

Abstract
Lateral flow or dipstick assays (e.g., home pregnancy tests), where an analyte solution is drawn through a porous membrane and is detected by localization onto a capture probe residing at a specific site on the flow strip, are the most commonly and extensively used type of diagnostic assay. However, after over 30 years of use, these assays are constrained to measuring one or a few analytes at a time. Here, we describe a completely general method, in which any single-plex lateral flow assay is transformed into a multiplex assay capable of measuring an arbitrarily large number of analytes simultaneously. Instead of identifying the analyte by its localization onto a specific geometric location in the flow medium, the analyte-specific capture probe is identified by its association with a specific optically encoded region within the flow medium. The capture probes for nucleic acids, antigens, or antibodies are attached to highly porous agarose beads, which have been encoded using multiple lanthanide emitters to create a unique optical signature for each capture probe. The optically encoded capture probe-derivatized beads are placed in contact with the analyte-containing porous flow medium and the analytes are captured onto the encoded regions as the solution flows through the porous medium. To perform a multiplex diagnostic assay, a solution comprising multiple analytes is passed through the flow medium containing the capture probe-derivatized beads, and the captured analyte is treated with a suitable fluorescent reporter. We demonstrate this multiplex analysis technique by simultaneously measuring DNA samples, antigen–antibody pairs, and mixtures of multiple nucleic acids and antibodies.
Introduction
Lateral flow assays (LFAs), such as home pregnancy or drug tests, comprise the most widespread diagnostic assay format and, because of low cost, portability, no reagent handling and use by untrained personnel, have been called the most successful microfluidic application.1 Many lateral flow assays are sandwich immunoassays2 with an antigen binding to detection antibodies conjugated to nanoparticles such as gold,3 which subsequently binds through a second antigenic epitope to capture antibodies at a specific location on the flow strip,4 indicating a positive test. It would be highly desirable to perform lateral flow assays in a multiplexed manner so that many diagnostic targets (e.g., proteins, antibodies, and nucleic acids) could be measured simultaneously, but difficulties in manipulating and detecting large numbers of different nanoparticles have prevented deep multiplexing. We report here multiplex flow assays (MFAs) that can measure arbitrarily large numbers of analytes in a flow format using only molecular species without nanoparticle reporters. Porous agarose beads, which are optically encoded with multiple lanthanide emitters and derivatized with capture probes for the analyte targets, contact the analyte-containing flow medium whereupon each capture probe binds its target. After reporter-staining the captured target, the lanthanide code and reporter intensity are measured for each bead giving the amount of each analyte. We demonstrate this multiplexing by measuring antibodies (against human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), influenza A (Inf A), and troponin I), multiple DNA sequences, or mixtures of antibodies and DNA in one sample. Using passive (no active fluid pumping) lateral flow, flow through, or capillary channel flow, the samples are analyzed with a portable (<500 g) instrument. With the analysis time, manufacturability, and required reagents similar to those of a single-plex assay, MFAs will increase the number of disease analytes screened, their screening rate, and diagnostic access for resource-challenged environments. MFAs represent the first steps toward self-screening for any disease.
Lateral flow and flow-through assays are the most common type of diagnostic assay performed outside of a clinical setting, and more than 200 companies are involved in the U.S. $ > 2 billion market.5 Most current LFAs, which are relatively unchanged since their introduction 30 years ago, share the common characteristics of (a) detecting an antigen (Ag)–antibody (Ab) interaction, (b) passive flow (no active fluid pumping) of an analyte-containing fluid by capillary action, (c) use of nanoparticles as reporters, and (d) localization of the nanoparticles onto a specific geometric location to indicate a positive test (Figure 1a). LFAs exploiting Ag–Ab interactions for diagnostic purposes had their beginnings in early agglutination studies,6−8 radioimmunoassays in 1960,9 the immobilization of the Ag–Ab moieties onto a solid support.10The first example of ELISA assays11 combined with capillary flow12−14 gave lateral flow devices similar to those widely used today. The advantages of the LFA, including passive flow-based fluidics, minimal reagent handling, small portable format, ease of use by clinically inexperienced practitioners, and low manufacturing cost, would be considerably expanded if LFAs could be used in a multiplexed fashion to measure large numbers of samples simultaneously in a single assay (i.e., MFA).
Figure 1.

Comparison between (a) traditional single-plex lateral flow assay, where the analyte is identified by localization onto a specific location (the capture line), and the new (b, c) MFAs where an analyte is identified via its optical code rather than onto a specific geometric location. A traditional lateral flow assay has a detection Ab attached to an Au nanoparticle and a capture Ab bound to the test line at a specific location (a1). The antigen within the analyte solution moves through the flow medium (purple arrows), bonds to the capture Ab (a2), and the tripartite antigen/capture Ab/Au nanoparticle moiety localizes onto a second Ab at the test line location (a3), indicating a positive antigen test. In contrast, the MFA assay for antigens (b) or antibodies (c) instead of nanoparticles uses a multitude of lanthanide-encoded beads contacting the flow medium to identify the captured analyte, and only molecular species move within the flow. To identify Ags using sandwich ELISA in a multiplexed fashion (b), the beads are derivatized with capture probe Abs, with each capture probe bound to a bead set with a unique optical code (b4), exposed to the Ag analyte solution, treated with the detection Ab (b5), and stained with antispecies stain or streptavidin–phycoerythrin (SAPE) to measure the amount of Ag (b6). To identify Abs instead of Ags in a multiplexed assay (c), a recombinant protein, which acts as an Ag against the Ab of interest, is attached to the lanthanide-encoded bead set (c7), exposed to the Ab analyte solution (c8), and labeled with an antispecies stain against the captured Ab (c9) to give the amount of analyte.
However, the preparation and manipulation of many different nanoparticle reporters, lack of covalent attachment methods for bonding capture probes to nanoparticles such as Au,3 use of unnatural substrates such as nitrocellulose that display irreversible protein binding, and the need to identify the target by localization onto a specific geometric location have precluded development of deeply multiplexed lateral flow assays with only two to three analytes reported in most studies to date.
All known LFA examples identify the analyte by its localization onto a specific location (capture line) on the flow strip.15−18
Results
We report here a new type of MFA, which (a) can multiplex and analyze arbitrarily large combinations of protein, antibody, and DNA analytes, (b) does not employ any nanoparticles but uses only soluble molecular reagents (Figure 1b,c), (c) contains >50 individual beads/mm2 (see number of beads/mm2 in Figure 2a or mask holes/mm2 in Figure 2f), (d) does not require localization of a reporter onto a line for analysis and may be randomly located within the flow medium, (e) uses familiar, low-binding and porous agarose as the capture probe substrate, (f) is inexpensively fabricated from polymer, paper, and tape components (Figure 2), and (g) may be rapidly analyzed with an inexpensive, portable, and wireless instrument.
Figure 2.
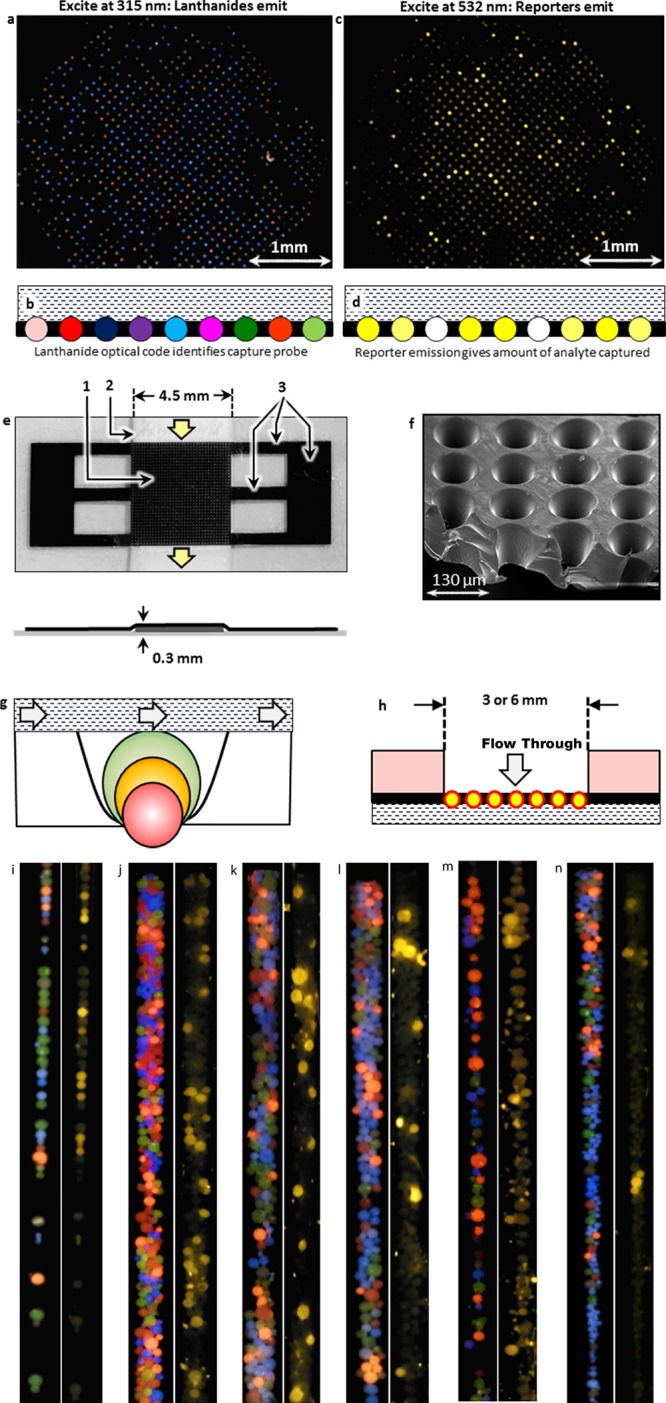
MFA uses ratiometrically encoded lanthanide emitters to optically encode and identify the capture probe-derivatized beads using lateral flow (a–g), flow-through (h), or capillary channel flow (i–n) geometries. Each capture probe is attached to a different lanthanide bead code (a) and the beads are in contact with the analyte-containing solution within the flow medium (b), whereupon the target analyte binds to the bead-bound capture probe. Only beads protruding through the well bottoms (g) are imaged (a, c). After the lanthanide optical code is read (a, b), the captured target is stained (c, d) with a labeled reporter (Alexafluor 555), and the reporter signal intensity provides the amount of analyte captured. The laser-machined, opaque polymer mask (e; where 1 = 37 × 37 grid, 2 = paper flow medium, and 3 = tethers to attach grid to flow medium) holds the beads in contact with the analyte solution within the paper flow medium (e) or the flow-through device shown in (h). The wells (f) are micromachined into a cup shape to accommodate beads of any size (g). A 6-plex MFA may be performed using a capillary tube geometry (110 μm i.d. borosilicate tube) where the analyte-containing solution flows over the capture probe-derivatized, encoded beads within the tube or channel. In panels (i)–(n), where the left figure in each of the six panels (i–n) is the lanthanide image taken under 320 nm excitation and the right image is the reporter image under 532 nm excitation, six different DNA capture probes are present on six different bead codes in all reactions. The 10 nM dye-labeled targets are added one or two at a time according to the protocols given in the Methods and Experimental Procedures section and the hybridization results showed selectivities comparable to the lateral flow DNA hybridization assays in Figure 7a–f.
The MFA measures analytes by (a) covalently attaching the capture probe to porous lanthanide-encoded beads (Figure 1b,c), (b) placing the beads into one of the MFA devices (Figure 2e–n), (c) passing the analyte-containing target solution through the flow medium and encoded regions, (d) staining the beads with a reporter if the target is unstained, and (e) imaging the beads to determine both the capture probe-identifying optical code (Figure 2a,b) and the amount of target analyte captured on each bead code (Figure 2c,d).
The most important enabling component of the MFA technology is the capture probe-derivatized regions (Figure 1b,c), each of which has been uniquely ratiometrically optically encoded with multiple lanthanide emitters19−22 (the so-called Parallume lanthanide optical encoding technology). The deep optical multiplexing is enabled by unique lanthanide optical properties, which include (a) very narrow (2–10 nm), nonoverlapping emission peaks (Figure 3), which allow thousands of optical codes to be resolved,19,20 (b) no photobleaching, (c) no overlap among the excitation spectra of lanthanides, proteins/DNA, and any organic reporter dye (Figures 2a,c and 3 inset), (d) very bright and stable phosphor type. Therefore, each encoded capture probe (Figures 1 and 2a) is associated with a unique Parallume optical code (Figure 4) emission (Figures 2a and 3), and (e) unlike organic dyes, all lanthanide colors are excited by a single excitation source. With >300 ratiometric lanthanide optical codes resolved using two colors,19 ∼1012 codes are statistically resolvable for six lanthanide emitter colors. It would be difficult to encode beads similarly with organic dyes instead of lanthanides because the extensive spectral overlap of the broad (compared to lanthanides) organic emission peaks (full-width at half-maximum ≈ 30–50 nm) (Figure 3) precludes resolving numerous optical codes, the organic dyes are unstable toward code-changing photobleaching, and the emission of the organic reporter dyes overlaps with that of the organic encoding dyes, concomitantly decreasing code resolution. Previous reports of using lanthanide materials in lateral flow assays used only lanthanide nanoparticles as reporters (analogous to Au nanoparticles) localized onto a line.23−25
Figure 3.

Plot of emission intensity vs wavelength for potential encoding fluorophores shows that lanthanide emission peaks (purple line) are much narrower than emission peaks from organic dyes or quantum dots. These narrow lanthanide emission peaks, which display less spectral overlap than any other fluorophore, allow their relative emission intensities to be measured more accurately thereby giving rise to more resolvable and numerous optical codes. Unlike the organic dyes and quantum dots, which are susceptible to code-altering photo-oxidation, the lanthanide material cannot be photochemically altered. The inset shows that nucleic acids and proteins are not excited at the lanthanide excitation wavelength used and any autofluorescence is eliminated. The 315 nm excitation also does not overlap with the absorption of the visible-light-excited, colored reporter dyes and, conversely, the colorless lanthanide materials are not excited by visible light eliminating any optical crosstalk between encoding and reporter fluorophores.
Figure 4.
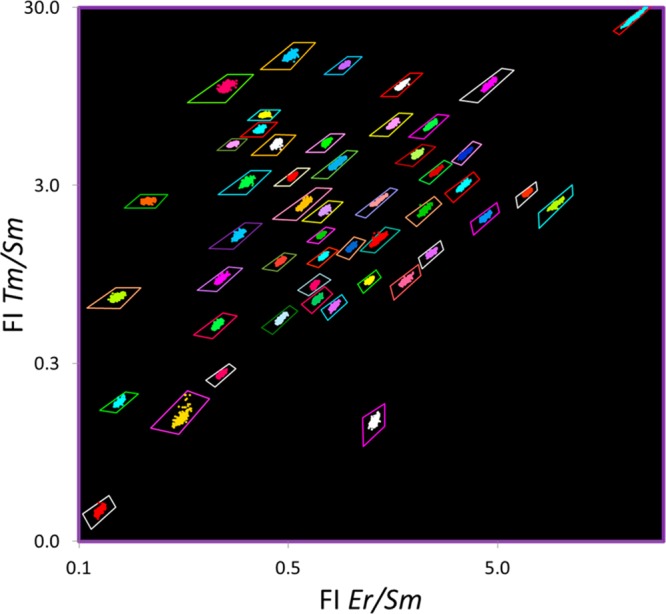
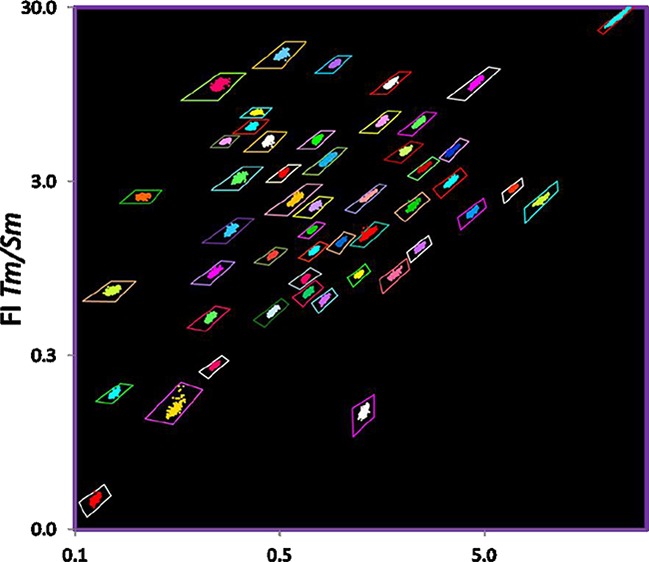
Lanthanide-encoded beads used for the MFA, where the capture probe is identified by the lanthanide-derived optical code of the bead to which it is attached, are optically encoded ratiometrically with multiple lanthanide emitters (in this case Er, Sm, and Tm) creating “optical bins” into which the beads are sorted for identification. The individual beads are shown enclosed by a tetragon representing the optical bin. As the beads are identified from the ratio of emission intensity of multiple lanthanide emitters, and not the absolute intensity of the emission, the measured ratios are independent of the bead size, excitation source brightness, angle of illumination, sample–detector distance, detector efficiency, etc.; thus, many bead sizes may be used (Figure 2g). The optical bins and bead data points are falsely colored for clarity as most encoded beads with three or more lanthanide emitters appear whitish to the eye.
The ∼40–80 μm Parallume-encoded (Figure 4) agarose beads (Figures 1b,c and 2a) are synthesized at a rate of millions of beads per minute by spraying26 and are (a) carboxylated to allow covalent attachment of the capture probe and (b) crosslinked to allow polymerase chain reaction (PCR) thermal cycling without decomposition. Multiplex titrations investigating bead pore size versus target size show that proteins and antibodies diffuse rapidly (Figure 5) into the beads, and species up to 1.4 MDa are accommodated within the bead pores (Figures 5 and 6).
Figure 5.

Target species diffuse rapidly into the capture probe-derivatized, Parallume-encoded beads without agitation. Two bead types, which contain DNA capture probes homologous (red) or nonhomologous (green) toward a 60-nt dye-labeled target (a), are placed into a flow-through device (Figure 2h), imaged at 315 nm (a), and subsequently treated with an excess of 10 nM dye-labeled target without agitation. Images in panels (b) and (c) show the reporter signal of the same beads imaged with 532 nm excitation at ∼1 min (b) and 60 min (c) after target addition. The chart (bottom) shows the time course of the static target diffusion into and captured by the beads with the homologous capture probe.
Figure 6.
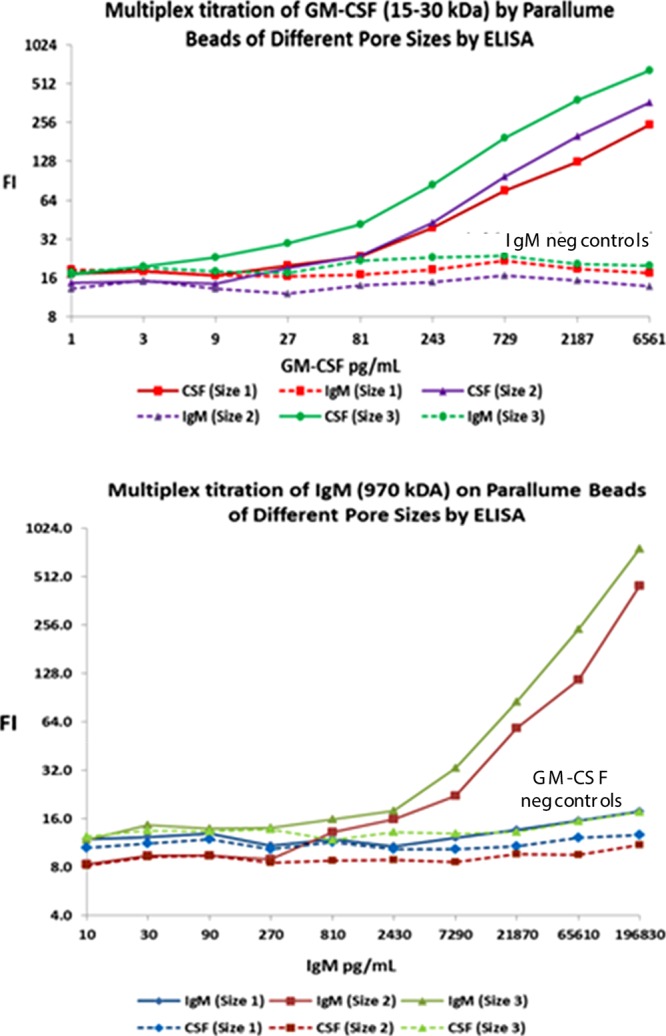
Six-plex ELISA titrations as a function of target size and percentage agarose in the beads show the effect of agarose concentration on bead pore size (pore size 1 = 4 wt % agarose, pore size 2 = 2 wt % agarose and pore size 3 = 1 wt % agarose; lower agarose percentage gives larger average pore size). An IgG Ab against GM-CSF Ag is attached to each of the three different pore size beads and an IgG Ab against IgM Ag is likewise attached to each of the three different pore size beads. The six pore size–capture probe combinations, each of which has its own unique bead optical code, are pooled together and titrated with increasing concentrations of either 15–30 kDa GM-CSF antigen (top) or ∼970 kDa IgM (bottom). The beads are treated with a biotinylated monoclonal Ab against the target species and then stained with SAPE. The top plot shows that none of the GM-CSF titrant is captured by the three codes with the anti-IgM capture probes, but the GM-CSF is taken up by the anti-GM-CSF capture Ab bead codes with larger pores capturing more protein target. The bottom plot shows that none of the IgM titrant reacts with either the bead codes with the anti-GM-CSF capture Ab or the small-pore 4 wt % agarose beads with anti-IgM capture Abs, but the large IgM target is taken up by the larger pore (1 and 2% agarose) beads.
These beads may be used in lateral flow (Figure 2a–d), flow-through (Figure 2h), or capillary channel flow (Figure 2i–n) configurations where the flowing analyte solutions contact the encoded target-sensing regions. In the lateral flow geometry, the beads are held in contact with the analyte solution by a laser-drilled, light-opaque polymer mask (Figure 2e). The opaque mask, which immobilizes the beads for imaging while allowing facile reagent flow, optically isolates the beads (Figure 2a–d). The mask eliminates bead-to-bead crosstalk from the lanthanide or reporter emission, allowing a very high sample density. The assembled device consists of a tape substrate to which the paper fluid medium and the bead-loaded mask are attached and is sealed by a UV-transparent tape for the lateral flow format or flow-through format or is used in a capillary flow channel where the beads directly contact the flowing analyte within a ∼100 μm channel (Figure 2i–n). In a manufactured device, the beads would be embedded directly into the flow medium thereby abrogating the need of the mask.
We demonstrate the multiplexing utility of this system by simultaneously analyzing, both individually and with all samples pooled, several Abs and nucleic acid sequences. Multiplex DNA detection (all capture probes present) was performed with both directly labeled targets and tripartite probe–target–reporter assemblies, and the results summarized in Figure 7a–f showing high target selectivity and a low and uniform background for probes with no targets in both lateral-flow and flow-through systems.
Figure 7.


MFA allows selective multiplex detection of nucleic acids (a–f), antibodies (g–k), or both nucleic acids and antibodies simultaneously (l). In panels (a)–(e), six different DNA capture probe sequences on six different optical codes, one of which is a universal negative control (S3), are present in all reactions set up as a lateral flow experiment (Figure 2a–f). Five dye-labeled targets (∼10 nM), each of which is complementary to one of the five bead-bound capture probes (a–e), are added one at a time to the six bead codes present in each of the five assays (a–e). Each target rapidly binds to its complementary capture probe with high selectivity (Figure 5) and all noncomplementary capture probes display low backgrounds without washing (a–e). When all five labeled targets are added, all beads except the negative control bind their respective targets (f). Ab analytes may be detected in a multiplexed fashion as shown in Figure 1c. The Abs against the bead-bound recombinant protein antigens troponin I (g), HBV (h), HCV (i), and Inf A (j) are added in a flow-through geometry (Figure 2h) to the five bead-bound recombinant protein capture probes one at a time (g–j) or all (troponin I, HBV, HCV, HIV, and Inf A) at once (k) and subsequently stained with dye-labeled rabbit anti-mouse (troponin I, HBV, HCV, and Inf A) or labeled rabbit anti-human (HIV). It is also possible to detect simultaneously four DNA sequences, three Abs, and a negative control within the same assay (l). The number of beads measured for each analyte is given at the lower right of each bar.
In addition to the multiplex sandwich ELISA (Figure 1b) format used to investigate the pore size access of various Abs (Figure 6), “antigen down” assays with the antigen as the capture probe (Figure 1c) have been performed to detect Abs against HIV, HCV, HBV, Inf A, and troponin I. With all antigenic capture probe-derivatized beads present, the Abs are added individually (Figure 4g–j) and then together (Figure 4k), clearly demonstrating selective and multiplexed target capture.
It is also possible to detect selectively and simultaneously both nucleic acids and Abs. When all nucleic acid targets and all Ab targets are pooled and mixed with the pooled capture probes, all added targets are selectively detected, indicating the capability to analyze both antibodies and nucleic acids within the same assay (Figure 4l).
Discussion
As the LFA diagnostic methods are currently one of the most useful and widespread assay varieties, expanding the number and scope of assays that may be performed simultaneously has obvious advantages in terms of cost, logistics, and convenience. In this section, we discuss why embedding lanthanide-encoded capture probes within a porous solid represents a general, straightforward, and particularly well-suited means to perform a MFA. This is followed by a discussion of the responsibility of the multiplexed assays shown in Figure 7.
Multiplexing
Multiplexing refers to the ability to screen each component within a multicomponent target analyte mixture simultaneously against multiple capture probes. To accomplish this multiplexing, the capture probes are pooled and added to the multicomponent target analyte mixture, and it is obvious that it must be possible to distinguish all of the different capture probes used from one another. Two common methods for differentiating capture probes are as follows: (a) spot the capture probe onto a planar substrate and identify the probe by its x, y location on the substrate or (b) attach the capture probe to a bead that has been optically encoded to differentiate it from other bead-bound capture probes. The below discussion emphasizes why porous agarose beads that have been optically encoded with multiple lanthanide emitters form a particularly well-suited platform for creating a diagnostic assay.
The multiplexing scheme used here to build an MFA to detect either antigens or antibodies is schematically illustrated in Figure 1b,c, respectively, depending on the whether an antibody or antigen acts as the capture probe to detect its corresponding antigen or antibody.
The aspects of assay multiplexing that may be facilitated by our method are contrasted to those omnipresent, recurring difficulties and difficult to control variables, such as matrix effects and nonspecific cross-reactivity, which are independent of the analysis platform.
Planar Arrays versus Beads
There are many reasons why porous encoded beads are superior to planar arrays for most diagnostic applications including the following:
Kinetics: assay time to completion. The diffusion rates for mixing surface-bound and bulk solution species directly on a planar surface are very low compared to bulk diffusion rates, and planar arrays often contact target mixtures for 12–18 h. Because the porous agarose beads are typically >95 wt % water and the beads can be agitated and stirred, reactant diffusion rates approach those of bulk solution. Small oligonucleotide targets reach equilibrium with bead-bound targets in <2 min and larger oligos within minutes.27 A typical LFA takes 10–30 min to perform.
Amount of capture probe per unit image area. The beads have a surface area ∼10 000× greater than that of a planar array spot of the same diameter; thus, for a given image size used for analysis, the beads can capture orders of magnitude more target in the same image area. Consider a qualitative comparison of a 50 μm planar spot and a 50 μm diameter bead with both appearing as a 50 μm object when analyzing the image for the assay. The surface area of the planar spot is about 2 × 103 μm2, whereas an agarose sphere of 50 μm diameter, with a typical agarose surface area of ∼250 m2/g beads, ∼106 beads/mL, and 5 wt % agarose, has a surface area of about 107 μ2. Obviously, the bead can capture far more target than a planar spot. As the Parallume beads are >95 wt % water, they are essentially transparent and much of the emitted light may be captured and measured.
Fabrication. Although each spot must be placed onto each individual array at a carefully defined location using expensive robotic equipment, the encoded beads may be placed at any random location within the flow assay. The beads could possibly be cast within the porous media during device fabrication. The porous Parallume beads are synthesized by spraying a mixture of Parallume nanoparticles in agarose at a rate of millions of beads per minute in a completely scalable process.
Probe substrate density. Although it is easy to place 50 μm beads at a distance of 10 μm from one another within a porous substrate, printing 50 μm liquid spots onto a porous membrane is more challenging. The beads could easily be much smaller than 50 μm but printed spots on a porous substrate cannot made as small.
Nonspecific binding. Agarose is widely used as a gel and is known for very low nonspecific binding of proteins and nucleic acids. Unnatural substrates such as glass have much higher nonspecific binding per unit area than agarose.
Self-similarity. Because beads are synthesized in millions at a time in a single batch, the assays performed on the beads synthesized at the same time would be expected to have a lower sample-to-sample variance than the spots deposited one at a time.
Lower operational costs. Bead handling requires no special equipment such as spotters, scanners, incubators, or mixers, and beads may be handled in standard PCR tubes.
Ease of use. Beads may be handled in everyday laboratory containers and transferred with standard pipets, whereas slides are more difficult to manipulate. For example, it is straightforward to perform PCR or primer extension on beads but quite difficult to thermally cycle or store slides.
Lanthanide Encoding
The advantages of lanthanide encoding over optical encoding with organic dyes are numerous and substantial. Two important advantages are the larger number of optical codes available with lanthanides because of the narrow emission peak width (Figure 3) and the exceptional chemical stability and photostability of the solid-state inorganic lanthanide materials, which provide stable and immutable optical codes.
The host compound used to create the multilanthanide Parallume materials in this study is YVO4. As prepared with a final calcination at >1000 °C, the surface of the YVO4 is dehydroxylated and hydrophobic and difficult to wet. In this state, YVO4 tenaciously and irreversibly binds DNA and proteins. These calcined particles are mixed with agarose, and the molten agarose is sprayed from a nozzle to form the beads,26 which are typically 1–4 wt % agarose and 1–2 wt % lanthanide-doped YVO4. The fact that the extremely low-binding level on agarose beads does not change with and without entrained YVO4 particles, and bare YVO4 instantly and strongly binds DNA, indicates that no YVO4 within the agarose beads is exposed to the target-containing solution. We have never observed any change in the Parallume codes (i.e., any change in the relative amounts of the multiple lanthanide emitters) once synthesized, as indicated by the facts that (a) the Parallume optical bins into which the encoded beads are sorted (Figure 4) are the same before and after any reaction, (b) the Parallume codes did not change upon storage in water for over five years, and (c) we have never observed a change in optical code when Parallume beads are thermally cycled under PCR conditions for 100 cycles. There is no indication of any lanthanide leakage from the beads under any conditions.
Unlike most organic reporters and dyes, the lanthanides in the YVO4 host used here have excitation spectra that do not overlap with the excitation spectra of either DNA/proteins (Figure 3 inset) or organic dyes absorbing in the near UV or visible (vis). Therefore, an image taken at 315 nm excites only the lanthanides and the white YVO4 is transparent to the visible excitation used to excite the reporter dyes (e.g., 532 nm).
Unlike organic dyes, which have very limited photostability especially in air, the lanthanide-doped YVO4 cannot be photobleached under any circumstances. These materials are photostable enough to form laser cavities, as evidenced by the popular green laser pointers (532 nm), which is the frequency-doubled 1064 nm output from Nd3+-doped YVO4.
Pore Properties of the Parallume Beads
The pore structure of beads and diffusion of analyte targets into the beads were studied in two different ways. The agarose is chemically crosslinked so that the pore structure is fixed and stable. As shown in Figure 5, a static diffusion experiment with a dyed target and a bead-bound capture probe provides information about the kinetic diffusion rate into the bead. As there is no mixing or agitation provided in this experiment, this represents the slowest diffusion rate possible. As seen in Figure 7b, the dyed target begins to be concentrated onto the correct capture probe within 1 min. A typical minimally acceptable 3:1 positive:negative control is achieved within minutes. When the samples are agitated (e.g., on a plate shaker), target–probe binding is very rapid and reaches a steady state within a few minutes.27
It is well known that agarose forms smaller pores and a higher surface area as the weight percentage of agarose increases upon forming a gel. The pore size sieving effect on target analyte size as a function of pore size is strong, sharp, and clear for the Parallume beads. As shown in Figure 6, the access of a series of antibody–antigen pairs and the SAPE stain is allowed or not allowed depending on the relative target and pore sizes. Note that the pores in the 1–2 wt % beads are very large and can accommodate species up to 1.4 MDa in size (probe + target + SAPE).
Multiplex Nucleic Acid and Antigen–Antibody Detection: Matrix Effects
Three types of multiplex target detection experiments were performed: (a) multiple antibody–antigen pairs, (b) multiple complementary strands of nucleic acids, or (c) both antigens and nucleic acid analytes, which are shown in Figure 7. An important general point to note is that LFA are used nearly exclusively to determine the presence or absence of an analyte and not often used for quantitative measurements.
It should be noted that in Figure 7, the signals from the same target are weaker when present within the multiplexed assay than when in the control assay with only one species present. This is because all targets are of a fixed concentration and when combined are necessarily diluted. For example, if six antibodies of concentration 1 mg/mL are mixed together, the concentration of each is 1/6 mg/mL. Of course, this situation would never occur in an actual assay as there is only one target mixture to be analyzed and is only relevant while preparing standards.
Common to all multiplex reactions are nonspecific cross-reactivity between targets and probes and matrix effects. It is seen that the cross-reactivity is higher with the antigens and antibodies (Figure 7g–j) than with DNA. This is because (a) the DNA is 10–50× more concentrated than the commercially obtained antibodies and (b) the antibodies are much less stable than the DNA and the amount of fully reconstituted and completely functional protein present is not known. Therefore, other interfering species are more likely to be present within the unstable protein samples than the chemically pure synthetic DNA samples.
In the case of the antibodies, cross-reactivity may be seen even when there is only one target added. For example, there is obvious cross-reactivity between the HBV and HCV antigens but very little with Inf A and Trop I.
Summary
We have shown that the use of optically encoded regions instead of a known geometric location on a flow medium for analyte identification can transform the most commonly used diagnostic assay format from a single-plex assay to a multiplex assay with the multiplexing enabled by the very narrow optical emission from the multicolor Parallume encoding materials. As the MFA employs the same reagents and similar manufacturing methods as conventional lateral flow assays, rapid introduction into the existing POC diagnostic area will lower the cost and increase availability in the short term for many types of assays. Comprehensive and wide-ranging MFA screenings reliably performed by anyone could provide the private, rapid, and detailed knowledge to participate in one’s own critical health decisions.
Methods and Experimental Procedures
General Bead Handling Procedures
These procedures are suitable for either (a) covalently attaching a capture antibody to the beads or (b) covalently attaching a recombinant protein to the beads using the well-known 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS) protocol. Smaller peptides often bind spontaneously to the beads without chemical crosslinking, but larger proteins and all antibodies require the crosslinking procedure given here to covalently link them to the beads.
-
1.
The virgin Parallume beads, which have never been treated with any proteins, surfactants, detergents, cell extracts, or nucleic acids, that is, as received from Parallel, are reactive and may adhere to the walls of the pipet tip and reaction tube or clump together. If the underivatized beads cling to the pipet tip while using your buffer, the use of commercially available “low-adhesion” pipet tips is recommended. If the beads clump or adhere to the tube during protein conjugation or coating, just continue with the protocol. Once the buffer contains surfactant, proteins, antibodies, etc., or the PBST used here, in subsequent steps of the assay, the beads will no longer adhere to surfaces and should become free floating once again.
-
2.
Stirring means agitation on an orbital plate shaker (suggested: 5 mm orbit). The speed of shaking required to best suspend the beads depends on the size and shape of the tube and the amount of beads used. For 0.1–50 μL of beads in 10–1000 μL of buffer, a 1.5 mL round-bottom centrifuge tube (Eppendorf) is shaken at 800 rpm, a 1.5 μL conical-bottom tube at 1200 rpm and a 200 μL PCR tube at >1700 rpm. In general, the beads are the easiest to stir in round-bottom tubes. Bead volumes of several microliters or larger may be conveniently handled in a 1.5 mL Pall Nanosep with a 500 μL 0.45 μm filter insert. Visually inspect the beads when on the plate shaker to make sure the liquid vortex rises up out of the bottom of the tube and coats the size of the tube—but not so fast that liquid adheres uniformly to the entire tube wall from centrifugal force. Rotation at too fast or too slow a speed results in less efficient mixing and potentially compromised detection limits. If the beads are not mixed as vigorously as required, the kinetic diffusion of the material in and out of the porous beads, as well as the detection limits, could suffer.
-
3.
To obtain more uniformity among beads during a reaction, do not add a concentrated solution of a reagent to the beads without agitation, but rather immediately aspirate the solution in and out of the pipet tip as rapidly as possible to expose all beads as quickly as possible to the same concentration of the reagent at the same time.
-
4.
Unless otherwise specified, washing means stirring for 5 min on the plate shaker.
-
5.
For isolation, the beads may be quickly centrifuged on a small centrifuge, pulled down with a magnet if magnetic beads are used or just allowed to settle by gravity.
-
6.
Settled beads refer to beads that have been allowed to settle for at least 10 min or centrifuged; 1 μL of settled beads is about 1 mg of beads and there are approximately 1500–3000 beads/μL. In general, 10 or more beads are needed per assay depending on the particular assay.
-
7.
To measure a small volume of beads, pipet a volume of beads into the appropriate dilution volume, quickly aspirate the beads in and out of the pipet tip to completely and uniformly suspend them, and then transfer the desired volume of suspended (diluted) beads. Accurate aliquotting requires a uniform and homogeneous bead suspension.
-
8.
Store bare, unused Parallume beads at room temperature or 4 °C in 0.03% NaN3. Beads are always supplied in 0.03% NaN3 unless otherwise specified.
Protocol for Conjugating Antibodies or Recombinant Proteins to Encoded Agarose Beads
Materials and Reagents
-
(a)
Carboxylated Parallume beads.
-
(b)
Capture antibody or recombinant protein.
-
(c)
Stain for antibody (usually dye-labeled antispecies antibody).
-
(d)
Zeba 7K MWCO spin columns (if needed).
-
(e)
Activation buffer: 2-(N-morpholino)ethanesulfonic acid (MES) buffer = 100 mM MES, 150 mM NaCl, pH 6.0.
-
(f)
1× phosphate-buffered saline (PBS), pH 7.4.
-
(g)
1× PBST, pH 7.4 (PBST is 0.05% v/v Tween-20 in PBS).
-
(h)
NHS or Sulfo-NHS (Thermo Scientific).
-
(i)
EDC (Thermo Scientific) [Do not substitute other brands of EDC]. When the EDC is received, place small samples into individual containers with an O-ring or equivalent seals and store at −20 °C. Always allow the EDC to warm to room temperature for at least 15 min before opening. Use only if the EDC appears as a fluffy powder—any sign of clumping or a damp appearance is indicative of decomposition and such material should be discarded.
-
(j)
Quench for EDC activation = 50 mM hydroxylamine in 1× PBST.
-
(k)
ELISA buffer (1% bovine serum albumin (BSA) in 1× PBST, pH 7.4).
-
(l)
Blocking buffer (appropriate normal serum, ELISA buffer or 5% w/v non-fat milk powder or other preferred blocking reagent).
-
(m)
Nanosep spin columns, 1.5 mL tubes or 200 μL PCR tubes.
-
(n)
Centrifuge for 1.5 mL or 200 μL PCR tubes.
-
(o)
Orbital plate shaker.
-
1.
Important: Materials that are often present in protein or antibody preparations, which could interfere with the EDC coupling, such as nucleophiles like Tris or azide, surfactants, urea, Tween and other materials, must be removed before commencing the bead–protein coupling reaction. Before coupling, the protein or antibody is passed through a 7K MWCO Zeba column to remove materials reactive toward EDC and to exchange the protein shipping/storage buffer for 1× PBS buffer, pH 7.4.
-
2.
Transfer 2 mg of settled agarose beads (∼2 μL) to a 200 μL tube, 1.5 mL tube, or Nanosep column.
-
3.
Add 200 μL (or more if using a 1.5 mL tube) of 100 mM MES, 150 mM NaCl buffer, pH 6.0. Wash the beads by stirring at room temperature for 5 min.
-
4.
Pellet the beads and aspirate the supernatant and discard without disturbing the pellet.
-
5.
Add 200 μL of 100 mM MES, 150 mM NaCl buffer, pH 6.0, and repeat the washing procedure twice for a total of three washes.
-
6.
Resuspend the beads in 20 μL of 100 mM MES, 150 mM NaCl, pH 6.0.
-
7.
In two separate tubes, prepare in 100 mM MES, 150 mM NaCl buffer, pH 6.0, a 15 mg/100 μL Sulfo-NHS solution (or a 6 mg/100 μL solution of NHS) and 5 mg/100 μL EDC solutions.
-
8.
Add 20 μL of the NHS solution to the beads, briefly mix, then add 20 μL of the EDC solution to the beads and stir at room temperature for 30 min to activate the carboxylate COOH groups.
-
9.
Pellet and wash the beads once with 200 μL of 100 mM MES, 150 mM NaCl buffer, pH 6.0, and twice with 200 μL of 1× PBS, pH 7.4, to remove excess coupling reagents.
-
10.
Resuspend the beads in 20 μL of 1× PBS, pH 7.4.
-
11.
Add capture antibody or recombinant protein (1–5 μg/mg of beads) in 1× PBS to the activated beads, adjust the final volume with 1× PBS, pH 7.4, to 50 μL and stir the reaction mixture at room temperature for 2 h. Note: use 5 μg protein for the first derivatization protocol and subsequently optimize.
-
12.
Wash the beads twice with 200 μL of 1× PBST, pH 7.4, by stirring, centrifuging, and removing the supernatant.
-
13.
Quench any excess coupling reagent by treating beads with 50 mM hydroxylamine in 100 μL 1× PBST.
-
14.
Stir beads for 15 min at room temperature.
-
15.
Wash the beads thrice with stirring in 200 μL 1× PBST.
-
16.
Pellet the beads in a mini centrifuge and discard the supernatant.
-
17.
The beads are now ready for target capture.
Protocol for Blocking Antigen or Antibody-Derivatized Beads
When using an antispecies reporter stain, it is best to use serum from the species that produces the antibody for the stain for blocking if possible. For example, if the detection antibody is a monoclonal mouse antibody, block with normal rabbit serum if the antispecies antibody reporter stain is rabbit anti-mouse. Use 1–5% v/v normal serum/PBST for blocking.
-
1.
Prepare a blocking buffer in PBST using normal serum, ELISA buffer (1% BSA in 1× PBST, pH 7.4), milk, or other preferred blocking buffer.
-
2.
Add 200 μL of the blocking buffer to the pelleted beads and stir at room temperature for 1 h.
-
3.
Wash in PBST, pellet the beads, and discard the supernatant.
-
4.
The capture probe-derivatized beads, which should be stored at 4 °C if not used immediately, are now ready for use in capturing antigens or antibodies.
Protocol for Preclearing Human Serum for Immunoassays on Agarose Beads
If human serum is part of the assay, there appears to be a human antibody against agarose present in all human sera that must be removed from the serum before any assay is performed.
-
1.
Prepare the serum for preclearing by placing 1 μL serum in 50 μL ELISA buffer. (This is the amount for a single assay.)
-
2.
To preclear the serum, add 1 mg of “preclearing” beads (∼1 μL settled beads) to the above serum/buffer solution. Any agarose beads that have never been previously used are suitable for preclearing.
-
3.
Stir for 45 min, then spin down beads and collect the supernatant. These preclearing beads should be discarded, but if these beads are stained with rabbit anti-human IgG, they stain very intensely.
-
4.
As, for example, the human HIV antibodies against the HIV coat protein gp41 are easily detected in human serum after this preclearing treatment, the preclearing does not remove other antibodies.
-
5.
This supernatant is the precleared serum.
Protocol for ELISA or Antibody Capture
Depending on whether an antigen (antigen down assay) or antibody (ELISA) was coupled to the beads as the capture probe, the following protocol is used to capture and stain the target antibody or antigen, respectively.
Target Capture
-
1.
Bead volume: use 0.05–0.2 mg of the above coupled and blocked beads per assay (i.e., per optical code). See Note 7 in General Bead Handling Procedures (above), for measuring and dispensing small bead volumes.
-
2.
Centrifuge and remove excess blocking buffer from the capture probe-derivatized and blocked beads.
-
3.
Add 50 μL of the previously precleared serum or other sample in ELISA buffer to the bead pellet.
-
4.
Stir for 1 h.
-
5.
Wash the beads thrice using 1× PBST and remove supernatant.
-
6.
If the target is reporter-labeled, the sample is ready for fluorescent measurement of the reporter.
Target Staining
If the target is not labeled, then the bead-bound target or detection antibody must be stained with a reporter antibody.
-
1.
Important: To obtain more uniform staining, do not add concentrated stain to a tube that has beads in a small volume of buffer. First, dilute the stain in a separate tube with buffer and mix thoroughly. Then quickly add this diluted stain to the beads and mix by aspiration or stirring as rapidly as possible.
-
2.
Stain the bound target by adding the reporter antibody in PBST. If the antibody is 1 mg/mL, then use a 1:500 dilution as a starting point. If the detection antibody is biotinylated, use ∼50 μL of 10 μg/mL SAPE in PBST.
-
3.
Stir the diluted reporter-labeled antibody in PBST with the beads containing the bead-bound targets.
The sample is ready for fluorescence measurement of the reporter.
Protocol for Coupling Amine-Modified Oligonucleotides to Carboxylated Agarose Beads
Materials and Reagents
-
(a)
Carboxylated Parallume beads
-
(b)
Amino-oligonucleotide1 (100 μM)
-
(c)
EDC (Thermo Scientific)
-
(d)
NHS (Thermo Scientific)
-
(e)
MES buffer 0.1 M, pH 4.5
-
(f)
1× PBS, pH 7.4
-
(g)
1× PBST, pH 7.4 (PBST is 0.05% v/v Tween-20 in PBS)
-
(h)
Tween-20 (Concentrated)
-
(i)
Tween-20 (0.05% w/v in H2O)
-
(j)
Tris–HCl 50 mM, pH 8.0
-
(k)
1.5 mL round-bottom centrifuge tubes or 200 μL PCR tubes or, for bead volumes of several microliters or larger, Pall Nanosep spin columns (500 μL)
-
(l)
Plate shaker for stirring
-
(m)
Thermal cycler
-
1.
Remove EDC from the freezer and let it warm to room temperature for at least 15 min.
-
2.
Vortex or repeatedly aspirate the stock beads for 10 s. Quickly aspirate the desired amount (2–4 mg)2 from the suspension and dispense into a 1.5 mL or 200 μL PCR tube or spin column.
-
3.
Spin down the beads for 15 s and remove the supernatant without disturbing the pellet or centrifuge in a filter spin column.
-
4.
Add 200 μL of 0.1 M MES buffer, pH 4.5, and 1 μL of concentrated Tween-20 and stir for 5 min at room temperature, pellet the beads, and remove the supernatant. Repeat once, wash once with 200 mL of 0.1 M, pH 4.5, MES buffer, spin down the beads, and remove the supernatant.
-
5.
Resuspend the pelleted beads in 20 μL of 0.1 M MES buffer, pH 4.5.
-
6.
For each mg of the beads, add 0.1 nmol (1.0 μL of a 100 mM solution) of amino-oligo capture probe.
-
7.
Stir the beads for 2 min.
-
8.
Prepare a 58 mg/mL solution of NHS in 0.1 M MES buffer, pH 4.5.
-
9.
Add 10 μL of the NHS solution to the tube and stir the beads for 1 min.
-
10.
Prepare the required volume of a 50 mg/mL solution of EDC in 0.1 M MES buffer, pH 4.5.
-
11.
Add 20 μL of the freshly prepared EDC solution to the tube.
-
12.
Stir the beads for 30 min at room temperature.
-
13.
Prepare another fresh solution of 50 mg/mL EDC in 0.1 M MES buffer, pH 4.5.
-
14.
Add a second aliquot of 20 μL of freshly prepared EDC solution to the tube.
-
15.
Stir the beads for an additional 30 min at room temperature.
-
16.
Pellet the beads in a mini centrifuge for 10 s. Remove as much solution as possible without disturbing the bead pellet.
-
17.
Wash the beads with 200 mL of 0.1 M MES buffer, pH 4.5, then 200 μL of H2O with 0.05% Tween-20, pellet the beads, remove the supernatant, and repeat the washing procedure once more.
-
18.
Wash the beads once with 200 μL of 50 mM hydroxylamine hydrochloride in PBST, pH 7.4, or 50 mM Tris, pH 8.0, for 5 min to quench any unreacted EDC or activated carboxylate groups, pellet the beads, and remove the supernatant.
-
19.
Wash the beads with 200 μL of 50 mM Tris buffer, pH 8.0, for 2 min at 95 °C and 5 min at 55 °C in a thermal cycler, pellet the beads, and remove the supernatant. Repeat the washing once more. These capture probe-derivatized beads are ready for target capture.
-
20.
If very low detection limits are required, it is possible to remove a small amount of bead background fluorescence generated during the EDC procedure using a borohydride procedure. This step is designed to reduce bead autofluorescence generated from the EDC coupling. This step is not usually necessary, especially when analyzing concentrated solutions such as PCR products. Prepare a fresh 0.25% sodium borohydride solution in 2× standard saline citrate (SSC), 0.05% sodium dodecyl sulfate (SDS). Add 200 μL of the NaBH4 solution to the washed beads from step 19. Incubate at 42 °C for 30 min with stirring. Leave the lid of the tube slightly open to allow the hydrogen gas generated during the NaBH4 treatment to escape. Pellet the beads and remove the NaBH4 solution. Repeat this treatment with 200 μL of freshly prepared NaBH4 solution. Pellet the beads to remove the supernatant. Wash the beads twice in 1× SSC, 0.05% Tween-20 at 42 °C for 10 min with stirring. Wash the beads two more times in 0.0.2× SSC, 0.05% Tween-20 at 42 °C for 10 min with stirring. Resuspend the beads in appropriate amount of 0.2× SSC, 0.05% Tween-20 so that the final bead concentration is between 0.01 and 0.02 mg/μL.
-
21.
Store oligo-derivatized beads at 4 °C in 0.03% NaN3 for long-term storage.
Protocol for Hybridization to Bead-Bound Oligonucleotide Capture Probes
This section contains the protocols for hybridizing the lanthanide-encoded beads in bulk, whereas the section Protocols for Lateral Flow, Flow-Through and Capillary Flow Assays below provides protocols for performing hybridizations in the lateral flow and flow-through geometries.
Materials and Reagents
-
1.
Parallume beads with immobilized capture probes
-
2.
Hybridization buffer
-
(a)
Ficoll (Sigma, 2 mg/mL)
-
(b)
Poly(vinylpyrrolidone) (Sigma, 2 mg/mL)
-
(c)
BSA (10 mg/mL)
-
(d)
3× SSC
-
(e)
SDS (0.25%)
-
(a)
-
3.
Wash buffer 1: 2× SSC, 0.03% SDS
-
4.
Wash buffer 2: 1× SSC, 0.05% Tween-20
-
5.
Wash buffer 3: 0.2× SSC, 0.05% Tween-20
-
6.
1.5 mL tubes or 200 μL PCR tubes
-
7.
Centrifuge for 1.5 mL or 200 μL tubes
-
8.
Thermal cycler
-
9.
Orbital plate shaker
Prehybridization Blocking Procedure
-
1.
Transfer the appropriate amount of capture probe-immobilized beads (0.1–4 mg) to a 200 μL PCR tube or 1.5 mL round-bottom tube, pellet the beads, and remove the supernatant.
-
2.
Add 200 μL of hybridization buffer to the beads and block the beads by incubating at 95 °C for 2 min and then at the hybridization temperature overnight with stirring.
Hybridization
-
1.
Transfer the desired amount (0.02–0.2 mg) of preblocked beads to a 200 μL PCR tube.
-
2.
Pellet the beads and remove the supernatant.
-
3.
Prepare 30–50 μL of labeled target DNA (direct hybridization) or unlabeled target plus labeled reporter (probe–target–reporter sandwich hybridization) in hybridization buffer, add the target DNA/reporter solution to the beads.
-
4.
Hybridization is carried out in a thermal cycler for 2 min at 95 °C and 30–45 min at the hybridization temperature (usually capture probe Tm −10 °C).
Posthybridization Washing
-
1.
Pellet the beads and remove as much supernatant as possible.
-
2.
Add 200 μL of 2× SSC, 0.03% SDS to the beads, incubate at 54 °C for 10 min with stirring.
-
3.
Pellet the beads and wash again in 200 μL of 1× SSC, 0.05% Tween-20 at 48 °C for 10 min with stirring.
-
4.
Pellet the beads and wash again in 200 μL of 0.2XSSC, 0.05% Tween-20 at 38 °C for 10 min.
-
5.
Pellet the beads and remove the wash buffer.
Imaging and Analysis of the Assays
Image Acquisition
The optical imaging data was collected on one of two the commercially available Multiplex Assay Reader System (MARS) instruments. A portable system (http://www.parallume.com/mmm.html) is based on a Dinolite color USB microscope where the color separation required to measure the intensity ratios of the lanthanide emitters derives from the color filters on each pixel of the microscope’s color CMOS sensor. A tri-pass notch filter, which has 25 nm wide transmission windows centered at 464/542/639 nm, is optionally placed in front of the color sensor during acquisition of the UV-excited image. The samples may also be measured with a MARS that has a CCD camera combined with seven notch filters to separate the lanthanide and reporter color intensity (http://www.parallume.com/mars.html).
-
(a)
For the UV image, a UV LED (∼100 μW) with 310–320 nm output is used and any visible light emitted removed with a UG-11 filter.
-
(b)
For the vis reporter image, the vis LED (5 W) has 532 nm output. The excitation light above 532 nm is removed using a short pass filter and the emitted reporter image collected using a 532 nm long pass filter.
-
(c)
Reporter images are taken with exposures times that are entered in the “Acquire UV Images” and “Acquire Reporter Images” dialog.
-
(d)
All images are saved as bitmap (.bmp) files with 2592 × 1944 resolution.
-
(e)
All images are saved in 24 bit RGB color.
Bead Identification
-
(a)
Bead locations are identified using the total (no emission filters) UV image.
-
(b)
AForge.NET Framework-2.2.4 is used for bead analysis.
-
(c)
The minimum acceptable width and height for bead identification values are set in the Exposure tab of the Instrument Settings dialog.
Bead Analysis
-
(a)
For each image, each identified bead location is used for initial analysis.
-
(b)
The bead is examined for size, shape, separation distance to nearest bead neighbors, brightness, and color to determine whether the bead is acceptable for analysis
-
(c)
If two beads are closer together than a given minimum pixel distance, then both beads are rejected. The distance value is set in the Exposure tab of the Instrument Settings dialog.
-
(d)
If the ratio of the bead width versus bead height is too large or small, it is rejected. The ratio is set in the Exposure tab of the Instrument Settings dialog.
-
(e)
At each location, all pixel values that are within an annulus around the bead center are analyzed. The bead height and width are examined and the larger of the two values is used as the initial radius. A percent of this radius is then included in the analysis. The percent value is set in the Exposure tab of the Instrument Settings dialog.
-
(f)
The number of any saturated pixels in the UV image is counted. A pixel is considered to be saturated if any of the RGB channels is saturated. If more than 30% of the pixels are saturated, the bead is rejected.
-
(g)
For the UV image, pixels with red, green, or blue signals above a threshold are included. The threshold value is set in the Exposure tab of the Instrument Settings dialog. If the red, green, or blue value for a pixel is saturated, the pixel is excluded. The red, green, and blue signals of included pixels are summed, individually.
-
(h)
For the reporter image, pixels with red, green, or blue signals above a threshold are included. The threshold value is set in the Exposure tab of the Instrument Settings dialog. The red, green, and blue signals of these pixels are summed, individually.
-
(i)
For the reporter image, a minimum number of accepted pixels are required for the reporter signal to be accepted. The minimum value is set in the Exposure tab of the Instrument Settings dialog.
-
(j)
Two ratios are calculated from the UV image sums. The red sum is divided by the green sum, and the blue sum is divided by the green sum R/G and B/G.
-
(k)
All bead locations, the number of pixels included for the UV images, and the summed values for the UV and reporter images are saved in an Excel file named RawData.xlsx
-
(l)
All bead locations, the number of pixels included for the UV images, the number of saturated pixels found, the summed values for the UV and reporter images, and the ratios are saved in an Excel file named BeadData.xlsx.
Optical Bin Creation and Analysis
-
(a)
The desired number of ratiometrically encoded lanthanide bead sets is prepared, with each set containing >200 beads of the same optical code, and imaged to obtain the RGB intensity values at each pixel in the image measured individually for each individual code.
-
(b)
The ratiometric intensities of R/G and B/G are plotted and a quadrilateral enclosing the beads of each code is drawn. This creates an optical bin for each ratiometric lanthanide code, which is used to sort the beads in a given image into their proper optical bin. The bins are sufficiently separated in ratiometric color space such that beads from a given code reside in only one bin.
-
(c)
For each bead in an experimental image, the computed R/G and B/G ratios are compared to the bin locations of each bead type in the well.
-
(d)
For each bin, linear regression is used to fit a line to the ratios.
-
(e)
The ratios are rotated by the slope from the linear regression and translated by the intercept of the linear regression to position the ratios along the x axis.
-
(f)
The bead ratios are also rotated by the slope from the linear regression.
-
(g)
It is then determined whether the rotated bead ratios are within the rotated bin areas (i.e., the optical bins) and, if so, the bead code is recorded.
-
(h)
For each set of beads with the same code, outliers are calculated and removed using the fourth spread method. The BeadData.xlsx is updated to include the outlier and bin information for each bead.
Reporter Analysis
-
(a)
For each bead associated with a code, the red, green, and blue reporter values are summed, and the summed value is divided by the integration time of the reporter images to normalize to a 1 second integration time.
-
(b)
If the user has chosen to normalize the reporter values to the bead size, an average is calculated for the sum of the red, green, and blue UV image values for all the beads with the given code, with outliers excluded. Each integration time normalized reporter value is then multiplied by this averaged UV value and divided by the sum of the red, green, and blue UV image values for the given bead.
-
(c)
A reporter data Excel file is created. This file includes the code numbers, associated probes, the averages and standard deviations of the normalized reporters for each code, the exposure time for the reporter images, the number of outlier beads for the code, and the number of beads associated with the code. The number of beads in the well that were not associated with a bead code is also recorded. The reporter values are also graphed. This file is named ReporterData.xlsx.
Protocols for Lateral Flow, Flow-Through and Capillary Flow Assays
General
The assays may be performed in a flow-through or lateral flow geometry using the devices shown in Figure 2, where the analyte-containing fluid contacts the capture probes in the encoded regions as it passively flows from a fluid source through a porous flow medium to a fluid sink by capillary action. The rate of fluid transport through the sample and porous flow medium is determined by the volume of the solution applied and especially the absorbent capacity and rate of the sink. In the devices here, the analyte-containing solution is applied to the source side of the device with a pipet and the sink comprises dry filter paper. The time for which the analyte-containing solution remains in contact with encoded regions may be controlled by the size of the sink, its capacity, and when it is applied to the flow medium. If the relative humidity is too low, the lateral flow and flow-through devices are placed in a closed container to prevent evaporation.
For imaging, the samples are treated with a solution of 10–20% glycerin in 1× PBST to slow evaporation or imaged directly in the device.
Materials
The devices shown in Figure 2 are made from a combination of hydrophobic, double-sided and single-sided adhesive pressure-sensitive adhesive (PSA) tape, a bead-masking grid, which is laser cut from a 50 μm thick black polymer film and Whatman filter paper. All grids contain an array of 37 × 37 holes. The tape and PSA are very hydrophobic, so the solution flow is completely confined to the porous flow medium and samples.
The lateral flow devices are fabricated from a length of filter paper strip (∼5 mm wide) either mounted directly onto the PSA tape serving as backing or mounted to a substrate with double-sided tape. The masking grid containing the encoded beads is placed in firm and intimate contact with the flow medium by taping the grid over the flow medium strip (Figure 2), and a UV-transparent PSA (3M) tape forms the top layer.
The flow-through devices are fabricated from 8 mm o.d. × 6 mm i.d. × 6 mm long UV-transparent acrylic tubing with a 37 × 37 bead-masking grid sealed to the acrylic with double-sided tape (Figure 2).
Lateral Flow Assays
An aliquot of the analyte-containing solution (1–20 μL depending on the sample size and concentration) is placed on the source region, travels through the capture probe-derivatized beads and into the sink. The time for which the solution is in contact with the bead samples may be controlled, from <1 s to indefinitely, by the location, dimensions, and capacity of the sink region.
Flow-Through Assays
The solution sample is placed onto the bead-containing grid, allowed to remain in contact with the beads for the desired time, and the solution removed by contacting the grid with the sink material. The rate at which the solution moves through the bead samples may be controlled by the location, dimensions, and capacity of the sink region.
Acknowledgments
We thank the U.S. National Institutes of Health and the U.S. National Science Foundation for financial support and Dr. Jun Wang for obtaining the data in Figure 6.
Author Contributions
All authors performed experiments and discussed the manuscript. R.C.H. conceived the project and wrote the manuscript.
The authors declare the following competing financial interest(s): R.C.H. and S.V. are employees of Parallel Synthesis Technologies, Inc. R.C.H. is an inventor on U.S. Patents 8,927,892, 8,796,030, and 8,676,107 and U.S. Patent Applications 20150192522 and 20140336061.
Footnotes
It is recommended that the amino-oligonucleotide contain a linker group of six poly(ethylene glycol) (−CH2CH2O−) linker groups, that is, (−CH2CH2O−)6, between the amino functionality and the oligo itself. A hexamethylene (−CH2−)6 linker may be used but is shorter and hydrophobic.
There are ∼1–2 × 103 beads/mg and ∼10 or more beads are required for each sample analysis.
References
- Mukhopadhyay R. Microfluidics: On the Slope of Enlightenment. Anal. Chem. 2009, 81, 4169–4173. 10.1021/ac900638w. [DOI] [PubMed] [Google Scholar]
- Gan S. D.; Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. J. Invest. Dermatol. 2013, 133, e12. 10.1038/jid.2013.287. [DOI] [PubMed] [Google Scholar]
- Chun P.Colloidal Gold and Other Labels for Lateral Flow Immunoassays. In Lateral Flow Immunoassays; Wong R. C., Tse H. Y., Eds.; Springer, 2009; pp 75–94. [Google Scholar]
- Posthuma-Trumpie G. A.; Korf J.; van Amerongen A. Lateral flow (immuno)assay: its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 2009, 393, 569–582. 10.1007/s00216-008-2287-2. [DOI] [PubMed] [Google Scholar]
- Rosen S.Market Trends in Lateral Flow Immunoassays. In Lateral Flow Immunoassays; Wong R. C., Tse H. Y., Eds.; Springer, 2009; pp 35–50. [Google Scholar]
- Durham H. E. On a special action of the serum of highly immunised animals. J. Pathol. 1897, 4, 13–44. 10.1002/path.1700040104. [DOI] [Google Scholar]
- Marrack J. Nature of Antibodies. Nature 1934, 133, 292–293. 10.1038/133292b0. [DOI] [Google Scholar]
- Landsteiner K. Zur Kenntnis der antifermentativen, lytischen und agglutinierenden wirkungen des blutserums und der Lymphe. Zentbl. Bakt. Orig. 1900, 27, 357–362. An English translation may be found in Camp, F. R., Jr., Ellis, F. R., Ed.; Selected Contributions to the Literature of Blood Groups and Immunology; Vol. 1. www.dtic.mil/get-tr-doc/pdf?AD=AD0662074.. [Google Scholar]
- Yalow R. S.; Berson S. A. Immunoassay of endogenous plasma insulin in man. J. Clin. Invest. 1960, 39, 1157–75. 10.1172/JCI104130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wide L.; Axen R.; Porath J. Radioimmunosorbent assay for proteins. Chemical couplings of antibodies to insoluble dextran. Immunochemistry 1967, 4, 381–386. 10.1016/0019-2791(67)90097-3. [DOI] [Google Scholar]
- Engvall E.; Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. 10.1016/0019-2791(71)90454-X. [DOI] [PubMed] [Google Scholar]
- Campbell R. L.; Wagner D. B.; O’Connel J. P.. Solid-Phase Assay with Visual Readout. U.S. Patent 4,703,017, 1987.
- Rosenstein R. W.; Bloomster T. G.. Solid-Phase Assay Employing Capillary Flow. U.S. Patent 4,855,240, 1989.
- May K.; Prior M. E.; Richards I.. Capillary Immunoassay and Device Therefore Comprising Mobilizable Particulate Labeled Reagents. U.S. Patent 5,622,871, 1997.
- Cohen M. H.; Gabriel K. A.; Loomis L. J.. Multiplexed Lateral Flow Assay Arrays. U.S. Patent 8,715,590, 2014.
- Swanson C.; D’Andrea A. Lateral flow assay with near-infrared dye for multiplex detection. Clin. Chem. 2013, 59, 641–648. 10.1373/clinchem.2012.200360. [DOI] [PubMed] [Google Scholar]
- Xu Y.; et al. Fluorescent probe-based lateral flow assay for multiplex nucleic acid detection. Anal. Chem. 2014, 86, 5611–5614. 10.1021/ac5010458. [DOI] [PubMed] [Google Scholar]
- Yonekita T. J.; et al. Development of a novel multiplex lateral flow assay using an antimicrobial peptide for the detection of Shiga toxin-producing Escherichia coli. J. Microbiol. Methods 2013, 93, 251–256. 10.1016/j.mimet.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Haushalter R. C.Methods for Fabricating Optically Encoded Particles and Methods for Optically Encoding Objects with such Particles. U.S. Patent 8,673,107, 2014.
- Haushalter R. C.Methods for Optically Encoding an Object with Upconverting Materials and Compositions Used Therein. U.S. Patent 8,796,030, 2014.
- Haushalter R. C.Rare Earth Downconverting Phosphor Compositions for Optically Encoding Objects and Methods and Apparatus Relating to Same. U.S. Patent 8,927,892, 2015.
- Zhang F.; et al. Rare-Earth Upconverting Nanobarcodes for Multiplexed Biological Detection. Small 2011, 7, 1972–1976. 10.1002/smll.201100629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedbala R. S.; et al. Detection of analytes by immunoassay using upconverting phosphor technology. Anal. Biochem. 2001, 293, 22–30. 10.1006/abio.2001.5105. [DOI] [PubMed] [Google Scholar]
- Hampl; et al. Upconverting Phosphor Reporters in Immunochromatographic Assays. Anal. Biochem. 2001, 288, 176–187. 10.1006/abio.2000.4902. [DOI] [PubMed] [Google Scholar]
- Corstjens P. L.; et al. Tools for diagnosis, monitoring and screening of Schistosoma infections utilizing lateral-flow based assays and upconverting phosphor labels. Parasitology 2014, 141, 1841–55. 10.1017/S0031182014000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook R. B.Process of Making Microparticles of a Thermally-Gelled Polysaccharide. U.S. Patent 6,248,268,1998
- Diffusion Data for Labeled Targets Diffusing into the Parallume Beads Containing Complementary, Bead-bound Captures Probes. http://parallume.com/beads.html#3-g.