

Abstract
Advanced or recurrent low-grade serous ovarian cancers (LGSC) are resistant to conventional systemic treatments. LGSC carry mutations in RAS or RAF, leading to several clinical trials evaluating MEK inhibitors (MEKi). As LGSC cell lines and xenografts have been difficult to establish, little is known about the efficacy and on-target activity of MEKi treatment in this disease. We compared four different MEKi (trametinib, selumetinib, binimetinib and refametinib) in novel LGSC patient-derived cell lines. Molecular characterization of these cells included copy-number variation and hotspot mutational analysis. Proliferation, apoptosis and cell viability assays were used to study drug efficacy. MEKi on-target efficacy was measured using western blotting and isoelectric point focusing for ERK1/2 phosphorylation. Ten LGSC cell lines were derived from 7 patients with advanced/recurrent disease. Copy number variation showed significant heterogeneity among cell lines, however all samples showed deletions in chromosome 9p21.3, and frequent copy number gains in chromosomes 12 and 20. Mutations in KRAS/NRAS were identified in 4 patients (57%) and RAS mutation status was not associated with higher baseline levels of ERK phosphorylation. Different degrees of MEKi sensitivity were observed in the LGSC cell lines. Two cell lines, both with KRAS mutations, were highly sensitive to MEKi. Drug anti-proliferative efficacy correlated with the degree of inhibition of ERK phosphorylation, with trametinib being the most potent agent. Differences in MEKi efficacy were observed in LGSC cell lines. Trametinib showed the greatest anti-proliferative effects. This study serves as a basis for much needed future research on MEKi drug efficacy in LGSC.
Keywords: Low-grade serous ovarian cancer, cell line models, gene mutations, copy-number alterations, MAPK signalling, MEK inhibitors, ERK1/2 phosphorylation
Introduction
LGSC is much less common than its high-grade serous counterpart (HGSC), however the 10-year survivals are similar for advanced/recurrent cases [1,2]. LGSC is often diagnosed in pre-menopausal women and the malignancy has distinct clinical, pathological and molecular features compared to HGSC [3-5]. As ovarian cancer is a heterogeneous disease, it is important that the various molecular subtypes are studied as distinct diseases [6]. In cases of advanced and/or recurrent disease, tumor growth and progression is more indolent than HGSC, however the disease usually proves to be fatal [2]. LGSC responds poorly to anti-hormone therapy (9%) or chemotherapy, with response rates of less than 5% with chemotherapy treatment in the neoadjuvant or relapsed setting [7-12].
The clinical precursors that lead to the development of LGSC have been characterized. Serous ovarian borderline tumors (SBOT) are known precursor lesions. Certain pathological features associated with SBOT, such as the micropapillary variant (MPSBT), or the presence of invasive implants are risk factors for developing LGSC [13,14]. Alternatively, some cases arise as primary LGSC presenting with either localized or metastatic disease. Molecular characterization of SBOTs, MPSBT and LGSC [15,16] has established a linkage between LGSC and its precursors, resulting in the pathological classification of LGSC as a distinct entity. Whole exome sequencing of LGSC has been reported and shown that these tumors contain few point mutations when compared to HGSC [17].
The Gynecologic Cancer InterGroup (GCIG) Consensus Conference recently affirmed an urgent need to develop specific treatment options for patients with advanced/recurrent LGSC [18,19]. Investigational studies using MEKi in LGSC are ongoing and previously predicated on experimental evidence showing activated MAPK signalling as a consequence of either BRAF or RAS mutations, often found in LGSC [20]. Activating mutations in RAS genes are one of the most common mutations found in cancer and multiple allosteric MEK inhibitors (MEKi), have been developed and used in many cancer types [21,22]. Recently, an important phase II clinical trial evaluated the MEKi selumetinib, reporting response rates of 15% in patients with recurrent LGSC [23]. These results compare favourably with a previous cohort study that reported response rates of 4% using conventional chemotherapy [12]. Several MEKi are now being tested in clinical trials in patients with advanced/recurrent LGSC [NCT00551070, NCT01849874, NCT02101788]. Remarkably, preclinical testing of MEKi in LGSC has been limited due to the absence of representative cell line and xenograft models [24]. Thus, the aim of our study was to establish and molecularly characterize novel LGSC cell lines derived from patients with advanced/recurrent disease in order to compare the efficacy of four different MEKi agents (trametinib, selumetinib, binimetinib and refametinib).
Materials and methods
Patients, tumor samples and clinical information
Advanced or recurrent LGSC samples (tumor and ascites) were obtained from tumor banks (Vancouver General Hospital and the British Columbia Cancer Agency (BCCA); London Regional Cancer Program, London, Ontario, Canada). Tumor bank protocols, cell line derivation, and the research relating to this study, was conducted according to institutional human ethics review board approvals at the BCCA and the University of British Columbia (H14-02859 and R05-0119), and Western University (HSREB 12668E). Clinical information was extracted retrospectively from patient medical records. Tumor bank pathology was reported by certified gynecological pathologists to ensure diagnostic accuracy.
Establishment and maintenance of patient-derived LGSC cell lines
LGSC patient-derived cell lines were established in-house through continuous in vitro culture of primary patient material (tumor tissue or ascites) obtained through the OvCaRe Tumor bank or the London Translational Ovarian Cancer Research Program (iOvCa241 and iOvCa250). LGSC cells were established and maintained in M199:MCDB105 (1:1) media (Cat. No. M5017, Cat. No. M6395, Sigma-Aldrich, Oakville, Ontario, Canada) supplemented with 10% fetal bovine serum (dFBS; Cat. No. SH30070.03, Hyclone, GE Life Sciences, Logan, UT, USA) at 37°C and 5% CO2. No immortalization methods were used.
Cell line authentication
Microsatellite Analysis of Short Tandem Repeats (STRs) was performed for cell line authentication. STR analyses of 10 markers/loci were performed by Genewiz Inc. (South Plainfield, NJ) (Supplementary Table 1).
Mutation testing and copy number analysis
DNA was extracted from all cell lines using All Prep DNA/RNA Mini kit (Cat. No. 80204, Qiagen, Toronto, ON, Canada) according to protocol instructions, and quantified using a NanoDrop 2000TM UV-Vis instrument (Thermo-Scientific, Burlington, ON, Canada). Sequencing libraries were created from cell DNA for molecular characterization using Ion Torrent AmpliSeqTM Cancer Hotspot Panel Version 2 (Life Technologies, Grand Island, NY, USA) as per manufacturer’s protocols. A total of 50 common oncogenes and tumor suppressor genes were screened (Table 2). Sanger sequencing was performed to confirm missense mutations using methods previously described [25]. Primer sequences used for verification are listed in Supplementary Table 2. Priming sites for -12 M13 forward and -27 M13 reverse were added to the 5’ ends to allow direct Sanger sequencing of amplicons [26]. CNV analysis was done using Illumina® HumanOmni 2.5M-8 Array or CytoScan® HD array (Affymetrix, Inc) according to manufacture’s protocols. Nexus Copy NumberTM (BioDiscovery, Inc.) software was used to analyze the copy number data from these two platforms.
Table 2.
Missense mutations identified in LGSC cell lines using Ion AmpliSeq Cancer Hotspot Panel v2 Analysis
| PATIENT 1 | PATIENT 2 | PATIENT 3 | PATIENT 4 | PATIENT 5 | PATIENT 6 | PATIENT 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| Gene Name | Mutation ID | VOA-1312 | iOvCa241 | VOA-3667# | VOA-1056 | VOA-3993 | VOA-3448 | VOA-3723 | VOA-4627 | VOA-4698 | iOvCa250 |
| APC (Chr.5) | COSM19099 | c.3949G>C (Hm) | |||||||||
| FGFR3 (Chr.4) | COSM1539830 | c.1156T>C (Ht) | c.1156T>C (Ht) | ||||||||
| JAK3 * (Chr.19) | COSM34213 | c.2164G>A (Ht) | c.2164G>A (Ht) | ||||||||
| KDR (Chr.4) | COSM149673 | c.1416A>T (Ht) | c.1416A>T (Ht) | c.1416A>T (Ht) | c.1416A>T (Ht) | ||||||
| KIT (Chr.4) | COSM28026 | c.1621A>C (Ht) | c.1621A>C (Ht) | c.1621A>C (Ht) | |||||||
| KRAS * (Chr.12) | COSM520, COSM521 | c.35G>T (Ht) | c.35G>A (Ht) | c.35G>T (Ht) | |||||||
| MET (Chr.7) | COSM5020653 | c.1124A>G (Ht) | c.1124A>G (Ht) | ||||||||
| NRAS * (Chr.1) | COSM584 | c.182A>G (Ht) | c.182A>G (Ht) | ||||||||
| PIK3CA (Chr.3) | COSM328028 | c.1173A>G (Ht) | c.1173A>G (Ht) | c.1173A>G (Ht) | c.1173A>G (Ht) | ||||||
| TP53 * (Chr.17) | COSM99729 | c.701G>A (Ht) | c.701G>A (Ht) | ||||||||
Means missense mutations reported as oncogenic;
transient primary cell culture.
A total of 207 amplicons covering approximately 2,800 COSMIC mutations from 50 oncogenes and tumor suppressor genes: ABL1, EGFR, GNAS, KRAS, PTPN11, AKT1, ERBB2, GNAQ, MET, RB1, ALK, ERBB4, HNF1A, MLH1, RET, APC, EZH2, HRAS, MPL, SMAD4, ATM, FBXW7, IDH1, NOTCH1, SMARCB1, BRAF, FGFR1, JAK2, NPM1, SMO, CDH1, FGFR2, JAK3, NRAS, SRC, CDKN2A, FGFR3, IDH2, PDGFRA, STK11, CSF1R, FLT3, KDR, PIK3CA, TP53, CTNNB1, GNA11, KIT, PTEN, VHL.
In vitro drug sensitivity assay
Trametinib (GSK1120212; Sellekchem, Cat. No. S2673), selumetinib (AZD6244; Cat. No. S1008), binimetinib (MEK162; Cat. No. S7007), and refametinib (Bay 86-9766; Cat. No. S1089) were purchased from Selleck Chemicals (Houston, TX, USA). Dimethylsulfoxide or DMSO (Sigma, Cat. No. D2650) was purchased from Sigma-Aldrich (Oakville, Ontario, Canada). LGSC cells were seeded in 96-well plates at 30-40% confluence. At 24 hours cells were treated with DMSO (control) or MEKi (trametinib, selumetinib, binimetinib, or refametinib) using a range of different drug concentrations. Inhibitory concentration (IC50, or concentration for 50% of maximal inhibition of cell proliferation) was determined using crystal violet assay after 72 h drug treatment.
Immunoassays to detect ERK1/2 phosphorylation
Western blot analysis
LGSC cell lysates were prepared according to previously published protocols [27,28]. Cell lysates (20 μg) were separated on 8% SDS-PAGE gel and transferred to nitrocellulose membranes. Primary antibodies included ERK1/2 (Millipore, Cat. No. 06-182), p-MAPK (p-ERK1/2, Cell signaling, Cat.No. 4376S), MEK1/2 (Santa Cruz Biotechnologies, Cat. No. 436), p-MEK1/2 (Cell Signaling, Cat. No. 9154S), c-PARP (Cell Signaling, Cat. No. 9541S). Vinculin (V9131, Sigma) was used as a protein loading control. The appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (goat-anti-mouse or goat-anti-rabbit, Sigma Cat. No. A9917 and A0545) were used accordingly. Western blots were imaged using Immobilon HRP reagent (Cat. No. WBKLS0500, Millipore, Etobicoke, ON, Canada) and developed by autoradiograph.
Capillary isoelectric point focusing proteomic analysis
Native capillary isoelectric point focusing (cIEF) was performed to assess total and phosphorylated ERK isoform expression using NanoPro1000™ System (ProteinSimple™, Santa Clara, CA) according to manufacturer protocols [23]. G2 premix gradient (pH 5-8) (Cat. No. 040-972, ProteinSimple™), ERK 1/2 (Cat. No. 040-474, ProteinSimple™) primary antibody and Goat anti-Rabbit (Santa Cruz sc-2054) Human Absorbed IgG secondary antibody was used to identify the protein isoforms. For both cIEF and Western blot studies, cells were treated with 20 ng/ml of EGF (Cat. No. PHG0315, Invitrogen) for 10 minutes.
Cell proliferation assays
Cell proliferation was measured using the IncuCyte™ (Essen Biosciences, Ann Arbor, MI, USA). LGSC cell lines were plated at 15-20% confluence on 96 well plates. After 24 hours, cells were treated once with DMSO (control) or MEKi (trametinib 0.1 and 0.5 μM; selumetinib, binimetinib, and refametinib 1 and 5 μM). The IncuCyte, a real-time imaging system using a non-labeled cell monolayer confluence approach, was used to monitor the cell growth by taking phase contrast images of cells at six hour intervals for 5-6 days. Data analysis was performed using IncuCyte™ cell proliferation assay software. Results are representative of three independent experiments, containing five technical replicates.
Cell viability assay
Cell viability was measured using the MTS-Cell Titer 96R Aqueous Non-Radioactive Cell Proliferation Assay following the manufacturer’s recommendations (Cat. No. G5430, Promega, Madison, WI, USA). LGSC cells were seeded at 15-20% confluence in 96-well plates. Cells were treated the next day with DMSO control or drug (trametinib -0.1 and 0.5 μM, or selumetinib, binimetinib, and refametinib at 1 and 5 μM) and incubated for 5-6 days. Then, the media in each well was replaced with 100 μL of fresh media and 20 μL of MTS reagent. Plates were incubated for 3.5 hours at 37°C in humidified 5% CO2. Absorbance at 490 nm was measured using microplate reader (BioTek Epoch SN257811). Cell viability after MEKi treatment was compared to DMSO treated cells.
Apoptosis assay
Apoptosis was measured using a Caspase-Glo® 3/7 Assay Kit (Cat. No. Cat. No. G8090, Promega, Madison, WI, USA). Briefly, iOvCa241, VOA-1056 and VOA-4627 LGSC cells (8×103, 5×103, 4×103 cells/96-well for 24 h treatment and, 6×103, 4×103, 3×103 cells/96-well for 72 h treatment) were incubated for 24 and 72 hours with vehicle (DMSO) or MEKi (trametinib 0.1 μM and 0.5 μM; or selumetinib, binimetinib or refametinib 1 μM and 5 μM). At the end of each treatment, luminescence of each well was measured in a plate-reading luminometer (Tecan Infinite M200Pro). Experiments were performed in triplicate for all conditions and cell lines.
Results
LGSC cell lines have unique cellular and molecular characteristics
Patient clinical data and cell line information used in this study are summarized in Table 1. All cell lines were derived from tumor tissues or ascites from patients with invasive LGSC (advanced or recurrent), though the original patient diagnosis varied as expected (borderline, micropapillary borderline and LGSC). A total of 10 LGSC cell cultures, derived from 7 different patients, were studied and molecularly characterized, though one became senescent (VOA-3667) and another (iOvCa250) grew too slowly to be used for most experiments. Three cell line pairs were derived from the same patient at different time-points in their disease (VOA 1056;VOA-3993, VOA-3448;VOA-3723, and VOA-4627;VOA-4698). Compared to other ovarian cancer cell lines, LGSC cell lines (Supplementary Figure 1) show vacuolization, strong cell adhesion properties (resistance to trypsinization), and low proliferation rates. STR analyses were used to define microsatellite loci for each new cell line and results were compared to the DSMZ STR profile website database (https://www.dsmz.de/services/services-human-and-animal-cell-lines/online-str-analysis.html), which confirmed the uniqueness of these cell lines.
Table 1.
Clinical information LGSC patients and samples
| Patient Number | Age at Diagnosis | Primary Pathology | Disease Stage | Sample Number | Cell Line | Pathology at Collection | Treatment Status | Sample Origin | Mutation Status |
|---|---|---|---|---|---|---|---|---|---|
| Patient #1 | 58 | SBOT with non-invasive implants | IIB | 1 | VOA-1312 | LGSC | Naïve | Ascites | KRAS |
| Patient #2 | 51 | SBOT with LGSC | IIIA | 2 | iOvCa241 | LGSC | Post-chemotherapy | Ascites | KRAS |
| Patient #3 | 53 | SBOT with LGSC | IIC | 3 | VOA-3667* | LGSC | Naïve | Tumor Tissue | KRAS, KDR |
| Patient #4 | 62 | Micropapillary SBOT with invasive implants | IIIC | 4 | VOA-1056 | LGSC | Naïve | Tumor Tissue | NRAS, FGFR3, JAK3 |
| 5 | VOA-3993 | Recurrent LGSC | Post-chemotherapy | Tumor Tissue | NRAS, FGFR3, JAK3 | ||||
| Patient #5 | 42 | Micropapillary SBOT | IC | 6 | VOA-3448 | Recurrent LGSC | Post-chemotherapy | Tumor Tissue | KDR, MET, PIK3CA |
| 7 | VOA-3723 | Recurrent LGSC | Post-chemotherapy | Tumor Tissue | KDR, MET, PIK3CA | ||||
| Patient #6 | 42 | LGSC | IIIC | 8 | VOA-4627 | Recurrent LGSC | Post-chemotherapy | Ascites | KIT, PIK3CA, TP53 |
| 9 | VOA-4698 | Recurrent LGSC | Post-chemotherapy | Ascites | KIT, PIK3CA, TP53 | ||||
| Patient #7 | 61 | Primary Peritoneal LGSC | IIIB | 10 | iOvCa250 | LGSC | Naïve | Ascites | APC, KDR, KIT |
Note: SBOT = Serous Borderline Ovarian Tumor.
Primary cell culture.
Hotspot analysis in our LGSC cells revealed missense mutations in 10 oncogenes: KDR (4/10 cell lines; 3 patients), PIK3CA (4/10; 2), KRAS (3/10; 3), KIT (3/10; 2), NRAS (2/10, 1), MET (2/10; 1), FGFR3 (2/10; 1), JAK3 (2/10; 1), TP53 (2/10, 1), and APC (1/10; 1). As confirmed by Sanger sequencing, 1 to 3 missense mutations were detected per case (Table 2). Only 3 of these missense mutations (KRAS, NRAS, and TP53 genes) are known to be pathogenic variants according to COSMIC and NCBI ClinVar classification (Table 2). Mutations in RAS (KRAS, NRAS) genes were the most frequent and they were detected in 5 out of 10 cell lines derived from 4 different patients (Table 1). No mutations in BRAF were found in the 10 cell lines, and KRAS and NRAS mutations were found to be mutually exclusive. Two cell lines (VOA-4627, VOA-4698), which were derived from the same patient, carried a TP53 missense mutation. This uncommon finding has been confirmed histologically and recently reported as an unusual LGSC of the peritoneum, potentially arising from a low-grade serous tubal intra-epithelial carcinoma (STIC) in a patient harbouring a BRCA1 mutation [29]. All missense mutations identified in the paired samples tested were stable over time.
Subsequent analysis of gene copy-number variation (CNV) was performed in 7 of the cell lines (derived from 6 patients) as shown in Figure 1. There were remarkable differences in the percentage of genomic aberrations (2-40%) among them, but interestingly all cell lines shared copy number loss affecting the chromosome region 9p21.3. This loss was homozygous in all but one cell line (VOA-4627). Amplification of chromosomes 8, 12 (containing the KRAS locus) and 20 were also commonly detected. Copy number gains in chromosomes 12 and 20 were detected in 5 out of 7 cell lines. Of note, chromosomes 4 and 13 were very well conserved. CNV changes were analyzed over time in one of our paired sample (VOA-1056 and VOA-3993). In this case, both cell lines shared similar copy number alteration profiles, however there were significant copy number gains in chromosomes 2, 12, and 20 in the recurrent sample (VOA-3993) following treatment with chemotherapy (Supplementary Figure 2). VOA-4627 showed loss of chromosome 1p, a finding that has been previously observed in LGSC [30] and also showed high levels of chromosomal copy number abnormalities and deletions than the other cell lines (Figure 1). Interestingly, all LGSC cell lines showed copy number changes (either gain or loss) in common MAPK-pathway related genes (Supplementary Table 3).
Figure 1.

Graphical representation of copy number variation according to chromosome number. The top graph is a compilation of all cell lines with increased copy number in blue and decreased in red. For each individual cell line, regions of loss of heterozygosity are represented by a yellow line (below the copy number changes), whereas a purple line represents regions of allelic imbalance.
LGSC cell lines that carried oncogenic mutations in KRAS (iOvCa241, VOA-1312) and NRAS (VOA-1056, VOA-3993) genes were found to have copy number gain with allelic imbalance in the same chromosomal regions containing these loci (chr12:25,357,722-25,403,865 for KRAS, and chr1:115,247,084-115,259,515 for NRAS). Furthermore, iOvCa241 showed copy number gain in BRAF, and VOA-3993 showed copy number gain in KRAS. In the case of the unusual cell line carrying mutations in TP53 and BRCA1, allelic imbalance in multiple regions without clear copy number change was identified. Copy number changes in relation to the genes tested for hot spot analysis, as shown in Supplementary Table 3.
LGSC cell lines have different sensitivities to MEK inhibitors
Drug sensitivity experiments were conducted using four different MEKi (trametinib, selumetinib, binimetinib and refametinib), the first three of which are investigational drugs for the treatment of advanced/recurrent LGSC patients. We determined the IC50 of each drug in each LGSC cell line (Figure 2). Figure 2 shows a comparison of the IC50 for each drug, and all of the cell lines tested were most sensitive to trametinib with IC50 values in the nM range (versus μM for all other drugs). Next, the same cell lines were used to study the impact of all four MEKi’s on proliferation. Using a live cell-imaging instrument (Incucyte®), we assessed the effect of a single drug dose on cell proliferation over a 5 day period. Based on our IC50 data and drug dosing from previous studies [31-33], we chose the following MEKi doses for these experiments: 0.1 μM trametinib, and 1 μM selumetinib/binimetinib/refametinib. MEKi treatments showed the greatest inhibitory effects on cell lines harbouring oncogenic mutations in KRAS (iOvCa241, VOA-1312). In keeping with the IC50 results, trametinib showed the strongest inhibitory effect across all LGSC cell lines (Figure 3).
Figure 2.
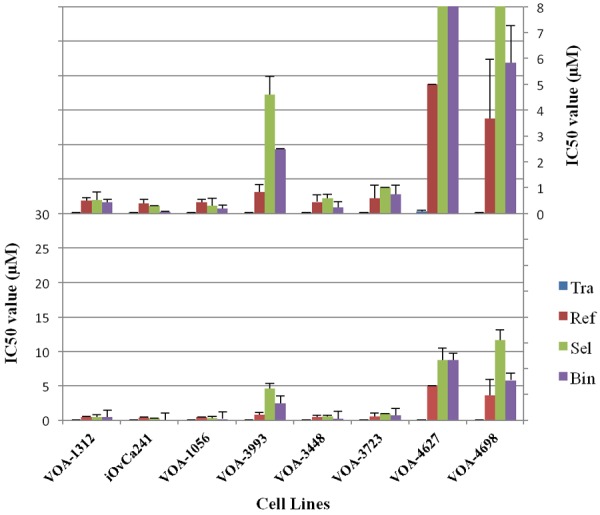
Inhibitory Concentration (IC50) values of LGSC cell lines in response to different MEKi treatments. Tra = trametinib, Sel = selumetinib, Bin = binimetinib, Ref = refametinib. The top graph is an expanded scale of the bottom graph in order to better show the lower IC50 values. Four replicates were performed for each treatment condition. All experiments were performed in triplicate.
Figure 3.

MEKi effects on LGSC cell proliferation. Single dose (at time 0) MEKi proliferation curves for each cell line starting at 20% confluence. Proliferation was measured using Incucyte®. Five replicates were performed for each experimental condition. Experimental results were confirmed by independent experiments performed in duplicate.
Notable differences in proliferation were observed between cell lines in response to MEKi treatment (Figure 3). Complete inhibition of proliferation was observed in iOvCa241 (KRAS G12D) and VOA-1312 (KRAS G12V) in response to treatment with any MEKi, and most of the cells remaining on the plate were visibly non-viable when the images were examined. Therefore, at the doses tested, drug efficacy was comparable for all MEKi in these two highly sensitive cell lines. In contrast, MEKi treatment in the other cell lines showed less anti-proliferative effects and the observed effects depended on the specific MEKi used for treatment. In the less sensitive cells (VOA-1056, VOA-3993, VOA-3448, VOA-3723, VOA-4627, VOA-4698), trametinib showed the greatest anti-proliferative activity (Figure 3, patients #3, #4, #5). Using an alternative assay to measure cell viability (tetrazolium assay reagent; data not shown) we confirmed the differential cell sensitivity and MEKi efficacy results detected in the proliferation assays (data not shown).
MEK inhibitors vary in their efficacy at blocking ERK1/2 phosphorylation
After detecting different proliferation and viability effects among the four MEKi’s, we analyzed the degree of on-target efficacy by measuring the levels of ERK1/2 phosphorylation (p-ERK1/2), a downstream effector of MEK. To analyze p-ERK1/2 we used two protein-based techniques: Western blot (WB) and capillary isoelectric point focusing technology (cIEF). As seen in Figure 4A, basal levels of p-ERK1/2 varied among cell lines, being lower in iOvCa241, VOA-1056, VOA-3448, VOA-4627, and higher in iOvCa250, VOA-1312, VOA-3723, VOA-3993, and VOA-4698. Of note, iOvCa241 cells only expressed the ERK1 isoform (ERK2 homozygous copy loss was detected in the CNV array), and VOA-1056 cells predominantly expressed ERK2. Interestingly, an increase in p-ERK1/2 was detected in all three of the recurrent cell line samples compared to their patient-matched initial cell line samples (VOA-1056 vs VOA-993; VOA-3448 vs. VOA-3723; VOA-4627 vs. VOA-4698). This increase in p-ERK1/2 was observed over time independently of patient treatment.
Figure 4.

MEKi on-target effects on ERK1/2 phosphorylation in LGSC cells. A: Baseline ERK and p-ERK1/2 by western blot is shown for each cell line. B: ERK and p-ERK1/2 levels in response to MEKi treatment. Tra = trametinib, Ref = refametinib, Sel = selumetinib, Bin = binimetinib. Films were intentionally overexposed to detect signal in lanes with low p-ERK1/2 expression. For each cell line all treatment conditions were performed as part of the same experiment. All experimental findings were confirmed in independent experiments performed in triplicate. Note: membranes incubated with p-ERK1/2 antibody were overexposed to capture all remaining signal after MEKi treatment. WB images have been cut to keep the same sample order. All membranes incubated with the same antibody have been exposed for the same period of time.
We then treated each cell line with the different MEKi (trametinib 0.1 μM, selumetinib 1 μM, binimetinib 1 μM, or refametinib 1 μM), and analyzed the levels of p-ERK1/2 expression at 24 hours. For most cell lines, 1 μM selumetinib or binimetinib only partially inhibited p-ERK1/2, there was greater inhibition using 1 μM refametinib, and complete inhibition with 0.1 μM trametinib (Figure 4B). EGF treatment was used to determine if each MEKi could still suppress phosphorylation after MAPK pathway stimulation. Only trametinib was able to completely block the induction of p-ERK1/2. To further characterize drug effect on p-ERK1/2 isoforms on native protein lysates, we selected 4 cell lines for further study using cIEF: the sensitive iOvCa241 cell line, and one cell line from each of the paired samples (VOA-1056, VOA 3723, and VOA-4627). The results from these experiments are shown in Supplementary Figure 3. LGSC cells treated with 0.1 μM trametinib showed little detectable levels of p-ERK1/2 across cell lines. Refametinib (1 μM) was the second most effective inhibitor of p-ERK1/2. Using cIEF we could not observe any drug-related differences in p-ERK1/2 in the treated sensitive cell line (iOvCa241) as treatment with any of the MEKi resulted in complete inhibition of p-ERK1/2 signal. Only partial inhibition of p-ERK1/2 was observed after treatment of the resistant cell lines using 1 μM selumetinib or binimetinib. As observed by WB, iOvCa241 cells only expressed ERK1, and VOA-1056 cells predominantly expressed ERK2.
MEK inhibitor anti-proliferative effects correlate with MEKi on-target efficacy over time
To further explore the inhibitory effects of MEKi treatment in LGSC cells, we performed dose-response experiments on the 4 selected cell lines. Each cell line was treated with low (0.1 or 1 μM) and high (0.5 or 5 μM) doses of each MEKi for periods of 24 and 72 hours. p-ERK1/2 expression levels were measured as a marker for MEK activity. In the sensitive iOvCa241, all drugs completely inhibited p-ERK1/2 at any dose or time (Figure 5A). In the other 3 cell lines (VOA-1056, VOA-3723, and VOA-4627) there were similarities in drug-dependent suppression of p-ERK1/2 levels. Trametinib completely blocked p-ERK1/2 even at low dose (0.1 μM) over 72 hours, however selumetinib or binimetinib at high dose (5 μM) failed to completely inhibit p-ERK1/2. In contrast, refametinib showed complete inhibition of p-ERK1/2 when used at 5 μM over 72 hours. In general, MEKi treatment resulted in an increase in MEK1/2 phosphorylation (p-MEK1/2). Interestingly, in the sensitive iOvCa241 line, no clear increase was observed after trametinib treatment.
Figure 5.
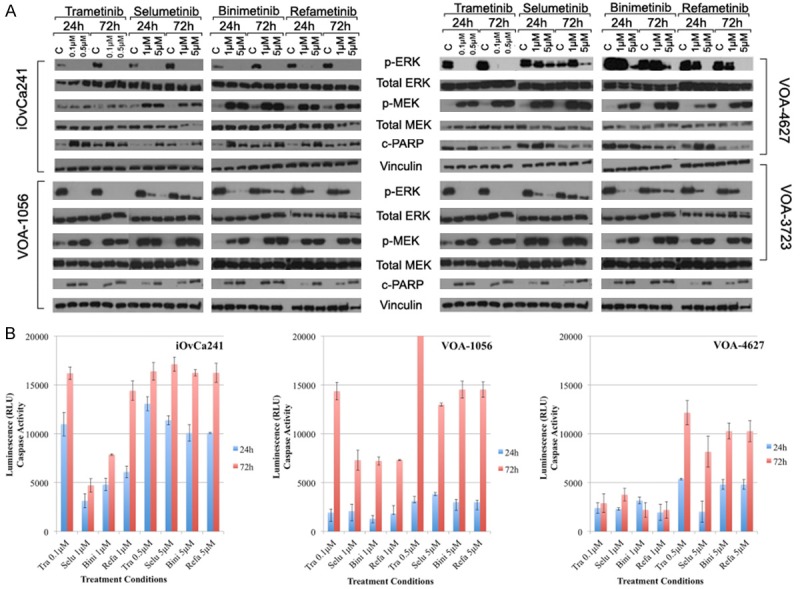
Time course and dose-response effects of MEKi treatment in LGSC cell lines. A: WB expression levels of ERK1/2, MEK and PARP after MEKi treatments with different dosing (low versus high) over time (24 h versus 72 h). B: Apoptosis induction measured by caspase 3/7 after MEKi treatment in LGSC cell lines. All experiments were repeated. Note: Caspase 3/7 control measurements at 24 h and 72 h were subtracted from all treatment values in the graphs shown.
We then examined the effects of high-dose MEKi treatment on cell proliferation and p-ERK1/2 expression. In iOvCa241, higher dosing (Figure 6) did not further reduce proliferation rates, as low doses completely inhibited proliferation (P>0.5; t-test, high vs. low dose of each drug; 120 h ours post-treatment). Notably, in the 3 resistant cell lines (VOA-1056, VOA-3723, and VOA-4627) there were drug-dependent differences in proliferation after higher drug dosing. For example, in the most resistant cell line, VOA-4627, each MEKi significantly decreased proliferation rates 120 hours post-treatment (P<0.05; t-test, high dose vs. low dose of each drug) (Supplementary Table 4). In contrast, for VOA-1056, statistically significant decreases in proliferation were observed at 120 hours post-treatment for all drugs (P<0.05; t-test) except trametinib (P = 0.09; t-test, high dose vs. low dose). In 3 more resistant cell lines, dose dependent reductions in p-ERK1/2 over time (Figure 5A) correlated with degree of anti-proliferative effect for all drugs except trametinib (Figure 6). Once again, trametinib and refametinib more effectively blocked p-ERK1/2 and show the greatest anti-proliferative activity across all tested LGSC cell lines.
Figure 6.
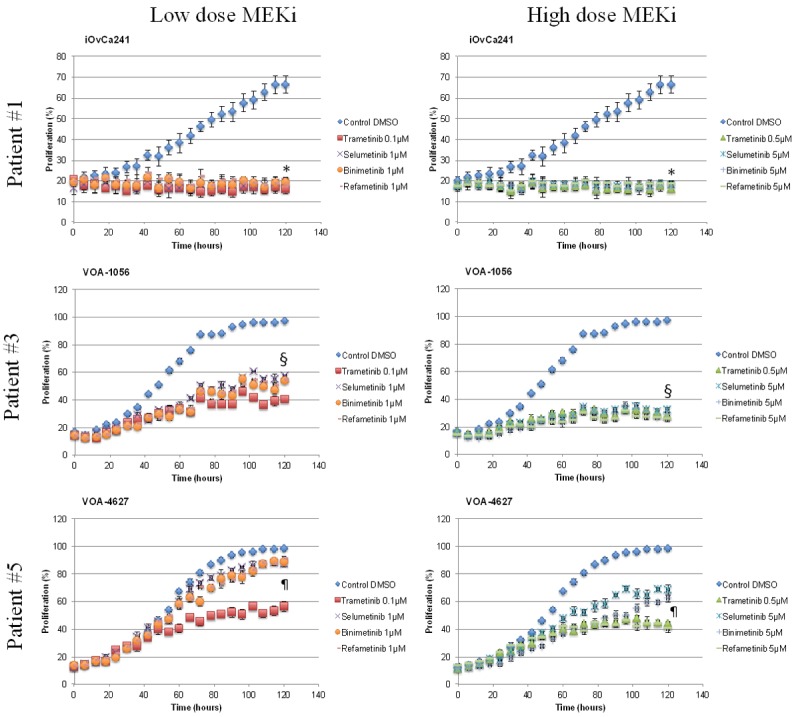
Anti-proliferative effects of MEKi treatment (low and high dose) in LGSC cells. Proliferation experiments showing the effects of low dose (1×) and high dose (5×) of MEKi treatment (trametinib, selumetinib, binimetinib, refametinib) in three different LGSC cell lines. (*) P-value >0.5 comparing high dose versus low dose for each drug in the iOvCa241 cell line. (§) P-value <0.05 comparing high dose versus low dose for each drug, except trametinib (p-value = 0.09), in the VOA-1056 cell line. (¶) P-value <0.05 comparing high dose versus low dose for each drug in the VOA-4627 cell line.
MEKi treatment induces apoptosis and the degree of apoptosis correlates with drug sensitivity
Using the same low and high doses of each MEKi, we then analyzed the levels of PARP activation (cleaved PARP or c-PARP) in four LGSC cell lines by WB (Figure 5A). Three cell lines (iOvCa241, VOA-1056 and VOA-3723) had little or no detectable baseline c-PARP activity, but considerable c-PARP induction after MEKi treatment. In contrast, VOA-4627 showed considerable baseline PARP activity without a corresponding increase after MEKi treatment. As another marker for apoptosis activation, we also measured the level of caspase 3/7 cleavage in 3 cell lines (iOVCa241, VOA-1056, and VOA-4627). Figure 5B shows the changes in cleaved caspase 3/7 (above substracted baseline control) pre-treatment and 24 and 72 hours post-treatment. All MEKi increased cleaved caspase levels at 24 and 72 hours. The MEKi sensitive iOvCa241 cell line showed the most robust apoptotic response to MEKi treatment and trametinib at low dose was as effective at inducing apoptosis as the high dose of any other MEKi (Figure 5B). The MEKi resistant VOA-4627 cell line showed no difference in caspase activity when treated with lower doses of MEKi over time (P = 0.08; t-test: 24 vs. 72 hours for all MEKi). In contrast, the less resistant VOA-1056 cells, showed significant increases in cleaved caspase after 72 h treatment (P<0.00001; t-test 24 vs. 72 hours, for all MEKi).
Discussion
We have established and molecularly characterized a cohort of advanced/recurrent LGSC cell lines. LGSC cells are difficult to culture and only a few LGSC cell lines (MPSC-1, HOC-7) are available and studied to date [24,34,35]. Clinical and molecular characterization (pathology at collection, tumor stage, treatment status, STR profiling, mutation and CNV analyses) of cell lines provides a basis for using these lines as experimental models. For example, previous tumor mutational studies have shown that BRAF mutations are found in LGSC, but are mostly confined to early-stage cases [30,36,37]. As such cases are often cured by surgery, the use of BRAF mutant LGSC cell lines for investigational drug studies will be of limited value. No BRAF mutations were identified in our cell line cohort in agreement with a previous study [38], though there has been one reported patient with a BRAF fusion who achieved a complete response to binimetinib in combination with paclitaxel [39]. KRAS/NRAS mutations are relatively frequent in advanced/recurrent LGSC [16], and in our study they were detected in 5 out of 10 cell lines from 4 different patients. Further study of LGSC cell lines with KRAS/NRAS mutations is very important to understand how these mutations affect pathway signalling and responses to MAPK pathway inhibition in LGSC. Successful targeted therapies against KRAS mutations have proven to be challenging [40,41] and it is not clear whether MEKi will effectively treat LGSC patients with KRAS mutations [23].
Cancer hotspot mutational analysis revealed that cell lines derived from tumor samples obtained from the same patient at different times during the course of the disease shared the same mutations (see mutations in cell line pairs VOA-1056 and VOA-3993; VOA-3348 and VOA-3723; and VOA-4627 and VOA-4698; Table 2). Copy number analysis showed marked heterogeneity among LGSC cell lines. Further genomic disruption was seen over time, or in response to treatment, in the paired cell lines that we analyzed, VOA-1056 and VOA-3993. Despite this heterogeneity, LGSC cell lines shared common regions of copy number change. Loss of chromosome 9p was detected in all cell lines, an event that has been observed in other recent genomic profiling studies done in LGSC tumors [42-44]. This genomic region is known to contain the CDKN2A/B locus, which encodes for three well-known tumor suppressor genes: p14 (Arf), p15 (INK4b), and p16 (INK4a). Loss of p16 has been described in LGSC tumors in comparison to their high-grade counterparts [2,45]. It is of interest that loss of chromosome 9 is frequently seen in LGSC but is rare in SBOT, suggesting a potential role for malignant transformation [42,43]. Other copy number changes were observed among cell lines included gains in chromosomes 8, 12 and 20. Gains in chromosome 12 have been described in both LGSC and SBOT [43,46]. Notably, KRAS is located on chromosome 8. More in-depth study of these regions may help further characterize LGSC in patients with relapsed or progressive disease.
We used our LGSC cell lines to test four MEKi and noted remarkable differences in the sensitivity of these lines to the individual drugs tested. We noted that the proliferation assays were helpful in terms of classifying sensitive versus resistant cells as the results from the IC50 experiments did not well characterized the efficacy of the different drugs over time. Based on patterns of growth inhibition and apoptosis, we identified two sensitive cell lines (VOA-1312 and iOvCa241). These two lines demonstrated marked inhibition of proliferation in response to all MEKi tested even at low doses. The iOvCa241, MEKi sensitive cells, also showed a marked apoptotic response after MEKi treatment when compared to two MEKi resistant cell lines (VOA-1056 and VOA-4627) (Figure 5). The remaining cell lines show varying degrees of resistance to MEKi with less anti-proliferative effects in response to drug. In most MEKi resistant cell lines, though a dose-response effect on proliferation was observed, proliferation continued despite higher doses of drug (See Figure 3). To confirm the significance of these findings it will be important to test additional LGSC cell lines derived from patients known to be sensitive or resistant to MEKi treatment.
Our evaluation of the biological effects of four different MEKi in LGSC cells showed that following a single treatment, trametinib is a more effective inhibitor of cell proliferation at 10-fold lower doses than the other drugs tested (refametinib, selumetinib, and binimetinib). Trametinib also showed the greatest reduction in cell proliferation and viability in the more resistant cell lines. These observations are similar to recent reports of MEKi testing in lung cancer cell lines and tumor models [32,47]. Trametinib most effectively induced apoptosis in our cell lines, which has been described as an important mechanism of MEKi action [31].
The study of on-target effects of each MEKi in our LGSC cell lines revealed interesting differences relating to cellular p-ERK1/2 at baseline and in response to drug treatment. In our study, there was no correlation between baseline p-ERK levels and MEKi sensitivity in keeping with a previous report [33]. We did observe a remarkable correlation between the degree of inhibition of p-ERK1/2 levels and the anti-proliferative effects of each MEKi. Interestingly, we observed that binimetinib, as well as selumetinib, was much less effective at inhibiting p-ERK1/2 and cell proliferation in LGSC cell lines than trametinib or refametinib. Dose dependent inhibition of p-ERK1/2 was observed in our study for all drugs except trametinib, which effectively blocked p-ERK1/2 even at low doses (0.1 μM). This finding is in keeping with a study on breast cancer cell lines showing no correlation between trametinib IC50 values and inhibition of ERK signaling as p-ERK1/2 is inhibited at trametinib concentrations not affecting proliferation [33]. Our results therefore suggest that using a particular MEKi dosing schedule or drug that fails to eliminate p-ERK1/2 will not result in maximal anti-proliferative effects in tumors from LGSC patients. On the other hand, cells with intrinsic resistance to MEKi continue to proliferate despite marked inhibition of p-ERK1/2. This observation was most apparent using trametinib as it so effectively inhibits ERK phosphorylation at low concentrations. Recently, an investigational drug trial using binimetinib in LGSC was closed following an interim analysis that failed to show a pre-defined benefit on progression-free survival compared to chemotherapy. A large randomized trial is currently open (NCT02101788) to evaluate trametinib as a treatment for LGSC and this will be a very informative trial relating to the use of MEKi in this disease.
In 2012, an important phase 2 clinical trial was reported evaluating selumetinib as a treatment for recurrent LGSC. There was a 15% response rate and KRAS/BRAF mutations were not a predictor of response. Copy number changes or other MAPK-pathway related aberations were not evaluated. As clinical responses to selumetinib were infrequent, it will be important to identify biomarkers of MEKi response in order to treat those patients who are most likely to benefit. In the present study, the most sensitive cell lines (VOA-1312 and iOvCa241) were found to have mutations in KRAS (G12V and G12D respectively) with associated gene copy-number gains. This finding suggests that these lines may be highly dependent on KRAS activity. Polh et al. found that primary ovarian cancer cell lines and cultures with BRAF (4 cell lines) and KRAS mutations (2 cell lines) were more sensitive to MEKi (CI-1040) than 6 wild type lines [24]. In a study of colon cancer cell lines, MEKi treatment impaired anchorage-independent growth of nearly all KRAS/BRAF mutants, but not wild type cells. However, neither increased baseline p-ERK1/2 nor a reduction of p-ERK1/2 secondary to MEKi treatment correlated with inhibition of anchorage independent growth [48]. Recently, it has been suggested that KRAS mutations may in fact result in a hyper-excitable KRAS rather than an altered protein in a statically active state [41]. MAPK signaling is very complex as there are copy number changes, other pathway mutations, and cross talk mechansims all of which can affect MAPK signaling and the response to MEKi drug treatment.
This study shows marked differences in LGSC cell line MEKi efficacy. We observed that trametinib is highly effective in blocking p-ERK1/2 at concentrations ten-fold lower than the other MEKi, and this on-target effect is reflected in its in vitro drug efficacy. As effective systemic treatments are lacking for this disease and no xenograft models for pre-clinical testing are available, these cell lines should be used to better understand drug- or tumor-related determinants of MEKi sensitivity. There is a need for this work as investigational MEKi (trametinib and pimasertib) are currently being tested in large randomized trials designed for drug registration and approval. These trials are proceeding with a very limited understanding of MAPK pathway function in LGSC. With recent advances in molecular medicine, pre-clinical investigational drug testing can play an important role in the design of clinical trials, molecular companion studies, and the selection of the most promising investigational drugs for clinical testing. Linking molecular research strategies with drug development and clinical trial design will be necessary in order to improve treatment outcomes in rare cancer types such as LGSC.
Acknowledgements
This work was supported by the British Columbia Cancer Foundation, the OvCaRe Program, and the London Run for Ovarian Cancer. We would also like to thank Janine Senz for all of her technical support and Megan Froats for her graph design expertise. The authors extend a special thanks to the MacKenzie family, and to all the patients, families, and donors for who have supported this research study.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Okoye E, Euscher ED, Malpica A. Ovarian Low-grade Serous Carcinoma: A Clinicopathologic Study of 33 Cases With Primary Surgery Performed at a Single Institution. Am J Surg Pathol. 2016;40:627–35. doi: 10.1097/PAS.0000000000000615. [DOI] [PubMed] [Google Scholar]
- 2.Ali RH, Kalloger SE, Santos JL, Swenerton KD, Gilks CB. Stage II to IV low-grade serous carcinoma of the ovary is associated with a poor prognosis: a clinicopathologic study of 32 patients from a population-based tumor registry. Int J Gynecol Pathol. 2013;32:529–535. doi: 10.1097/PGP.0b013e31827630eb. [DOI] [PubMed] [Google Scholar]
- 3.Chen M, Jin Y, Bi Y, Yin J, Wang Y, Pan L. A survival analysis comparing women with ovarian low-grade serous carcinoma to those with high-grade histology. Onco Targets Ther. 2014;7:1891–1899. doi: 10.2147/OTT.S67812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plaxe SC. Epidemiology of low-grade serous ovarian cancer. Am J Obstet Gynecol. 2008;198:459, e451–458. doi: 10.1016/j.ajog.2008.01.035. discussion 459, e458-459. [DOI] [PubMed] [Google Scholar]
- 5.Schmeler KM, Gershenson DM. Low-grade serous ovarian cancer: a unique disease. Curr Oncol Rep. 2008;10:519–523. doi: 10.1007/s11912-008-0078-8. [DOI] [PubMed] [Google Scholar]
- 6.Anglesio MS, Wiegand KC, Melnyk N, Chow C, Salamanca C, Prentice LM, Senz J, Yang W, Spillman MA, Cochrane DR, Shumansky K, Shah SP, Kalloger SE, Huntsman DG. Type-specific cell line models for type-specific ovarian cancer research. PLoS One. 2013;8:e72162. doi: 10.1371/journal.pone.0072162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmeler KM, Sun CC, Bodurka DC, Deavers MT, Malpica A, Coleman RL, Ramirez PT, Gershenson DM. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol Oncol. 2008;108:510–514. doi: 10.1016/j.ygyno.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Ansquer Y, Leblanc E, Clough K, Morice P, Dauplat J, Mathevet P, Lhomme C, Scherer C, Tigaud JD, Benchaib M, Fourme E, Castaigne D, Querleu D, Dargent D. Neoadjuvant chemotherapy for unresectable ovarian carcinoma: a French multicenter study. Cancer. 2001;91:2329–2334. [PubMed] [Google Scholar]
- 9.Gourley C, Farley J, Provencher DM, Pignata S, Mileshkin L, Harter P, Maenpaa J, Kim JW, Pujaide-Lauraine E, Glasspool RM, Ray-Coquard I, Gershenson D. Gynecologic Cancer InterGroup (GCIG) consensus review for ovarian and primary peritoneal low-grade serous carcinomas. Int J Gynecol Cancer. 2014;24:S9–13. doi: 10.1097/IGC.0000000000000257. [DOI] [PubMed] [Google Scholar]
- 10.Grabowski JP, Harter P, Heitz F, Pujade-Lauraine E, Reuss A, Kristensen G, Ray-Coquard I, Heitz J, Traut A, Pfisterer J, du Bois A. Operability and chemotherapy responsiveness in advanced low-grade serous ovarian cancer. An analysis of the AGO Study Group metadatabase. Gynecol Oncol. 2016;140:457–462. doi: 10.1016/j.ygyno.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Gershenson DM, Sun CC, Iyer RB, Malpica AL, Kavanagh JJ, Bodurka DC, Schmeler K, Deavers M. Hormonal therapy for recurrent low-grade serous carcinoma of the ovary or peritoneum. Gynecol Oncol. 2012;125:661–666. doi: 10.1016/j.ygyno.2012.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gershenson DM, Sun CC, Bodurka D, Coleman RL, Lu KH, Sood AK, Deavers M, Malpica AL, Kavanagh JJ. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant. Gynecol Oncol. 2009;114:48–52. doi: 10.1016/j.ygyno.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Harter P, Gershenson D, Lhomme C, Lecuru F, Ledermann J, Provencher DM, Mezzanzanica D, Quinn M, Maenpaa J, Kim JW, Mahner S, Hilpert F, Baumann K, Pfisterer J, du Bois A. Gynecologic Cancer InterGroup (GCIG) consensus review for ovarian tumors of low malignant potential (borderline ovarian tumors) Int J Gynecol Cancer. 2014;24:S5–8. doi: 10.1097/IGC.0000000000000282. [DOI] [PubMed] [Google Scholar]
- 14.Silva EG, Gershenson DM, Malpica A, Deavers M. The recurrence and the overall survival rates of ovarian serous borderline neoplasms with noninvasive implants is time dependent. Am J Surg Pathol. 2006;30:1367–1371. doi: 10.1097/01.pas.0000213294.81154.95. [DOI] [PubMed] [Google Scholar]
- 15.Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum Pathol. 2011;42:918–931. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Della Pepa C, Tonini G, Santini D, Losito S, Pisano C, Di Napoli M, Cecere SC, Gargiulo P, Pignata S. Low Grade Serous Ovarian Carcinoma: from the molecular characterization to the best therapeutic strategy. Cancer Treat Rev. 2015;41:136–143. doi: 10.1016/j.ctrv.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Jones S, Wang TL, Kurman RJ, Nakayama K, Velculescu VE, Vogelstein B, Kinzler KW, Papadopoulos N, Shih Ie M. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol. 2012;226:413–420. doi: 10.1002/path.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart GC, Kitchener H, Bacon M, duBois A, Friedlander M, Ledermann J, Marth C, Thigpen T, Trimble E participants of 4th Ovarian Cancer Consensus Conference (OCCC); Gynecologic Cancer Intergroup. 2010 Gynecologic Cancer InterGroup (GCIG) consensus statement on clinical trials in ovarian cancer: report from the Fourth Ovarian Cancer Consensus Conference. Int J Gynecol Cancer. 2011;21:750–755. doi: 10.1097/IGC.0b013e31821b2568. [DOI] [PubMed] [Google Scholar]
- 19.Nakayama N, Nakayama K, Yeasmin S, Ishibashi M, Katagiri A, Iida K, Fukumoto M, Miyazaki K. KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br J Cancer. 2008;99:2020–2028. doi: 10.1038/sj.bjc.6604783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 22.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 23.Farley J, Brady WE, Vathipadiekal V, Lankes HA, Coleman R, Morgan MA, Mannel R, Yamada SD, Mutch D, Rodgers WH, Birrer M, Gershenson DM. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14:134–140. doi: 10.1016/S1470-2045(12)70572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pohl G, Ho CL, Kurman RJ, Bristow R, Wang TL, Shih Ie M. Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Res. 2005;65:1994–2000. doi: 10.1158/0008-5472.CAN-04-3625. [DOI] [PubMed] [Google Scholar]
- 25.Tone AA, McConechy MK, Yang W, Ding J, Yip S, Kong E, Wong KK, Gershenson DM, Mackay H, Shah S, Gilks B, Tinker AV, Clarke B, McAlpine JN, Huntsman D. Intratumoral heterogeneity in a minority of ovarian low-grade serous carcinomas. BMC Cancer. 2014;14:982. doi: 10.1186/1471-2407-14-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McConechy MK, Anglesio MS, Kalloger SE, Yang W, Senz J, Chow C, Heravi-Moussavi A, Morin GB, Mes-Masson AM Australian Ovarian Cancer Study Group. Carey MS, McAlpine JN, Kwon JS, Prentice LM, Boyd N, Shah SP, Gilks CB, Huntsman DG. Subtype-specific mutation of PPP2R1A in endometrial and ovarian carcinomas. J Pathol. 2011;223:567–573. doi: 10.1002/path.2848. [DOI] [PubMed] [Google Scholar]
- 27.Wiegand KC, Hennessy BT, Leung S, Wang Y, Ju Z, McGahren M, Kalloger SE, Finlayson S, Stemke-Hale K, Lu Y, Zhang F, Anglesio MS, Gilks B, Mills GB, Huntsman DG, Carey MS. A functional proteogenomic analysis of endometrioid and clear cell carcinomas using reverse phase protein array and mutation analysis: protein expression is histotype-specific and loss of ARID1A/BAF250a is associated with AKT phosphorylation. BMC Cancer. 2014;14:120. doi: 10.1186/1471-2407-14-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson CL, Wang Y, Rustandi RR. Applications of imaged capillary isoelectric focussing technique in development of biopharmaceutical glycoprotein-based products. Electrophoresis. 2012;33:1538–1544. doi: 10.1002/elps.201100611. [DOI] [PubMed] [Google Scholar]
- 29.Chay WY, Horlings HM, Tinker AV, Gelmon KA, Gilks CB. Low grade serious carcinoma of the peritoneum in a BRCA1 carrier previously diagnosed with a “low-grade serous tubal intra-epithelial carcinoma” (STIC) on risk reducing surgery. Gynecol Oncol Rep. 2015;12:72–74. doi: 10.1016/j.gore.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emmanuel C, Chiew YE, George J, Etemadmoghadam D, Anglesio MS, Sharma R, Russell P, Kennedy C, Fereday S, Hung J, Galletta L, Hogg R, Wain GV, Brand A, Balleine R, MacConaill L, Palescandolo E, Hunter SM, Campbell I, Dobrovic A, Wong SQ, Do H, Clarke CL, Harnett PR, Bowtell DD, deFazio A Australian Ovarian Cancer Study (AOCS) Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin Cancer Res. 2014;20:6618–6630. doi: 10.1158/1078-0432.CCR-14-1292. [DOI] [PubMed] [Google Scholar]
- 31.Baranski Z, Booij TH, Kuijjer ML, de Jong Y, Cleton-Jansen AM, Price LS, van de Water B, Bovee JV, Hogendoorn PC, Danen EH. MEK inhibition induces apoptosis in osteosarcoma cells with constitutive ERK1/2 phosphorylation. Genes Cancer. 2015;6:503–512. doi: 10.18632/genesandcancer.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gannon HS Jr, Kaplan N, Tsherniak A, Vazquez F, Weir BA, Hahn WC, Meyerson M. Identification of an “Exceptional Responder” Cell Line to MEK1 Inhibition: Clinical Implications for MEK-targeted Therapy. Mol Cancer Res. 2016;14:207–15. doi: 10.1158/1541-7786.MCR-15-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung EY, Kim JE, Askarian-Amiri M, Rewcastle GW, Finlay GJ, Baguley BC. Relationships between signaling pathway usage and sensitivity to a pathway inhibitor: examination of trametinib responses in cultured breast cancer lines. PLoS One. 2014;9:e105792. doi: 10.1371/journal.pone.0105792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, van IJcken WF, Heine AA, Smid M, Koudijs MJ, Brenton JD, Berns EM, Helleman J. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS One. 2014;9:e103988. doi: 10.1371/journal.pone.0103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buick RN, Pullano R, Trent JM. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res. 1985;45:3668–3676. [PubMed] [Google Scholar]
- 36.Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, Shih Ie M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 37.Wong KK, Tsang YT, Deavers MT, Mok SC, Zu Z, Sun C, Malpica A, Wolf JK, Lu KH, Gershenson DM. BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. Am J Pathol. 2010;177:1611–1617. doi: 10.2353/ajpath.2010.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang YT, Deavers MT, Sun CC, Kwan SY, Kuo E, Malpica A, Mok SC, Gershenson DM, Wong KK. KRAS (but not BRAF) mutations in ovarian serous borderline tumour are associated with recurrent low-grade serous carcinoma. J Pathol. 2013;231:449–456. doi: 10.1002/path.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grisham RN, Sylvester BE, Won H, McDermott G, DeLair D, Ramirez R, Yao Z, Shen R, Dao F, Bogomolniy F, Makker V, Sala E, Soumerai TE, Hyman DM, Socci ND, Viale A, Gershenson DM, Farley J, Levine DA, Rosen N, Berger MF, Spriggs DR, Aghajanian CA, Solit DB, Iyer G. Extreme Outlier Analysis Identifies Occult Mitogen-Activated Protein Kinase Pathway Mutations in Patients With Low-Grade Serous Ovarian Cancer. J. Clin. Oncol. 2015;33:4099–4105. doi: 10.1200/JCO.2015.62.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316–29. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 42.Kuo KT, Guan B, Feng Y, Mao TL, Chen X, Jinawath N, Wang Y, Kurman RJ, Shih Ie M, Wang TL. Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 2009;69:4036–4042. doi: 10.1158/0008-5472.CAN-08-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew YE, Rowley SM, Doyle MA, Li J, Gilks CB, Moss P, Allan PE, Stephens AN, Huntsman DG, deFazio A, Bowtell DD Australian Ovarian Cancer Study Group. Gorringe KL, Campbell IG. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6:37663–77. doi: 10.18632/oncotarget.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, Krischek B, Ozduman K, Omay SB, Sorensen EA, Turcan S, Bakirciglu M, Carrion-Grant G, Murray PB, Clark VE, Ercan-Sencicek AG, Knight J, Sencar L, Altinok S, Kaulen LD, Gulez B, Timmer M, Schramm J, Mishra-Gorur K, Henegariu O, Moliterno J, Louvi A, Chan TA, Tannheimer SL, Pamir MN, Vortmeyer AO, Bilguvar K, Yasuno K, Gunel M. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48:59–66. doi: 10.1038/ng.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Neill CJ, McBride HA, Connolly LE, Deavers MT, Malpica A, McCluggage WG. High-grade ovarian serous carcinoma exhibits significantly higher p16 expression than low-grade serous carcinoma and serous borderline tumour. Histopathology. 2007;50:773–779. doi: 10.1111/j.1365-2559.2007.02682.x. [DOI] [PubMed] [Google Scholar]
- 46.Woo MM, Salamanca CM, Miller M, Symowicz J, Leung PC, Oliveira C, Ehlen TG, Gilks CB, Huntsman D, Auersperg N. Serous borderline ovarian tumors in long-term culture: phenotypic and genotypic distinction from invasive ovarian carcinomas. Int J Gynecol Cancer. 2008;18:1234–1247. doi: 10.1111/j.1525-1438.2007.01171.x. [DOI] [PubMed] [Google Scholar]
- 47.Mas C, Boda B, CaulFuty M, Huang S, Wiszniewski L, Constant S. Antitumour efficacy of the selumetinib and trametinib MEK inhibitors in a combined human airway-tumour-stroma lung cancer model. J Biotechnol. 2015;205:111–119. doi: 10.1016/j.jbiotec.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009;8:834–843. doi: 10.1158/1535-7163.MCT-08-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.