

Abstract
The cell cycle, an essential process leading to the cell division, is stringently controlled by the key cell cycle regulators, cyclin-CDK complexes, whose activity is further regulated by a variety of mechanisms. p27Kip1 is a cyclin-CDK inhibitor that arrests the cell cycle at the G1 phase by blocking the activation of cyclin E-CDK2 complex, preventing the improper entry to the cell cycle. Dysfunction of p27 has been frequently observed in many types of human cancers, resulting from p27 protein degradation and cytoplasmic mislocalization, which are highly regulated by the phosphorylation status of p27. Although the kinases that phosphorylate p27 have been extensively studied, phosphatases that dephosphorylate p27 remain to be elucidated. By using genomic phosphatase screening, we identified a PPM family phosphatase, PPM1G, which could reduce p27 phosphorylation at T198. We further confirmed that PPM1G is a novel p27 phosphatase by demonstrating that PPM1G can interact with and dephosphorylate p27 in cells and in vitro. Functionally, ectopic expression of PPM1G enhanced p27 protein stability and delayed cell cycle progression from G1 to S phase. In accordance, knockdown of PPM1G accelerated p27 degradation during G1 phase and rendered cells resistant to the cell cycle arrest induced by serum deprivation. Mechanistically, PPM1G inhibited the interaction of p27 to 14-3-3θ, a chaperone protein that facilitates p27 nuclear export. Knockdown of PPM1G promoted the cytoplasmic localization of p27. Taken together, our studies identified PPM1G as a novel regulator of p27 that dephosphorylates p27 at T198 site and, together with p27 kinases, PPM1G controls cell cycle progression by maintaining the proper level of p27 protein.
Keywords: PPM1G, protein phosphatase, p27, cell cycle
Introduction
The cell cycle, which comprises G1, S, G2, and M phases, is a series of ordered events that lead to the division of one cell into two daughter cells [1]. The transition between different phases of the cell cycle is tightly regulated by a variety of mechanisms to avoid any error that might result in the cell death or cancer, and the most important players are cyclin-CDK (cyclin-dependent kinase) complexes.
In mammalian cells, the initiation of the cell cycle is driven by the transcriptional activation of D-type cyclins, including cyclin D1, D2 and D3, and the activation of cyclin D-CDK4 and cyclin E-CDK2 [2,3]. As an important CDK inhibitor, p27 binds to and prevents cyclin E-CDK2 activation [4,5], and cooperates with other mechanisms to prevent the improper progression of the cell cycle from the quiescent state [3,6,7]. Deregulation of p27 is frequently observed in human cancers, including lung, breast, prostate, and colorectal cancers [4]. Understanding the regulation of p27 during the cell cycle will help to elucidate how cancer cells escape from cell cycle arrest.
In response to growth inhibitory signals that maintain normal cells in a quiescent state, p27 binds to and inactivates cyclin E-CDK2 in the nucleus, which prevents the subsequent transcription of genes required for G1-S transition [4,5,8]. Upon mitogen stimulation, p27 is either exported into the cytoplasm for proteasome-mediated degradation [9,12], or sequestered by increased amounts of cyclin D-CDK4 [2,7]. These events release p27 inhibition on cyclin E-CDK2. Active cyclin E-CDK2 phosphorylates p27 and induces its degradation in the nucleus [13,15]. The rapid decrease of p27 level leads to the accumulation of active cyclin E-CDK2, which drives the cell cycle into S phase. During this period, p27 shuttles between the nucleus and cytoplasm [9,10,16-21]. Nuclear p27 inhibits cell cycle progression by binding to cyclin E-CDK2, whereas cytoplasmic p27 promotes cell cycle progression by facilitating the assembly and nuclear import of cyclin D-CDK4 [22]. In addition, cytoplasmic p27 binds to RhoA to increase cell motility [23,25]. Therefore, decreased nuclear or increased cytoplasmic p27 level often suggests poor outcome to radiotherapy in cancer patients [26].
Subcellular distribution and protein stability of p27 during the cell cycle are mainly regulated by phosphorylation [27]. In quiescent cells, S10 and T198 phosphorylation interferes with the interaction between p27 and cyclin E-CDK2 and prevents p27 degradation, contributing to the nuclear accumulation of p27 [17,27-29]. At the G1 phase, Kinase Interacting with Stathmin (KIS) induces p27 phosphorylation at S10, which is required for p27 nuclear export and for Kip1 ubiquitination-promoting complex (KPC) E3 ligase-mediated p27 ubiquitination and degradation in the cytoplasm [10,12,30]. Similarly, T198 phosphorylation by AGC family kinases (e.g., Akt, RSK1/2) is required for 14-3-3θ-mediated p27 nuclear export and for p27-RhoA interaction [18,19,24]. Meanwhile, T198 phosphorylation of cytoplasmic p27 is also crucial for the assembly of cyclin D-CDK4/6 to promote cell cycle progression [22]. Many cancers, particularly breast cancer where PI3K/Akt signaling is frequently activated, often exhibit increased T157 and T198 phosphorylation of p27, and therefore increased cytoplasmic p27 [14,18]. These events are associated with poor prognosis [24,31-36]. Tyrosine phosphorylation by Src family kinases (e.g., Src, Abl) converts p27 from the inhibitor to the substrate of cyclin E-CDK2 [19,20], which further phosphorylates p27 at T187, and induces p27 polyubiqutination and degradation mediated by SCFSkp2 E3 ligase [13-15,37]. This mechanism also contributes to the dysfunction of p27 in cancer. Low p27 level is frequently observed in cancers with hyperactive Src or overexpression of Skp2, which results in resistance to cancer therapies [38,39].
Despite the critical role of phosphorylation in regulating p27 functions, we still do not understand how the dynamic p27 phosphorylation is achieved. Kinases that phosphorylate p27 have been identified [27]; however, whether protein phosphatases participate in regulating p27 phosphorylation is largely unknown. Although some evidence suggests that degradation of p27 helps control the level of phosphorylated p27, emerging evidence points to protein phosphatases in this process [40,41]. A member of PPM family phosphatases, PPM1H, has been recently reported to dephosphorylate p27 at T187 in breast cancer cells. Loss of PPM1H increases T187 phosphorylation of p27, decreases p27 protein level, and confers trastuzumab resistance in breast cancer patients [42].
p27 is phosphorylated at several sites and these phosphorylation, in turn, controls the stability and functions of p27. To identify additional p27 phosphatases other than PPM1H, we first analyzed the dynamic phosphorylation pattern of p27 during cell cycle progression, and found that one of the phosphorylation sites, T198, was potentially regulated by dephosphorylation. By using functional screening of known genomic Ser/Thr phosphatases, we identified another member of PPM family phosphatases, PPM1G, as a novel p27 phosphatase that specifically regulates T198 dephosphorylation. We also demonstrated that T198 dephosphorylation by PPM1G affects the subcellular localization and stability of p27 and the subsequent cell cycle progression. Our study not only identifies PPM1G as a novel p27 phosphatase, but also provides the rationale for the potential use of p27 phosphatase in cancer prognosis and treatment.
Materials and methods
Plasmids, mutants, and small hairpin RNA expression constructs
The p27, PP1MG, and 14-3-3θ cDNAs were obtained by PCR from human HaCaT genomic cDNA library and cloned into mammalian expression vectors. Point mutants (p27-T198A/V and PPM1G-D496N) and deletion mutants of p27 or PPM1G were also obtained by PCR. The pcDNA3.0-HA-AktDD was a kind gift from Dr. Gordon Mills. The shRNA against mouse p27 (target sequence 5’-GCCAACAGAACAGAAGAAA) was generated in pRight vector [43]. Lentiviral shRNAs against human PPM1G were kindly provided by Dr. Tianyan Gao from the University of Kentucky, KY, USA.
Antibodies
Antibodies against p27, p27pS10, cyclin A, cyclin D1 and Myc tag (9E10) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); p27pT157 and p27pT198 from R&D (Minneapolis, MN, USA); PPM1G from Bethyl Laboratories (Montgomery, TX, USA); Flag tag (M2) from Sigma (Santa Fe, NM, USA); HA tag from Babco (Richmond, California, USA), and GFP from Invitrogen (Grand Island, NY, USA).
Cell culture, cell transfection, immunoprecipitation and Western blot
Cell culture, transfection, immunoprecipitation, and Western blot were performed essentially as previously described [43,44]. HeLa, 293T and NIH3T3 cells were obtained from American Type Culture Collection (Manassas, VA). HeLa and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Corning, NY, USA) with 10% fetal bovine serum (FBS, Thermo Scientific Hyclone, Logan, UT, USA). NIH3T3 cells were cultured in DMEM with 10% calf serum (CS, Thermo Scientific Hyclone). To arrest cells at G0 phase, HeLa cells were cultured in serum-free media for 48 to 72 h, while NIH3T3 cells were cultured in DMEM containing 0.2% CS. Normal cell culture media were used to release cells into the cell cycle. Cells were harvested at indicated time points for subcellular fractionation, immunoprecipitation, and Western blot.
In vitro protein binding assay
Recombinant glutathione S-transferase (GST) fusion proteins were purified from E. coli as per manufacturer’s instruction (Amersham Biosciences, Piscataway, NJ, USA). In vitro translated proteins (TNT kit, Promega, Madison, WI, USA) were pre-cleared with GST protein for 1 h and then incubated with the indicated GST fusion protein for 2 h in the in vitro binding buffer (50 mM Tris pH 7.5, 120 mM NaCl, 2 mM EDTA, 0.1% NP40). After extensive washing with the in vitro binding buffer, proteins bound to GST fusion proteins were retrieved by incubation with glutathione sepharose beads and identified by Western blot with indicated antibodies.
In vitro phosphatase assay
In vitro phosphatase assay was performed as previously described [43,45]. Phospho-p27 was immunoprecipitated from 293T cells transfected with Flag-p27. PPM1G, WT, or DN mutant, was purified from E. coli BL21 strain as GST fusion protein. PPM1G was incubated with phospho-p27 in the in vitro phosphatase buffer for 1 h at 37°C. Dephosphorylation of p27 was analyzed by Western blot using p27pT198 antibody.
BrdU incorporation and immunofluorescence staining
Cells were grown on coverslips for 24 h, and then BrdU was added to the culture media for 4 h. Cells were then fixed with 4% paraformaldehyde at 4°C, treated with 2N HCl to denature DNA, and incubated with fluorescence-conjugated anti-BrdU antibody (Invitrogen) in 5% fat-free milk at room temperature for 4 h. Cells were examined under a Zeiss Axioplan II microscope (Thornwood, NY, USA).
Subcellular fractionation
Subcellular fractionation was carried out as previously described [11]. Cells were collected in isotonic buffer (20 mM HEPES, pH 7.9, 110 mM KAc, 5 mM NaAc, 2 mM MgAc, 1 mM EGTA, 2 mM DTT and 50 μg/ml Digitonin) containing protease and phosphatase inhibitors (Roche, Basel, Switzerland). The cell lysate was centrifuged at 3,000 rpm for 10 min, and the supernatant collected as the cytoplasmic fraction. The pellet was washed once with isotonic buffer, dissolved in 2x SDS Laemmli buffer, and saved as the nuclear fraction. Both fractions were analyzed using Western blot with indicated antibodies.
Results
PPM1G regulates endogenous p27 phosphorylation at T198 during early G1 phase
To investigate the regulatory roles of p27 phosphorylation during cell cycle progression, we first examined the profile of p27 phosphorylation during the G1-S transition in the cell cycle. HeLa cells were synchronized at G0 phase by serum starvation and released into the cell cycle by restoring the normal culture media, and the phosphorylation of p27 was determined by Western blot with phospho-specific antibodies. As shown in Figure 1A, phosphorylation of p27 at T198 site (p27pT198) was absent at 0 h, peaked 30 min after serum stimulation, and then declined rapidly to almost undetectable at 2 hours. However, the total p27 level did not change within the first 4-6 hours culture in serum-containing medium, suggesting that phosphatase activity was involved in regulating T198 phosphorylation. In contrast, the regulation in the levels of p27pT157 and p27pS10 exhibited a different pattern than that of p27pT198. p27pT157 and p27pS10 levels did not show a significant change during the first 2 hours of serum stimulation.
Figure 1.
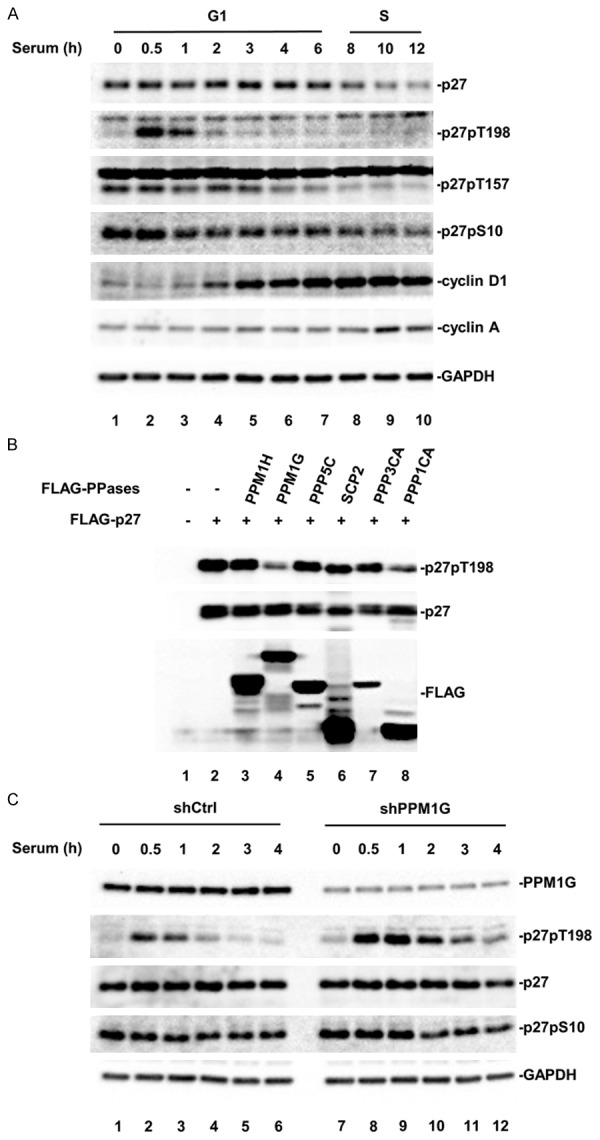
PPM1G regulates endogenous p27 phosphorylation at T198 during early G1 phase. A. Dynamic phosphorylation of p27 during G1 phase. HeLa cells were arrested at G0 phase and then released into the cell cycle. Cell lysates were collected at the indicated time points. Phosphorylation of p27 was analyzed by Western blot using specific antibodies. B. Phosphatase screening. 293T cells were transfected with YFP-p27 and Flag tagged phosphatase. Phosphorylation of p27 at T198 (p27pT198) was determined by Western blot. C. PPM1G knockdown increases p27pT198 levels at early G1 phase. Control and PPM1G-depleted HeLa cells stably expressing shRNA against human PPM1G or harboring empty vector were generated. Cells were treated as described in A, and collected at the indicated time points. Levels of PPM1G, p27, and p27pT198 were determined by Western blot.
To identify phosphatase(s) that targeted p27pT198 for dephosphorylation, we screened 40 protein serine/threonine phosphatases including 18 PPMs, 13 PPPs, 5 FCP/SCPs and 4 DUSPs [26]. Representative screening data (Figure 1B) showed that co-transfection of the phosphatase PPM1G or the catalytic subunit of PP1α reduced the level of p27pT198. To further validate this PPM1G or PP1α effect on T198 phosphorylation, we measured the level of p27pT198 at G1 phase in HeLa cells treated with calyculin A, a PP1/PP2A inhibitor [46,47]. We found calyculin A treatment failed to rescue the reduction of p27pT198 level at G1 phase (Supplemental Figure 1), suggesting that PP1α was not involved in this process. Because no inhibitor for PPM1G has been reported, we determined the level of endogenous p27pT198 at G1 phase in HeLa cells with PPM1G knockdown. PPM1G knockdown was achieved by stably expressing a specific shRNA against human PPM1G, which was verified by Western blot with anti-PPM1G antibody (Figure 1C, PPM1G blot). We found that, during G1 phase, the p27pT198 level in PPM1G knockdown cells (shPPM1G) was dramatically higher than that in control cells (shCtrl) (Figure 1C). A higher level of p27pT198 was also observed in MCF7 cells with PPM1G knockdown (Supplemental Figure 2). Together, these results suggested that PPM1G participated in the regulation of p27pT198 during the G1-S transition of the cell cycle.
PPM1G dephosphorylates p27 at T198
To further demonstrate that PPM1G controls p27 phosphorylation at T198 site, we first determined whether the phosphatase activity of PPM1G was required to decrease p27pT198 phosphorylation. For this purpose, we generated a catalytically inactive PPM1G mutant (PPM1G-DN), in which amino acid Asp 496 at the catalytic loop was mutated into Asn. Then, we tested the effect of PPM1G-DN on p27 phosphorylation in 293T cells. Whereas PPM1G-WT caused a dose-dependent reduction in the p27pT198 level, PPM1G-DN did not reduce the p27pT198 level at any dosage (Figure 2A), indicating that the phosphatase activity of PPM1G was essential for its effect on p27pT198.
Figure 2.

PPM1G dephosphorylates p27 at T198. A. The catalytically inactive mutant of PPM1G (D496N) fails to reduce p27pT198. 293T cells were transfected with Flag-p27 and increasing amounts of Flag-PPM1G, WT or DN mutant. PPM1G, p27 and p27pT198 levels were determined by Western blot. B. MG132 has no effect on PPM1G-mediated p27 dephosphorylation. Experiments were carried out similarly as described in A. Cells were treated with MG132 (10 µM) for 8 h. C. PPM1G dephosphorylates p27 in vitro. Phosphorylated p27 was immunoprecipitated from 293T cells, and mixed with PPM1G purified from E. coli as GST fusion protein in the phosphatase reaction buffer. Levels of p27pT198, p27pS10, p27 and PPM1G were determined by Western blot.
There are at least two possible mechanisms that may explain how PPM1G reduces the p27pT198 level: PPM1G-mediated pT198 dephosphorylation or PPM1G-enhanced p27pT198 protein degradation. To distinguish these two possibilities, we treated cells with MG132, a potent inhibitor of proteasomemediated protein degradation pathway, and determined the p27pT198 level in cells expressing ectopic PPM1G. We found that MG132 did not rescue the PPM1G-induced reduction of p27pT198 in 293T cells (Figure 2B), suggesting that PPM1G regulated the dephosphorylation of p27pT198.
To directly prove that PPM1G is a bona fide phosphatase of p27, we performed an in vitro phosphatase assay of PPM1G toward p27. Recombinant GST-PPM1G purified from E. coli was used as the phosphatase, and T198-phosphorylated p27 was immunoprecipitated from cells and used as the substrate. Figure 2C shows that the p27pT198 level was dramatically reduced after the addition of increasing amounts of PPM1G-WT, but not the DN mutant, indicating that PPM1G directly dephosphorylated p27 at T198. We also examined the effect of PPM1G on p27pS10, which was affected neither by PPM1G-WT nor the DN mutant, indicating that PPM1G specifically targeted p27 at T198 site.
PPM1G physically interacts with p27 in cells and in vitro
Because an enzyme-substrate complex is required for enzymatic reaction, and if PPM1G is a phosphatase of p27, we expect to observe a physical interaction between PPM1G and p27. To determine if PPM1G binds to p27, we first used a co-immunoprecipitation (Co-IP) assay in 293T cells that were co-transfected with YFP-p27, and Flag-PPM1G-WT or DN mutant. We found that both PPM1G-WT and DN bound to p27, indicating that the phosphatase activity of PPM1G was not required for their interaction (Figure 3A).
Figure 3.
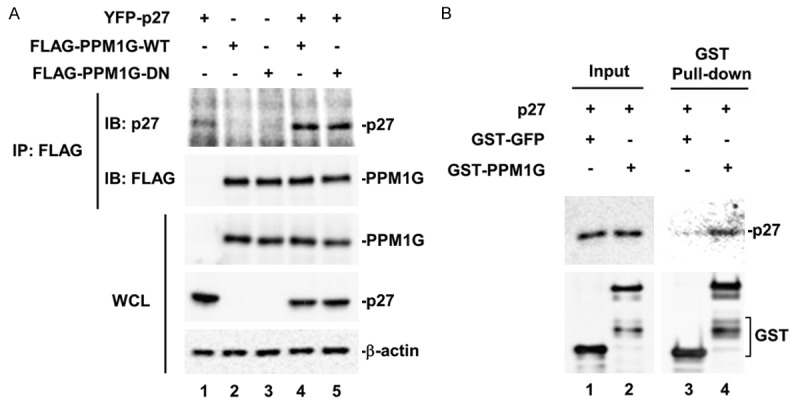
PPM1G physically interacts with p27 in cells and in vitro. A. PPM1G interacts with p27 in 293T cells. GFP-p27 and Flag-PPM1G (WT or DN) were co-transfected into 293T cells. PPM1G was immunoprecipitated (IP) with anti-Flag antibody and the presence of p27 in the IP complex was determined by Western blot. B. PPM1G interacts with p27 in vitro. In vitro translated Flag-p27 was incubated with GFP or PPM1G purified from E. coli as GST fusion protein in an in vitro binding buffer. GST pull-down product was then analyzed by Western blot using p27 antibody.
To exclude the possibility that other cellular proteins mediate the formation of PPM1G-p27 complex, we carried out an in vitro GST pull-down assay. GST-PPM1G and GST-GFP (used as negative control) were purified from E. coli, and Flag-p27 was obtained by in vitro coupled transcription/translation. Figure 3B shows that p27 was detected only in the pull-down product of GST-PPM1G (lane 4), but not in that of GST-GFP (lane 3), indicating that PPM1G interacted directly with p27. Thus, from both dephosphorylation and protein interaction assays we have identified PPM1G as a p27 phosphatase targeting T198 site.
PPM1G delays cell cycle progression in NIH3T3 cells by stabilizing p27
To allow cell cycle progression, the phosphorylation of p27 at T198 facilitates p27 cytoplasmic translocation at G1 phase where p27 is targeted for degradation mediated by KPC E3 ligase [17-19,24]. After identifying PPM1G as a p27 phosphatase to dephosphorylate pT198, we investigated the effect of PPM1G on p27 protein level and cell cycle progression. NIH3T3 cells stably expressing PPM1G or control vector were generated for this study. In control cells, the p27 level was highest at quiescent state, started to decrease 4 hours after serum stimulation, and reached minimum at 10 hours (Figure 4A). However, in PPM1G-expressing cells, the p27 level did not change throughout the G1 phase, indicating that PPM1G stabilized p27. qRT-PCR analysis of p27 mRNA levels in these cells confirmed that PPM1G did not affect p27 transcription (Supplemental Figure 3). Treating cells with MG132 to block protein degradation further confirmed that PPM1G stabilized p27 as p27 in control cells treated with MG132 was recovered to the same level as that in PPM1G-expressing cells (Figure 4B). These data suggested that p27 underwent a proteasome-mediated protein degradation process during G1-S transition, and that ectopic expression of PPM1G protected p27 from degradation.
Figure 4.

PPM1G delays cell cycle progression in NIH3T3 cells by stabilizing p27. A. PPM1G increases p27 protein level at G1 phase of the cell cycle. NIH3T3 cells stably expressing PPM1G or control vector were arrested at G0 phase and then released into the cell cycle. Cell lysates were collected and the p27 protein level was determined by Western blot. The results were quantified according to three independent experiments. The p27 protein levels were normalized to GAPDH protein. Values represent the mean of three independent experiments; error bars are ± standard deviation of the mean. P values were determined by student’s t-test. B. PPM1G inhibits the degradation of p27 protein. Experiments are carried out as described in A except that one set of cells was treated with MG132 (10 µM). C. PPM1G delays cell cycle progression in NIH3T3 cells. Cell proliferation was determined using BrdU incorporation assay. Upper panel: BrdU staining. Lower panel: Quantification of BrdU positive cells (total cell number >1,000). P values were determined by one-way ANOVA. D. Knocking down p27 rescues PPM1G-induced cell cycle delay. NIH3T3 cells stably expressing PPM1G or control vector were transfected with control or p27 shRNA. Cell proliferation was determined using BrdU incorporation assay. Left panel: Quantification of BrdU positive cells. Right panel: Western blot of p27. P values were determined by student’s t-test.
Because high p27 protein level is important in maintaining the cell cycle at G0/G1 phase [48,49], we hypothesized that overexpressing PPM1G would arrest or delay cell cycle progression by upregulating p27. We used BrdU incorporation assay to assess DNA synthesis as the indicator for cell cycle progression into S phase. Indeed, in NIH3T3 stable cells we found that PPM1G-expressing cells displayed significantly fewer BrdU-positive signals than control or PPM1G-DN-expressing cells (Figure 4C). Moreover, this delay could be completely reversed by knocking down p27 (Figure 4D). This demonstrated that PPM1G could inhibit G1-S transition by upregulating p27.
Knocking down PPM1G renders cells resistant to G1 cell cycle arrest
The physiological function of PPM1G in regulating p27 level and G1-S progression was further determined in PPM1G-depleted HeLa cells. Western blot confirmed the efficient knockdown of PPM1G by PPM1G-specific shRNA in these cells (Figure 5A, PPM1G blot). In PPM1G knockdown cells, the p27 level decreased 6 hours after serum stimulation and was barely detectable at 10 hours, whereas p27 level remained unchanged in control cells (Figure 5A, p27 blot), indicating that PPM1G is required to maintain the stability of p27 during G1 phase. Furthermore, we also confirmed that the reduction of p27 levels in PPM1G-depleted cells was due to decreased p27 protein stability, but not transcriptional repression (Supplemental Figure 4).
Figure 5.
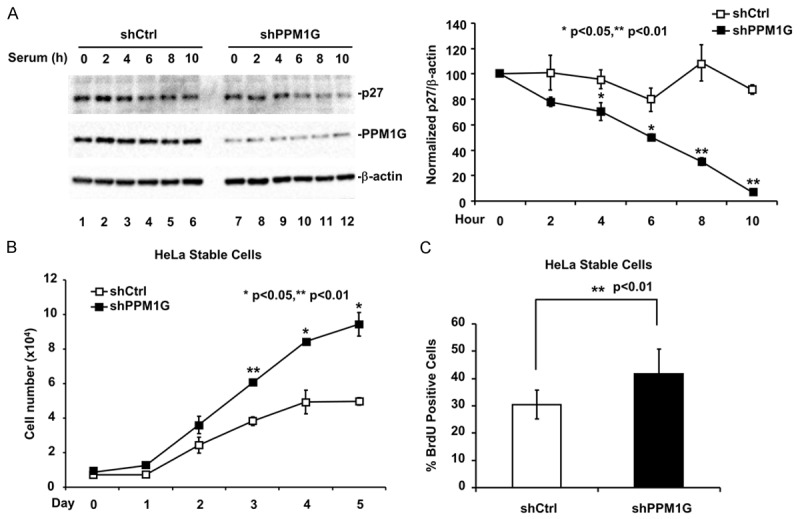
Knocking down PPM1G renders cells resistant to G1 cell cycle arrest. A. PPM1G knockdown decreases p27 protein level at G1 phase. The p27 level in HeLa cells stably expressing PPM1G shRNA or carrying control was examined as described in Figure 4A. B. PPM1G depletion increases cell proliferation. HeLa cells were cultured under serum derprivation condition. Cell number was determined every 24 h with a hemocytometer. C. PPM1G depletion promotes DNA synthesis. DNA synthesis in HeLa cells was measured by using BrdU incorporation assay.
Since knocking down PPM1G resulted in destabilization of p27, we speculated that PPM1G knockdown could render cells resistant to G1 phase arrest-inducing conditions, such as growth factor withdrawal [3,31-34]. Thus, we analyzed the effect of PPM1G knockdown on serum starvation-induced G1 arrest in HeLa cells. Control HeLa cells grew exponentially until day 4 and stopped at day 5. In contrast, PPM1G knockdown HeLa cells grew at a significantly higher rate at all time-points examined (Figure 5B). Similar results were obtained using the BrdU incorporation assay. As shown in Figure 5C, under serum deprivation condition, PPM1G-depleted HeLa cells exhibited more BrdU positive signals than control cells.
PPM1G regulates the interaction between p27 and 14-3-3θ, and subcellular distribution of p27
It has been reported that upon growth factor stimulation, nuclear p27 is exported to the cytoplasm by the chaperon protein 14-3-3θ to release its growth inhibitory effect. This process requires p27 phosphorylation at T198 site, as T198-phosphorylated p27 exhibits a higher binding affinity to 14-3-3θ. One of the kinases that phosphorylates p27 at T198 is Akt. Akt-mediated T198 phosphorylation leads to p27 binding to 14-3-3θ and the subsequent p27 cytoplasmic translocation at G1 phase [18,19,24]. Cytoplasmic p27 is then degraded through the KPC E3 ligase-mediated polyubiquitination/proteasome pathway [12]. Since we have observed that PPM1G co-expression enhanced p27 stability, we were wondering if PPM1G affected the p27 binding to 14-3-3θ and the subsequent nuclear export. We first determined the effect of PPM1G on Akt-induced p27 T198 phosphorylation. To exclude the direct effect of PPM1G on Akt or upstream of Akt, we used a constitutively active form of Akt (AktDD) in our study. A non-phosphorylatable mutant of p27, p27-T198A, was used as Akt-specific phosphorylation control. AktDD strongly enhanced p27 T198 phosphorylation (compare lane 3 to lane 1, Figure 6A), which was abolished by the co-expression of PPM1G-WT (Figure 6A, lane 5) but not PPM1G-DN (Figure 6A, lane 6), suggesting that PPM1G regulated Akt-induced p27 T198 phosphorylation.
Figure 6.
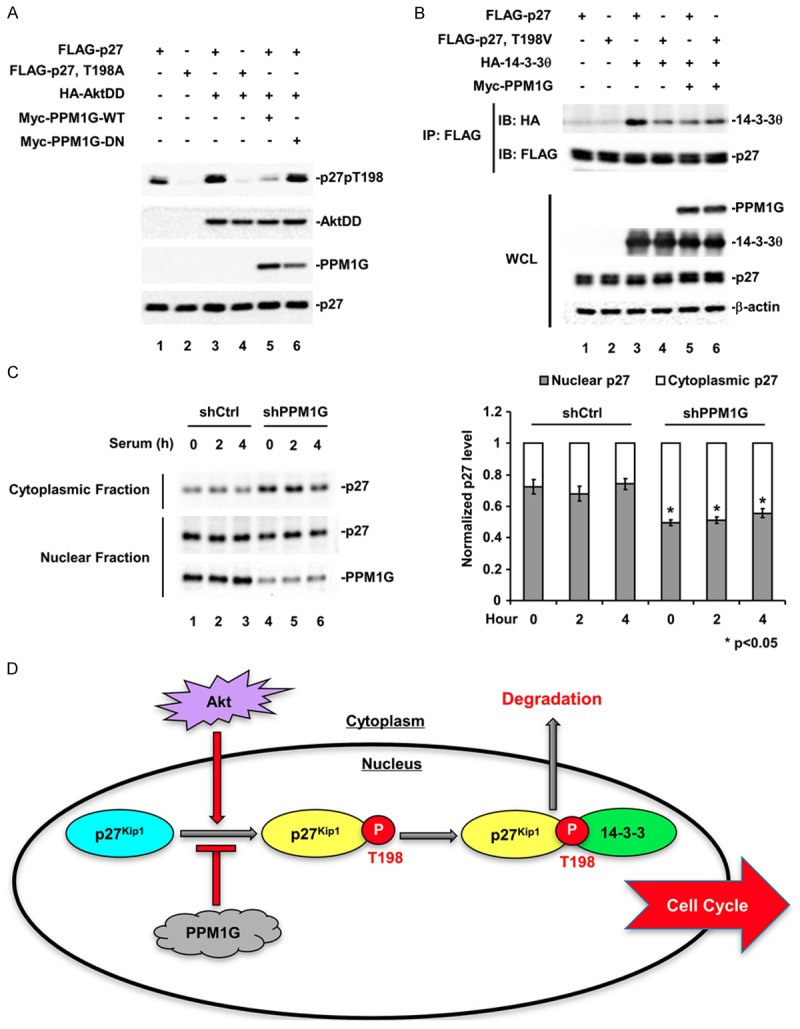
PPM1G regulates the interaction between p27 and 14-3-3θ, and the subcellular distribution of p27. A. PPM1G dephosphorylates Akt-phosphorylated p27pT198. Flag-p27, HA-AktDD, and Myc-PPM1G were co-transfected into 293T cells, and p27pT198 levels were determined by Western blot. B. PPM1G inhibits the interaction between p27 and 14-3-3θ. Flag-p27, HA-14-3-3θ, and Myc-PPM1G were co-transfected into 293T cells, and the interaction between p27 and 14-3-3θ was determined by Co-IP. IP: p27 was immunoprecipitated with anti-Flag antibody. 14-3-3θ levels in the IP complex were determined by Western blot using HA antibody. C. Knocking down PPM1G increases cytoplasmic localization of p27. The subcellular distribution of p27 in control and PPM1G-depleted HeLa cells was determined using subcellular fractionation assay. β-tubulin and Histone H3 were used as cytoplasmic and nuclear control. P values were determined by student’s t-test. D. A working model for T198 phosphorylated cytoplasmic translocation of p27 at early G1 phase. PPM1G counteracts Akt-induced p27pT198 to reduce interaction of p27 with 14-3-3, p27 nuclear export and subsequent degradation in cytoplasm.
Since T198 phosphorylation is essential for the interaction between p27 and 14-3-3θ [18], we next determined the effect of PPM1G on p27 binding to 14-3-3θ. For this purpose, we examined if p27 and 14-3-3θ could be co-immunoprecipitated from 293T cells in the presence or absence of PPM1G. Wild-type p27 strongly interacted with 14-3-3θ, as indicated by the presence of 14-3-3θ in the p27 immunoprecipitates (Figure 6B; lane 3), whereas the non-phosphorylatable mutant p27-T198V exhibited a much weaker interaction with 14-3-3θ (lane 4). In supporting our hypothesis, co-expression of PPM1G greatly inhibited the interaction of wild-type p27 with 14-3-3θ, as indicated by the decreased level of 14-3-3θ in the p27 immunoprecipitated complex (lane 5). PPM1G had no effect on the binding of p27-T198V to 14-3-3θ (lane 6), suggesting that PPM1G inhibited the interaction between p27 and 14-3-3θ by dephosphorylating p27 at T198.
Binding to 14-3-3θ facilitates p27 translocation to the cytoplasm at early G1 phase [18,19]. Thus, we examined the effect of PPM1G on the subcellular localization of p27 at G1 phase both in control and in PPM1G-depleted HeLa cells. Specifically, PPM1G- or scramble shRNA-expressing stable cells were serum-starved for 3 days, refed with serum-containing normal medium, and cells were collected at indicated time points. Subcellular fractionation assay was performed to measure the levels of nuclear and cytoplasmic p27. As shown in Figure 6C, comparing to the cytoplasmic level of p27 in control cells, knocking down PPM1G greatly increased the level of cytoplasmic p27 during early G1 phase (0-4 hours after serum stimulation). In consistence with this, nuclear p27 was decreased in PPM1G knockdown cells comparing to that in control cells. These data suggested that PPM1G knockdown promoted the cytoplasmic localization of p27 at G1 phase.
Discussion
p27Kip1 is one of the key regulators at the early stages of the cell cycle. High nuclear p27 level in quiescent cells is essential to prevent the initiation of the cell cycle, while nuclear export and degradation of p27 triggers cell cycle initiation [10-12,17-19]. Protein level and subcellular localization of p27 are precisely controlled, mainly by post-translational modifications, particularly phosphorylation [27].
Although dephosphorylation has been implicated in regulating p27 activities [40,50], p27 protein phosphatases have only been studied recently. A PPM family phosphatase, PPM1H, has been shown to dephosphorylate p27 at T187 site in breast cancer cells, to stabilize p27, and to inhibit cell cycle progression [42]. However, it was suggested that T187 phosphorylation tends to be rapidly removed by degradation, but not by dephosphorylation [14,15,38,51]. In contrast, the decrease of p27 phosphorylation at T198 level at early G1 phase was not concomitant with p27 degradation, suggesting the involvement of phosphatase(s) in this process. In this study, we identified a PPM family phosphatase, PPM1G, as a novel p27 phosphatase specifically and directly dephosphorylating p27 at T198 site. Through a series of biochemical and genetic experiments, we provided evidence that supported the regulation of p27pT198 by PPM1G phosphatase.
How selective is PPM1G-mediated p27 dephosphorylation? p27 is phosphorylated at various sites simultaneously during G1-S transition. However, knocking down PPM1G only enhanced T198 phosphorylation; it had no effect on S10 or T157 phosphorylation, which was also confirmed by the in vitro phosphatase assay (Figure 2C). Phosphorylation of T157 shows great structural and functional similarities with T198 phosphorylation [18,22,31-33,52,53], but cannot be targeted by PPM1G, suggesting that PPM1G is specific toward T198 dephosphorylation.
Our identification of PPM1G as a p27pT198 phosphatase is consistent with the roles of T198 phosphorylation in controlling subcellular distribution, protein stability and likely cytoplasmic functions of p27. With regard to the subcellular distribution, T198 phosphorylation is important for nuclear export of p27 at G1 phase [17,19]. Our results showed that PPM1G inhibited the interaction of p27 with 14-3-3θ and affected the nuclear and cytoplasmic levels of p27. Thus, our results support the notion that T198 dephosphorylation protects p27 from degradation. This appears to be contrary to a previous report showing that the p27-T198A mutant is more susceptible to degradation than wild-type p27 [17]. However, other studies have demonstrated that the decreased stability of p27-T198A is due to structural change but not to loss of phosphorylation [54]. In accordance, we observed that only p27-T198A mutant, but not p27-T198V mutant, was rapidly degraded when transfected into 293T cells (data not shown). Therefore, we believe that PPM1G-mediated dephosphorylation sustains p27 nuclear retention and increases p27 stability. In addition to regulating p27 subcellular distribution and stability, T198 phosphorylation is essential for cyclin D-CDK4 assembly and p27-RhoA interaction [22,24]. Indeed, we observed an increased ratio of cytoplasmic p27/nuclear p27 in PPM1G-depleted HeLa cells, which could potentially affect cell motility. Further experiments are needed to evaluate these functions of p27.
PPM1G participates in many essential physiological processes. Originally identified as a cell cycle inhibitor [55], PPM1G has been mainly studied in DNA damage processes [56,57], as it was first identified as a histone γ-H2AX phosphatase [58]. Recently, PPM1G has been found to dephosphorylate and inactivate deubiquitinase USP7S, stabilize Mdm2, and mediate p53-dependent DNA damage response [57]. PPM1G has also been shown to dephosphorylate survival motor neuron (SMN) for pre-mRNA splicing [59] and 4E-BP1 for protein synthesis inhibition [60]. Our study added p27 as a novel substrate of PPM1G and demonstrated that PPM1G-mediated cell cycle inhibition is at least in part through its effect on p27.
How the activity of PPM1G itself is regulated is still unknown. In our studies, PPM1G protein level showed no obvious change during the cell cycle (data not shown). It has been reported that PPM1G can be regulated by phosphorylation, acetylation, and ubiquitination [61,63], and that it is hyperphosphorylated, potentially by ATM kinase, after UV treatment [56]. Therefore, it is possible that post-translational modifications (PTMs) of PPM1G may affect how PPM1G selects its various substrates. However, how PTMs regulate the activity of PPM1G still needs further investigation.
In conclusion, PPM1G is a p27 phosphatase that specifically targets T198 dephosphorylation, a process that sustains p27 nuclear localization and inhibits cell cycle initiation. Since protein destabilization and cytoplasmic mislocalization of p27 are frequently observed in cancers, our identification of PPM1G as a p27 regulator warrants further studies.
Acknowledgements
We thank Drs. Pumin Zhang, Tianyan Gao, and Gordon Mills for providing essential reagents. We thank Ana Maria Rodriguez (Baylor College of Medicine) for assistance on paper editing. This research was supported by MOST (2012CB966600, 2015CB553800), NSFC (91540205, 31571447, 31090360), DoD (1W81XWH-15-1-0650), NIH grants (R01DK073932 to X.L., R01AR053591, R01GM063773 and R01CA108454 to X.-H.F.), Project 985, and the Fundamental Research Funds for the Central Universities.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Alberts B. Molecular biology of the cell. New York: Garland Science; 2002. [Google Scholar]
- 2.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 4.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 5.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 6.Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1:103–110. [PubMed] [Google Scholar]
- 7.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 8.Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:5291–5295. doi: 10.1073/pnas.91.12.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 11.Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengst L, Slingerland JM. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 13.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- 14.Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 16.Susaki E, Nakayama KI. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015–3020. doi: 10.4161/cc.6.24.5087. [DOI] [PubMed] [Google Scholar]
- 17.Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JS, Manns MP, Malek NP. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 2006;25:5159–5170. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 19.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- 20.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–2310. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 21.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larrea MD, Liang J, Da Silva T, Hong F, Shao SH, Han K, Dumont D, Slingerland JM. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008;28:6462–6472. doi: 10.1128/MCB.02300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106:9268–9273. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: an oncogenic function for CDK inhibitors? Nat Rev Cancer. 2004;4:948–955. doi: 10.1038/nrc1501. [DOI] [PubMed] [Google Scholar]
- 26.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 27.Vervoorts J, Luscher B. Post-translational regulation of the tumor suppressor p27(KIP1) Cell Mol Life Sci. 2008;65:3255–3264. doi: 10.1007/s00018-008-8296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotake Y, Nakayama K, Ishida N, Nakayama KI. Role of serine 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a serine 10 mutation. J Biol Chem. 2005;280:1095–1102. doi: 10.1074/jbc.M406117200. [DOI] [PubMed] [Google Scholar]
- 30.Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 33.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 34.Alkarain A, Slingerland J. Deregulation of p27 by oncogenic signaling and its prognostic significance in breast cancer. Breast Cancer Res. 2004;6:13–21. doi: 10.1186/bcr722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen S, So Y, Singh K, Slingerland JM, Resnick MB, Zhang S, Ruiz V, Moss SF. Promotion of cytoplasmic mislocalization of p27 by Helicobacter pylori in gastric cancer. Oncogene. 31:1771–1780. doi: 10.1038/onc.2011.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He S, Lu M, Xue W, Wang Y, Zhao Y, Gao S, Ke Q, Liu Y, Li P, Cui X, Cheng C, Shen A. Phosphorylated p27Kip1 on Thr157 is an important prognosis in human hepatocellular carcinoma in vivo and in vitro. Med Oncol. 2011;28:94–104. doi: 10.1007/s12032-009-9408-4. [DOI] [PubMed] [Google Scholar]
- 37.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 38.Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128:281–294. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Krek W. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci U S A. 2001;98:5043–5048. doi: 10.1073/pnas.081474898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Asa SL, Fantus IG, Walfish PG, Ezzat S. Vitamin D arrests thyroid carcinoma cell growth and induces p27 dephosphorylation and accumulation through PTEN/akt-dependent and -independent pathways. Am J Pathol. 2002;160:511–519. doi: 10.1016/S0002-9440(10)64870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. 1999;19:2416–2424. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee-Hoeflich ST, Pham TQ, Dowbenko D, Munroe X, Lee J, Li L, Zhou W, Haverty PM, Pujara K, Stinson J, Chan SM, Eastham-Anderson J, Pandita A, Seshagiri S, Hoeflich KP, Turashvili G, Gelmon KA, Aparicio SA, Davis DP, Sliwkowski MX, Stern HM. PPM1H is a p27 phosphatase implicated in trastuzumab resistance. Cancer Discov. 2011;1:326–337. doi: 10.1158/2159-8290.CD-11-0062. [DOI] [PubMed] [Google Scholar]
- 43.Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, Shi Y, Chen YG, Meng A, Feng XH. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006;125:915–928. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai F, Chang C, Lin X, Dai P, Mei L, Feng XH. Erbin inhibits transforming growth factor beta signaling through a novel Smad-interacting domain. Mol Cell Biol. 2007;27:6183–6194. doi: 10.1128/MCB.00132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wrighton KH, Willis D, Long J, Liu F, Lin X, Feng XH. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J Biol Chem. 2006;281:38365–38375. doi: 10.1074/jbc.M607246200. [DOI] [PubMed] [Google Scholar]
- 46.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 47.Resjo S, Oknianska A, Zolnierowicz S, Manganiello V, Degerman E. Phosphorylation and activation of phosphodiesterase type 3B (PDE3B) in adipocytes in response to serine/threonine phosphatase inhibitors: deactivation of PDE3B in vitro by protein phosphatase type 2A. Biochem J. 1999;341:839–845. [PMC free article] [PubMed] [Google Scholar]
- 48.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- 49.Rivard N, L’Allemain G, Bartek J, Pouyssegur J. Abrogation of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in fibroblasts. J Biol Chem. 1996;271:18337–18341. doi: 10.1074/jbc.271.31.18337. [DOI] [PubMed] [Google Scholar]
- 50.Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000;275:25146–25154. doi: 10.1074/jbc.M001144200. [DOI] [PubMed] [Google Scholar]
- 51.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 52.Motti ML, De Marco C, Califano D, Fusco A, Viglietto G. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle. 2004;3:1074–1080. [PubMed] [Google Scholar]
- 53.Yang H, Zhang Y, Zhao R, Wen YY, Fournier K, Wu HB, Yang HY, Diaz J, Laronga C, Lee MH. Negative cell cycle regulator 14-3-3sigma stabilizes p27 Kip1 by inhibiting the activity of PKB/Akt. Oncogene. 2006;25:4585–4594. doi: 10.1038/sj.onc.1209481. [DOI] [PubMed] [Google Scholar]
- 54.Schiappacassi M, Lovisa S, Lovat F, Fabris L, Colombatti A, Belletti B, Baldassarre G. Role of T198 modification in the regulation of p27(Kip1) protein stability and function. PLoS One. 2011;6:e17673. doi: 10.1371/journal.pone.0017673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guthridge MA, Bellosta P, Tavoloni N, Basilico C. FIN13, a novel growth factor-inducible serine-threonine phosphatase which can inhibit cell cycle progression. Mol Cell Biol. 1997;17:5485–98. doi: 10.1128/mcb.17.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 57.Khoronenkova SV, Dianova II, Ternette N, Kessler BM, Parsons JL, Dianov GL. ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol Cell. 2012;45:801–813. doi: 10.1016/j.molcel.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimura H, Takizawa N, Allemand E, Hori T, Iborra FJ, Nozaki N, Muraki M, Hagiwara M, Krainer AR, Fukagawa T, Okawa K. A novel histone exchange factor, protein phosphatase 2Cgamma, mediates the exchange and dephosphorylation of H2A-H2B. J Cell Biol. 2006;175:389–400. doi: 10.1083/jcb.200608001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petri S, Grimmler M, Over S, Fischer U, Gruss OJ. Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2Cgamma governs Cajal body localization and stability of the SMN complex. J Cell Biol. 2007;179:451–465. doi: 10.1083/jcb.200704163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J, Stevens PD, Eshleman NE, Gao T. Protein phosphatase PPM1G regulates protein translation and cell growth by dephosphorylating 4E binding protein 1 (4E-BP1) J Biol Chem. 2013;288:23225–23233. doi: 10.1074/jbc.M113.492371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 62.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–40. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.