolerability with this PD-1 blocking monoclonal antibody. In one study, the incidence of AEs involving the use of pembrolizumab was 12% [12]. These patients experienced grade 3-5 drug-related AEs. The most common AE was pneumonitis. Other patients experienced dermatologic rashes and pruritus, gastrointestinal diarrhea and colitis, endocrinopathies, and hepatitis. Fatigue was often experienced among patients receiving therapy with pembrolizumab. Most IRAEs occur during the first 2 to 6 months of treatment [12]. However, these can be effectively managed with the use of corticosteroid treatments. Long-term exposure to corticosteroids can lead to infection and gastrointestinal irritation. Endocrine disorders have been managed with hormone replacement. The use of combination therapy such as dual checkpoint blockades involving anti-CTLA-4, anti-PD-1, and anti-PD-L1 mAbs has led to stronger immune system stimulation and enhanced antitumor activity. However, combination regimens lead to higher AE incidence compared to single agent therapies alone. 62% of patients undergoing concurrent anti-CTLA-4 and anti-PD-1 treatment experienced grade 3-4 AEs. Ongoing studies are further evaluating the safety of pembrolizumab monotherapy and combination therapy.
Conclusions
PD-1 checkpoint inhibitors, such as pembrolizumab, are emerging as new promising treatments for advanced melanoma. Results of various clinical trials involving the use of different types of PD-1 and PD-L1 checkpoint inhibitors have shown that this immunotherapy is an effective and promising cancer treatment. Pembrolizumab, nivolumab, and MEDI4736, have shown impressive efficacy with less severe AEs compared to other cancer treatments. Trials are still underway to examine the efficacy and safety of these drugs in single agent therapy. Given the tolerability and efficacy of many of the anti-CTLA and anti-PD-1 monoclonal antibody monotherapies, immunotherapy combination trials are underway to determine the effectiveness of different combined treatments for melanoma. Clinical trials of using single agent pembrolizumab have shown positive results, with increased efficacy and durable responses. Further investigations are warranted to optimize the use of pembrolizumab, alone or combined with other modalities, in the treatment of additional tumors types.
Disclosure of conflict of interest
None.
References
- 1.Mahoney KM, Freeman GJ, McDermott DF. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin Ther. 2015;37:764–782. doi: 10.1016/j.clinthera.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 3.Legha SS, Gianan MA, Plager C, Eton OE, Papadopoulos NE. Evaluation of Interleukin-2 administered by continuous infusion in patients with metastatic melanoma. Cancer. 1996;77:89–96. doi: 10.1002/(SICI)1097-0142(19960101)77:1<89::AID-CNCR15>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Phan GQ, Rosenberg SA. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control. 2013;20:289–297. doi: 10.1177/107327481302000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee S, Margolin K. Tumor-Infiltrating Lymphocytes in Melanoma. Curr Oncol Rep. 2012;14:468–474. doi: 10.1007/s11912-012-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med. 1993;178:1223–1230. doi: 10.1084/jem.178.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, Hughes MS, Kammula US, Feldman SA, Toomey MA, Kerkar SP, Restifo NP, Yang JC, Rosenberg SA. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niezgoda A, Niezgoda P, Czajkowski R. Novel approaches to treatment of advanced melanoma: a review on targeted therapy and immunotherapy. Biomed Res Int. 2015;2015:851387. doi: 10.1155/2015/851387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 11.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson MH, Schrama D, Thor Straten P, Becker JC. Cytotoxic T cells. J Invest Dermatol. 2006;126:32–41. doi: 10.1038/sj.jid.5700001. [DOI] [PubMed] [Google Scholar]
- 13.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550–557. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, Nishimura MI. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. 2005;65:1570–1576. doi: 10.1158/0008-5472.CAN-04-2076. [DOI] [PubMed] [Google Scholar]
- 15.Ray S, Chhabra A, Chakraborty NG, Hegde U, Dorsky DI, Chodon T, von Euw E, Comin-Anduix B, Koya RC, Ribas A, Economou JS, Rosenberg SA, Mukherji B UCLA-CALTECH-CHLA-USC-UCONN Consortium on Translational Program in Engineered Immunity. MHC-I-restricted melanoma antigen specific TCR-engineered human CD4+ T cells exhibit multifunctional effector and helper responses, in vitro. Clin Immunol. 2010;136:338–347. doi: 10.1016/j.clim.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govers C, Sebestyén Z, Roszik J, Van Brakel M, Berrevoets C, Szoor A, Panoutsopoulou K, Broertjies M, Van T, Vereb G, Szollosi J, Debets R. TCRs genetically linked to CD28 and CD3ε do not mispair with endogenous TCR chains and mediate enhanced T cell persistence and anti-melanoma activity. J Immunol. 2014;193:5315–5326. doi: 10.4049/jimmunol.1302074. [DOI] [PubMed] [Google Scholar]
- 17.Elia AR, Circosta P, Sangiolo D, Bonini C, Gammaitoni L, Mastaglio S, Genovese P, Geauna P, Avolio F, Inghirami G, Tarella C, Cignetti A. Cytokine-induced killer cells engineered with exogenous T-cell receptors directed against melanoma antigens: enhanced efficacy of effector cells endowed with a double mechanism of tumor recognition. Hum Gene Ther. 2015;26:220–231. doi: 10.1089/hum.2014.112. [DOI] [PubMed] [Google Scholar]
- 18.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Mahvi AV, Helman LJ, Mackall CL, KAmmula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, LI YF, EL-Gamil M, Dchwarz SL, Laurencot C, Rosenberg SA. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luke JJ, Ott P. PD-1 pathway inhibitors: The next generation of immunotherapy for advanced melanoma. Oncotarget. 2015;6:3479–3492. doi: 10.18632/oncotarget.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bretscher P. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999;96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 23.Azoury SC, Straughan DM, Shukla V. Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr Cancer Drug Targets. 2015;15:452–462. doi: 10.2174/156800961506150805145120. [DOI] [PubMed] [Google Scholar]
- 24.McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S MDX010-20 Investigators. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20) Ann Oncol. 2013;24:2694–2698. doi: 10.1093/annonc/mdt291. [DOI] [PubMed] [Google Scholar]
- 25.Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother. 2009;58:823–830. doi: 10.1007/s00262-008-0653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, Esposito A, Paone M, Palla M, Cavalcanti E, Sandomenico F, Petrillo A, Botti G, Fulciniti F, Palmieri G, Queirolo P, Marchetti P, Ferraresi V, Rinaldi G, Pistillo MP, Ciliberto G, Mozzillo N, Ascierto PA. Immunological and biological changes and their correlation with Clinical Response and Survival during Ipilimumab in metastatic melanoma. Cancer Immunol Immunother. 2014;63:675–683. doi: 10.1007/s00262-014-1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ascierto PA, Marincola FM, Ribas A. Anti -CTLA-4 monoclonal antibodies: the past and the future in clinical application. J Transl Med. 2011;9:196. doi: 10.1186/1479-5876-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camacho L, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, Redman B, Pavlov D, Bulanhagui C, Bozon VA, Gomez-Navarro J, Ribas A. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J. Clin. Oncol. 2009;27:1075–81. doi: 10.1200/JCO.2008.19.2435. [DOI] [PubMed] [Google Scholar]
- 29.Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N, Honjo T, Fujii S. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104:3360–3365. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zielinski CC. A phase I study of MEDI4736, NNT-PD-L1 antibody in patients with advanced solid tumors. Transl Lung Cancer Res. 2014;3:406–407. doi: 10.3978/j.issn.2218-6751.2014.08.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deeks ED. Nivolumab: a review of its use in patients with malignant melanoma. Drugs. 2014;74:1233–1239. doi: 10.1007/s40265-014-0234-4. [DOI] [PubMed] [Google Scholar]
- 32.Jazirehi AR. Editorial: Functional complementation, molecular targeted strategies, and chemo/immuno-sensitization in cancer treatment: hurdles and solutions. Curr Drug Deliv. 2012;9:1–4. doi: 10.2174/156720112798376069. [DOI] [PubMed] [Google Scholar]
- 33.Poole RM. Pembrolizumab: first global approval. Drugs. 2014;74:1973–1981. doi: 10.1007/s40265-014-0314-5. [DOI] [PubMed] [Google Scholar]
- 34.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]