

Abstract
The growing importance of biologics and biosimilars as therapeutic and diagnostic agents is giving rise to new demands for analytical methodology that can quickly and accurately assess the chemical and physical state of protein-based products. A particular challenge exists in physical characterization where the proper fold and extent of disorder of a protein is a major concern. The ability of NMR to reflect structural and dynamic properties of proteins is well recognized, but sensitivity limitations and high levels of interference from excipients in typical biologic formulations have prevented widespread applications to quality assessment. Here we demonstrate applicability of a simple one-dimensional proton NMR method that exploits enhanced spin diffusion among protons in well-structured areas of a protein. We show that it is possible to reduce excipient signals and allow focus on structural characteristics of the protein. Additional decomposition of the resulting spectra based on rotating frame spin relaxation allows separate examination of components from aggregates and disordered regions. Application to a comparison of two different monoclonal antibodies and to detection of partial pH denaturation of a monoclonal antibody illustrates the procedure.
Graphical abstract
Proteins, including antibodies, are increasingly being used as therapeutic and diagnostic agents. Assuring the quality and stability of preparations is more complex than in the case of a small molecule drug. Not only composition, but folding into a specific three dimensional structure, and maintaining that structure, becomes an issue2. As some of these early biologic products come off patent, production of biosimilars raises similar challenges in comparing generic products to innovator products. Methods for rapidly assessing this three dimensional, or higher order structure (HOS), have therefore become important. One dimensional proton NMR methods are, in principle, capable of assessing both composition and HOS, and doing so rapidly on multiple samples. However, there are challenges that arise in reducing these methods to practice. High concentration of excipients used to stabilize preparations during storage give strong signals that can obscure parts of a protein spectrum. All parts of the protein spectrum are also not of equal interest. Signals from less ordered parts are likely to increase in intensity as the structure begins to degrade, or they may vary from sample to sample if production conditions are not well controlled. It would be desirable to separate HOS signals from excipient signals, as well as separate signals of more disordered regions of protein from HOS signals, so evidence for changes in formulations could be more easily detected and assessed. Here we present an approach to meeting these challenges that capitalizes on efficient spin diffusion of protons in well-structured areas to eliminate excipient signals and extract spectra from HOS regions. Additional deconvolution of spectra based on translational diffusion and transverse spin relaxation rates is used to improve the quality of spectra and allow separation into sub-spectra representing less ordered and more ordered parts. Using monoclonal antibodies as a test case, we show that this approach makes it possible to distinguish different antibody constructs and detect minor structural variations well in advance of accepted denaturation points.
Many potential approaches have been suggested for monitoring structural characteristics of proteins, including circular dichroism, NMR, and mass spectrometry3,4. Few, however, offer the potential of NMR for probing both structure and dynamics of proteins at the single residue level. Much recent consideration has focused on standard two dimensional NMR methods such as 13C-1H and 15N-1H heteronuclear single quantum coherence (HSQC) spectra as a means of providing a fingerprint of a properly folded protein that can be compared to those from a range of samples5. Normally these experiments are quite time consuming, particularly if applied to samples without isotopic enrichment, and they are usually feasible only for smaller, highly soluble, proteins. However, there are special cases, such as the observation of 13C-1H methyl correlations, where observations on whole antibodies have been achieved6. The length of acquisition is still long, and a recent analysis has suggested that, for applications to large numbers of samples, alternate methods that depend on one dimensional (1D) proton NMR should be considered7.
The use of 1D proton NMR to characterize structural properties of proteins has a long history8. It is well known that line widths (or equivalently, transverse relaxation rates) are dependent on levels of internal motion and the size of independently tumbling structures, whether they be whole proteins, domains within proteins, or protein complexes. The chemical shift dispersion of resonances also carries information about secondary structure. More recently the additional problems of separating protein spectra from excipient signals and separating the HOS components of spectra from those of more mobile regions, including glycans of glycoproteins, have been addressed9. The procedure, referred to as protein fingerprint by line shape enhancement (PROFILE), relies primarily on translational diffusion editing using a pulse gradient stimulated echo (PGSTE) sequence and spectral subtraction of a reference spectrum to remove signals from excipients. A sharp line fingerprint is extracted by post-acquisition processing of the resulting spectrum. The advantages of the simplicity of the process and the utility of the sharp line fingerprint are well demonstrated. However, it is often difficult to exactly reproduce sample conditions in a reference sample, and the existing procedure does not take advantage of a unique characteristic of a well-structured protein, namely rapid spin diffusion among protons in HOS regions.
Here we explore the use of spin-diffusion among protons in the protein's structured regions to select for HOS spectral components during acquisition. The use of spin-diffusion to differentiate highly structured from less structured proteins is not new. It was suggested some time ago as a means of distinguishing molten globule and random-coil proteins from well folded proteins10. Experiments rely on the fact that in rigid parts of a large protein, energy conserving, paired spin-flips (zero quantum transitions) of interacting protons are far more efficient than spin-flips that restore proton magnetization to equilibrium. Irradiation of a resonance anywhere in the spectrum that is unique to the protein then leads to saturation of all resonances from interacting protons, essentially resonances from the entire HOS component, and many from less ordered areas as well. Subtracting a spectrum acquired immediately after saturation from a control that is irradiated off-resonance removes resonances from all non-protein components in a sample, including those from excipients. The off-resonance control substitutes for, and is arguably superior to, a separately acquired reference spectrum; acquisition on the actual sample under investigation eliminates the mismatches that can occur in separately prepared samples.
The suggested use of spin diffusion effects is very similar to what is done in the Saturation-Transfer Difference (STD) spectra that are applied to epitope identification on ligands that bind to large proteins11. Here, resonances from protons on exchanging ligands that interact sufficiently with protein protons are also saturated by spin diffusion and survive in the difference spectrum. The main difference between the application proposed here and an STD experiment is that the latter uses an R1ρ (rotating frame transverse relaxation) filter, that exploits the rapid decay of broad lines from protein, to suppress protein resonances. By eliminating or minimizing this filter in our application we retain protein resonances.
The substantial interest in parts of the protein exhibiting less ordered structure (glycans added to proteins as posttranslational modifications, mobile parts in improperly folded segments) makes it important to decompose spectra into more ordered and less ordered parts. R1ρ (the inverse of T1ρ) is dependent on motional correlation times that are expected to decrease for more mobile or disordered parts of a protein. Separating spectra into sub-spectra for highly ordered and less ordered parts was done previously by taking differences of smoothed and un-smoothed spectra. Here we have taken an approach that decomposes spectra based on R1ρ values extracted from a series of spectra with different R1ρ delays.
Experimental Design
The NMR pulse sequence was designed starting from a standard STD sequence. It is depicted in Figure 1. In addition to the indicated phases, the pulses within the diffusion filter element were independently phase cycled, along with the receiver, to minimize artifacts. The Gaussian cascade (shaded shaped pulse) accomplishes saturation when RF is set to methyl or aromatic regions of the spectrum. Acquisitions with saturation in the protein region and alternately off-resonance (no protein saturation) are separately summed in a file and subtracted during processing of the data. Because there may be formulation components that interact with the protein and produce STD-like signals, and because complete suppression of excipient resonances with just the STD element is difficult, we include as an option a PGSTE diffusion filter element in the sequence12. It can be implemented as a filter using magnetic field gradients (g2) set to a single high value, or as an array set to a number of gradient strengths. In the latter case a MATLAB script is used to extract diffusion constants and these are used to decompose spectra into protein and excipient spectra. The sequence also includes an R1ρ (T1ρ) relaxation element that applies a strong rf field parallel to the magnetization produced by the preceding 90° pulse, as in the normal STD experiment (shaded square pulse), but here the R1ρ delay is implemented as an array and MATLAB scripts are used to decompose spectra into highly ordered and less ordered parts. The following pair of 90° pulses and intervening pulsed gradient (g1) serves as a z filter to eliminate phase artifacts. Some versions of STD sequences use an R2 (T2) filter in place of an R1ρ filter, but R2 delays cause J modulation that complicates use of decomposition scripts that assume a simple exponential decay of spectral elements. R1ρ elements also introduce some unwanted modulation, which is dependent on rf field strength and spectral offset, but this is more predictable and can be eliminated by choosing R1ρ delay times that occur near modulation maxima, or by compensating for modulation in the decomposition scripts.
Figure 1.
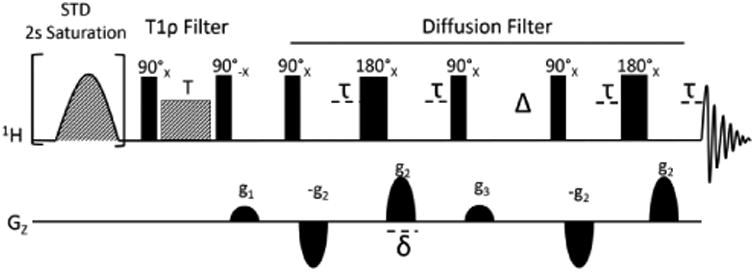
STD-PGSTE pulse sequence created to exploit spin diffusion in monoclonal antibodies.
The final signals acquired are functions of pulsed field gradient strengths (g2), gradient durations (δ), the translational diffusion delay (Δ), magnetogyric ratios (γ) and diffusion constants (D), as well as R1ρ delays (T) transverse relaxation delays (τ) and molecular relaxation rates (R2 and R1ρ). This dependency is detailed in equation 1. The equation can be normalized relative to the initial intensity and parameters collected in the constants, const1 and const2, as shown in equation 2. Const1 is linearly dependent on the translation diffusion constants of various spectral components, D, when g2 is used as an array variable. Alternatively, it is dependent on rotating frame relaxation rates, R1ρ, of various components when T is used as an array variable. Const2 contains other terms that remain constant over collection of arrays. These constants are fit, spectral point by spectral point, in MATLAB routines that fit data at various values of g2 or T to the multiple exponential decays of equation 2 or to the linear decay of equation 3.
| Eq. (1) |
| Eq. (2) |
| Eq. (3) |
The resulting diffusion constants, D, or rotating frame relaxation constants, R1ρ, found by fitting each spectral point in the 1D spectrum are clustered using a kmeans algorithm. The choice of number of clusters and extraction of representative constants from the clusters is somewhat subjective. However, a simple procedure in which constants are repeatedly clustered specifying an increasing number of clusters makes the process more objective. As the number of clusters rise, one frequently sees clusters with low populations appear; these can be ignored, and clusters with similar average values can be combined to minimize the number of clusters. Average constants from the 2-4 most representative clusters are then used to decompose spectra into excipient and protein spectra, or highly ordered and less ordered spectra respectively. A non-negative matrix factorization routine in MATLAB is used to accomplish this separation. Relevant MATLAB scripts are provided as supporting information.
In the results section below we describe application of this sequence to two distinct antibody preparations. Both have high levels of excipients. Suppression of signals from excipient components by factors approaching 1000 is demonstrated. Separation of protein spectra into what is believed to be representative of a HOS monomer and a HOS aggregate is shown for both proteins. Comparison of the spectra in the monomeric state proves very useful. In the case of one antibody we demonstrate an ability to detect partial denaturation by lowering the pH to a point about 1.5 pH units above the accepted denaturation pH and attempt to use the sub-spectrum for less ordered parts to identify segments in the protein sequence most amenable to pH denaturation. We conclude that the 1D proton NMR spin-diffusion approach will prove a viable and efficient means of screening antibody samples for consistency in production and stability over time.
Materials and Methods
Monoclonal antibody (mAb) samples, dissolved in or lyophilized from formulation buffer, were provided by Pfizer Inc. (St. Louis, MO, USA). The first, designated mAbT, is an IgG2 monoclonal antibody and the second, designated mAbB, is an IgG4 monoclonal antibody. Both mAbs are highly concentrated in the original formulation, being 75μM and 225μM respectively. Both are in high concentrations of excipients with the principal component being trehalose (245 mM) in the first case and sucrose (234 mM) in the second case. Solvents and other reagents were supplied by Sigma-Aldrich.
Sample Preparation
The mAb samples, which had not been previously lyophilized were lyophilized, all samples were then dissolved in 99.8% pure D2O to bring them back to their original concentrations. We found that dilution from this initial concentration greatly improved the spectral quality. Therefore, a75 μL aliquot was taken from the sample and 225μL D2O was added to provide a 300 μL sample with a four-fold dilution. The diluted sample was placed into a Shigemi NMR tube and the insert was lowered to produce a sample height of 1.5 cm.
NMR Experiments
All experiments presented were collected on a 900 MHz Agilent DD2 system at 25°C using a triple resonance cryoprobe. To generate a difference spectrum from acquisitions with and without spin diffusion among protons of the protein, the saturation pulses at the beginning of the pulse sequence were alternately centered on a spectral region that corresponds to the protein and another region that was far off resonance. To achieve this, the Pbox function in VNMRJ was used to create two Gaussian shaped pulses, one of which was set to saturate at 0.3 ppm and the other to saturate at 18 ppm. The rf level for this saturation sequence was set to 10 db (110Hz). R1ρ delays (T) ranged from 0 to 2 ms with the rf level set to 52 db (14900 Hz), and diffusion gradient pulses of 0.5 ms (δ) ranged from 2 to 40 G/cm (g2) with the translational diffusion delay (Δ) set to 200 ms. Other delays and pulse gradients were kept to a minimum (200-250μs, and 3-6 G/cm). Typical data collections employed a 2 s saturation period during which spin diffusion occurred and a 0.75 s acquisition time. 64-128 transients were collected over 8192 complex points covering 16 ppm at each of the specified set of R1ρ delays and diffusion gradients. For a fixed high gradient value and an array of ten R1ρ delays typical acquisition times were 1-2 hrs.
Once the data had been collected, the two separately stored (off and on resonance) spectra were subtracted to yield a difference spectrum with reduced excipient signals. Initial data processing was done with Mnova software (Mestrelab Research). Line broadening of 10 Hz preceded Fourier transformation, manual phasing, and baseline correction using the ablative function with 15 points and 15 passes. Parameters were set on the first spectrum of an array and used subsequently on all other spectra. The spectra were exported as an ASCII file and processed further in MATLAB as described in the text.
Results
We first explore the extent to which a spin diffusion element alone can eliminate excipient resonances and still reproduce the native protein spectrum. Figure 2C shows a spectrum of antibody B acquired with the spin-diffusion sequence, but with the gradients and delays in the diffusion element set to minimum values and the R1ρ element set to zero. It is compared to a spectrum acquired with a basic 90° single-pulse experiment on the same sample (2A) and to a spectrum similarly acquired on a sample that was dialyzed against a 10 fold excess of 20 mM sodium phosphate buffer at pH6.5 three times (2B). The 90° pulse spectrum is dominated by excipient resonances (mostly sucrose) to the extent that the protein spectrum is nearly unobservable. The spin diffusion element alone shows a suppression of the largest component of the formulation by a factor of 60, giving a very reasonable protein spectrum. Comparison to the spectrum of the dialyzed sample suggests that most components of the protein spectrum are well preserved. Using a single strong gradient to further suppress signals from small molecules, or a gradient array to separate spectra of rapidly diffusing small molecules from spectra of slowly diffusing proteins effectively suppresses the remaining signals from the excipients. In the single strong gradient case (39G/cm) suppression is by a factor of 350 (Figure 2D). Use of a gradient array to decompose spectra of rapidly and slowly diffusing species effectively eliminates remaining excipient signals. We present an example of spectral decomposition based on diffusion constants in supporting information. However, collecting the gradient array of spectra required for decomposition (6-8 points running from gradient values of 3.7 G/cm to 39 G/cm) is time consuming, particularly if combined with a second array of R1ρ delays. We therefore recommend this double array procedure only if small molecule components, perhaps those produced by proteolytic degradation, are of interest.
Figure 2.
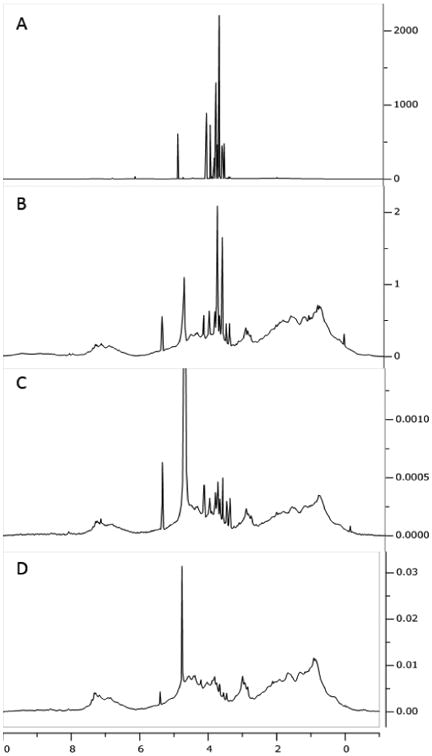
Comparison of NMR spectra for mAbB. 90° single-pulse experiment (with water presaturation) with excipient (A); 90° single-pulse experiment (with water presaturation) on a sample dialyzed against 20 mM sodium phosphate (B); spin diffusion experiment with a low gradient (C); and spin diffusion experiment with a high gradient (39 G/cm) (D).
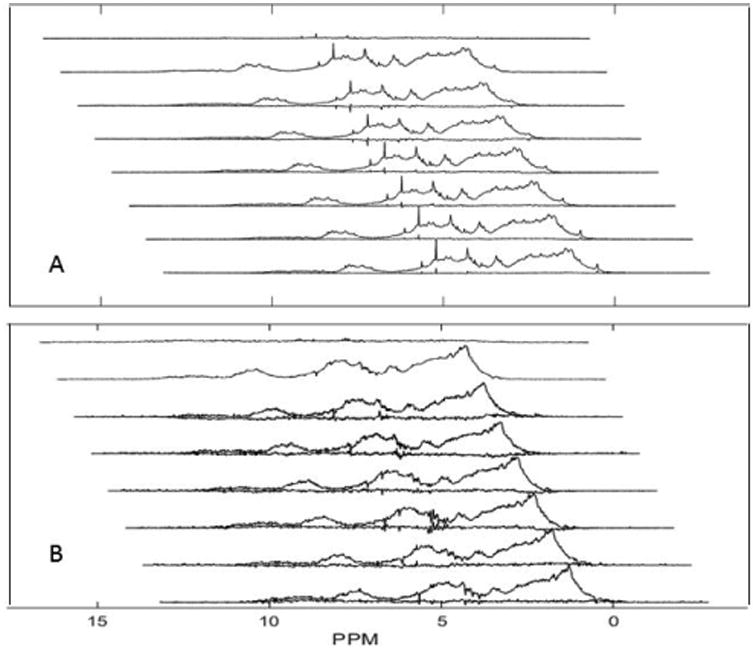
Figure 3 shows an array of spectra in which R1ρ delays vary from 0 to 2.05 ms. A relatively low fixed gradient value of 3.7 G/cm was chosen for this illustration so that the effect on resonances from residual excipients, which have small R1ρ values, can be seen. As delays increase excipient peaks decay relatively little, while resonances from protein decay substantially. Close examination suggests a slight sharpening of spectra at the longer R1ρ values, but deconvolution of these features is better done mathematically.
Figure 3.
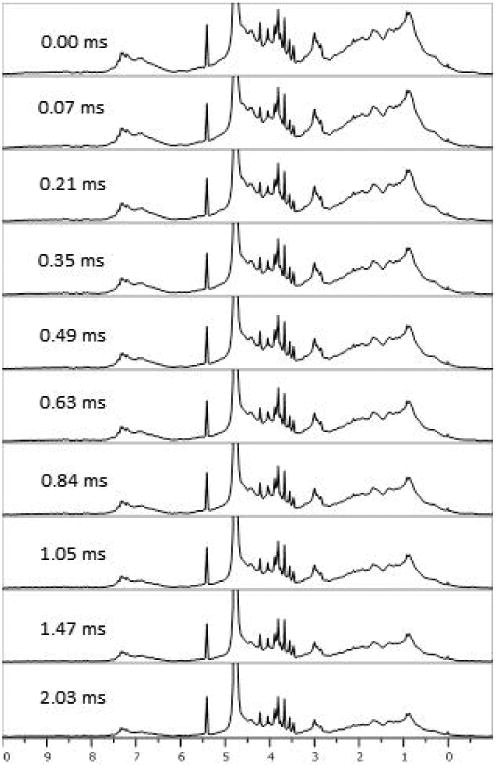
STD-PGSTE spectra of a 125 μM mAbB. A low gradient value was used (3.7 G/cm) while the R1ρ delays were arrayed from 0 to 2.05 ms.
Decompositions of similar sets of spectra at 10 different R1ρ delays, but with a high gradient value (39 G/cm) for better excipient suppression are shown for mAbT in Figure 4. Two R1ρ values were extracted at each spectral point by fitting data to a bi-exponential decay, and these values were clustered to extract a minimal set for spectral decomposition. Two large clusters were identified and average R1ρ values of 250 and 3000 s-1 were found. These values were then used to decompose the spectra into broad and narrow spectral components. The acquisition and processing was repeated 6 times with duplicate runs on three different samples of the same antibody preparation to provide a measure of reproducibility in the spectra. In each case the spectrum is shown with the residual compared to the mean under the spectra. The average residual and average spectrum are shown at the top. The largest components in the R1ρ spectra are from water (4.75 ppm) and the anomeric resonance from trehalose, the major component of the formulation buffer (5.19 ppm) for mAbT. It is not surprising that even a few tenths of a percent variation in signals, which are initially very intense, could result in observation in residuals of processed spectra. It is surprising that the anomeric resonance is the only trehalose resonance to show a substantial residual. It is possible that selective transfer through spin diffusion from the protein accentuates this resonance. Outside of these excipient and water signals the residuals are small, less than 4% of the maximum intensity in the protein spectrum. This value provides one basis for accessing validity when comparing spectra from different antibodies and antibodies prepared under different conditions.
Figure 4.
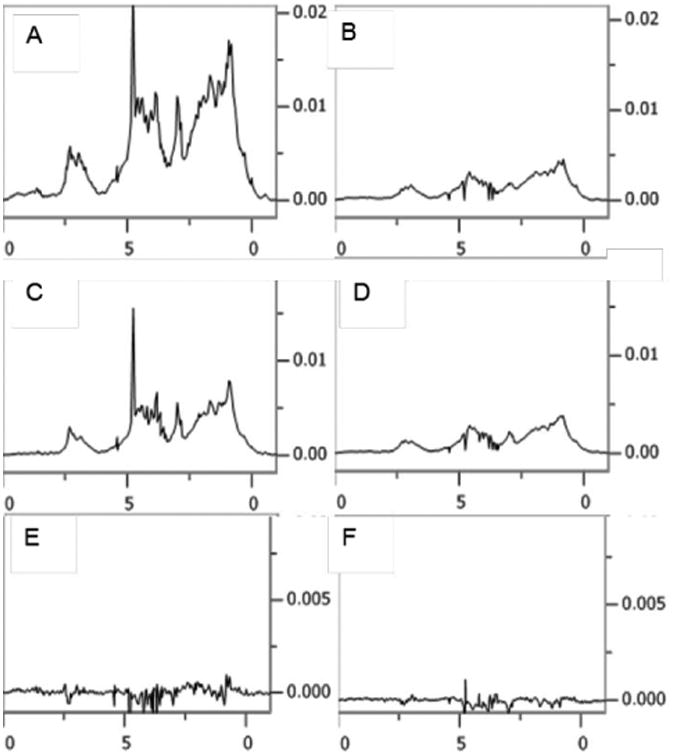
Residuals comparing individual decompositions to means for antibody T at 125 μM. (A) represents sharper resonances using an R1ρ of 250 s-1. (B) represents broader resonances using an R1ρ of 3000 s-1.
Poppe et al. have suggested an alternate measure of similarity that reduces differences across the entire spectrum to a single number that is independent of spectral amplitudes7. It is based on the deviation of a correlation coefficient from 1.0. 1.0 results when spectra are identical and zero results when spectra have no similarity. To expand the scale for very similar spectra, and still retain a measure that increases with more similar spectra, the measured coefficient is subtracted from one and 10 times the negative log of the resulting value is used to put measures on the familiar db scale. Masking residual excipient peaks and using the spectral area from -1 to 10 ppm, the average similarity score for spectra in Figure 4 (relative to their mean) is 32 db. This will give a sense of a score for spectra only differing only by experimental noise.
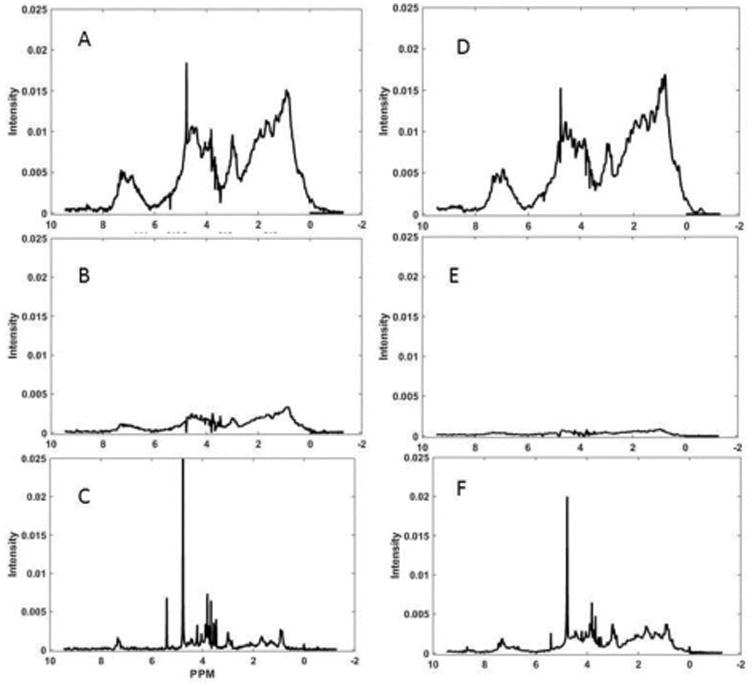
An R1ρ array for mAbB was analyzed using a similar set of R1ρ values. Figure 5 shows a comparison of decomposed spectra for mAbB and mAbT. The sharper and broader protein spectrum components for mAbB are shown in A and B. The sharper and broader spectrum components for mAbT are shown in C and D. Variations in amino acid composition and significant structural differences should be reflected in changes in the spectral profile for these components. While there are significant intensity differences, some of which are due to variations in antibody concentration, changes in the spectral profile are hard to see. To accentuate changes in the profile the differences between spectral components (A-C and B-D), scaled to minimize the sum of differences, is presented in E and F for the sharper and broader components respectively. This procedure will eliminate most differences arising from variation in sample composition and response to diffusion or relaxation filters, and leave differences arising from variation in chemical shift or line width profiles. The broader spectrum differences are smaller as might be expected for broader lines. However, there are a number of discrete features in the sharp component difference spectrum that are clearly significant compared to the residuals of Figure 4. In particular the downfield part of the aromatic regions near 7.3 ppm is more intense for mAbT with some contrasting increase in the more upfield part of the aromatic region for mAbB. In comparison, the upfield methyl region near 1 ppm is more intense for mAbB with some contrasting loss in intensity for the downfield methyl region of mAbT. There is also a general increase in the methylene region between 2 and 2.5 ppm for mAbB. In terms of similarity, the spectrum in A is similar to that in C by a score of 17 db, certainly much less similar than the controls in Figure 4. In principle, the departures in similarity can be interpreted in terms of differences in amino acid composition or alteration in structure.
Figure 5.
Comparison of spectral components of mAbB and mAbT. Sharper spectral components are in (A) and (C) respectively. Broader spectral components of mAbB and mAbT are in (B) and (D) respectively. The scaled differences between mAbT and mAbB in sharper (A-C) and broader components (B-D) are shown in (E) and (F) respectively (note the factor of 2 increase in vertical intensity scale for E and F). Horizontal axes are in ppm.
A question arises as to the interpretation of the broader components in the decomposition discussed above. If we consider R1ρ relaxation rates for most protons to arise from interactions with three other protons at 2 Å, an R1ρ value of 250 s-1 is near what one expect for protein domains of 50-100 kDa. This value of R1ρ should, therefore, extract a spectrum that we would associate with HOS of a monomeric protein. An R1ρ value of 3000 s-1 is well out of this range and can only correspond to the presence of HOS in an aggregate. The tendency to form some higher order assemblies would be consistent with our observed increase in signal strength as samples were diluted four fold. In recognition of this additional complication, subsequent analyses were conducted using a larger number of clusters for R1ρ, selecting the two most populated as representative of HOS in monomer and aggregate.
In an effort to explore our ability to detect spectral components from a less ordered region we examined mAbB at a pH of 4.5 in addition to the initial pH of 6.5. This antibody is known to undergo denaturation at ∼ pH 3.0. The question is whether the methods presented can detect some structural changes well before accepted denaturation conditions are reached. The larger set of clusters gave two well populated clusters with R1ρ values of 500 s-1 and 5500 s-1 that could be used to represent HOS in monomers and aggregates. A third R1ρ value was added to represent less ordered components in the decomposition (60 s-1). Figure 6 shows a comparison of spectra decomposed into sharper and broader HOS components and a less ordered component. There are again some differences in intensity between the two samples, the pH 4.5 spectrum being more intense. Therefore, the spectra for the pH 6.5 sample were scaled up to minimize differences between spectra at an R1ρ delay of zero (factor of 1.8). The profiles of the sharper HOS components (A and D) appear to be similar, but not identical. They have a similarity score of 22 db, one that is higher than for our comparison of different antibodies, but lower than our control score.
Figure 6.
Comparison of spectral components of mAbB at pH 6.5 and pH 4.5. Sharper HOS components at pH 6.5 and 4.5 are shown in A and D respectively. Broader HOS components at pH 6.5 and 4.5 are shown in B and E respectively. Comparison of less ordered components pH 6.5 and pH 4.5) are shown in C and F respectively.
Despite the similar profiles, the change in the intensity of the broad HOS component as pH is lowered to 4.5 is substantial (Figure 6E compared to 6B). This suggests less aggregate in the sample. Lowering the tendency to form aggregates can be understood in molecular terms. The pI of this protein is predicted to be 7.6. Lowering the pH to 4.5 would lead to increased positive charge on each element of an aggregate and a decreased tendency to aggregate. There is a small corresponding increase in the sharp HOS component as pH is lowered (Figure 6D in comparison to 6A), consistent with a shift in equilibrium toward a monomeric species. There are clearly some minor changes in the profiles, but these are difficult to separate from the overall changes in intensity.
A more distinct difference is seen on comparing less ordered components (60 s-1 R1ρ, Figure 6F to 6C). These spectral components contain strong signals from excipients and water, but also several signals that are clearly from the protein. In particular there is a relative increase in aromatic components around 7 ppm, methylene components in the 2-2.5 ppm region and components in the 3 and 4-4.5 ppm regions. This is in contrast to the relatively small change in the methyl region near 1 ppm. These features are well above the residuals shown in Figure 4 and are open to a molecular level interpretation in terms of parts of the protein that may preferentially unfold as pH is lowered.
Discussion
The proton 1D spectral deconvolution methods presented above would seem to provide an effective protocol for quality control of antibody samples and other protein based therapeutics. Applications to detecting variations in protein structure during production, as well as on storage, certainly seem possible. Comparisons of biosimilars to innovator products should also be possible. Similarity measures, based on correlation coefficients between 1D spectral vectors of a reference sample and a test sample as applied to data in Figures 5 and 6, could provide a means of quantitating these differences7.
Among the many advantages of proton NMR 1D methods is the minimal sample preparation required, the uniform distribution of detectable sites throughout a protein, the quantitative relationship between resonance intensity and sample content, and the potential for a molecular interpretation of spectral changes. The samples studied here had been lyophilized from their formulation buffer and then simply dissolved in D2O containing DSS for a chemical shift reference. However, it may be possible to eliminate the lyophilization step. Our procedures typically dilute the lyophilized sample to one fourth the formulation concentration. Some preliminary work dissolving samples in 75% D2O and 25% H2O suggests that is may be possible to collect similar data by just diluting an off-the-shelf sample, with a 3 fold excess of D2O.
The minimum variation that can be detected in the experiments presented is ultimately dependent on the signal to noise ratio of the spectra. This in turn is dependent on the length of time devoted to acquisition. For the data presented in Figure 4 10 different R1ρ delays were used with acquisitions requiring 1-2 hours. Sensitivity scales linearly with sample concentration and as the square root of time invested in acquisition. Under the conditions used we estimate that we could detect a change in spectral content of about 4% of maximum signal amplitude in a 1D protein NMR spectrum. For the comparison of antibodies B and T shown in Figure 5, the integral of the aromatic peak near 7.3 in the difference spectrum represents 12 % of the maximum intensity of the Figure 5A spectrum. Sensitivity also depends on the capabilities of instrumentation. Here we used a very high field spectrometer, primarily to more fully explore changes as a function of chemical shift, but also to maximize sensitivity. However, many comparisons do not require a high level of chemical shift resolution. In these cases lower field instruments can be used, and additional signal averaging can compensate for sensitivity differences.
While many applications require minimal spectral interpretation, for example assigning a single similarity score to alternate preparations or to different biosimilars, it is tempting to try to interpret some differences on a molecular basis. The changes in line shape and changes in particular chemical shift regions shown in Figure 6 for mAbB on decreasing pH are a good example. The sharp HOS (monomer) component spectra in 6A and 6D associated with R1ρ value of 500 s-1 show some indication of sharper lines for the pH 4.5 sample with little change in profile. Refitting and re-clustering R1ρs specifically for the pH 4.5 data do, in fact yield somewhat smaller R1ρ values. This may reflect a general loosening of the structure. Changes that occur between pH 5.5 and pH 3 have previously been described as a lower extent of interaction between domains13. This could result in a general increase in motion of individual domains and a decrease in resonance line widths with no significant changes in resonance positions.
The changes do, however, seem more complex when we consider the appearance of peaks in discrete regions of the spectral component associated with the smaller R1ρ value assigned to less ordered regions (Figure 6F). The increases in signals in the 7.0 ppm, 4.0 ppm, 3.0 ppm and 2.0 ppm regions in contrast to other regions is more indicative of selective unfolding of protein segments that have amino acid compositions that depart from those of the whole protein. It is well documented that antibodies do not unfold in a single cooperative event14-16. Several studies have focused on a region of the C2H domain as showing increased conformational sampling under, thermal and pH induced unfolding13,17, as well as with deglycosylation18. It would be of interest to see if the chemical shifts of the peaks showing changes in our difference spectrum correlate with amino acid compositions of specific regions in the peptide sequence of antibody B.
The peptide sequence for mAbB is provided in Figure 7B. IgG antibodies have two chains, a heavy chain and a light chain. Each is folded into domains of about 100 amino acids. The light chain has two, one variable (VL) domain and one constant domain (CL). The heavy chain has four, one variable domain (VH) and three constant domains (C1H, C2H, and C3H). A homo dimer is formed by association of two copies of the C2H, and C3H domains to make the Fc substructure and two heterodimers are formed by association of VL and CL with VH and C1H to make Fab substructures. Each domain differs somewhat in amino acid composition and one might expect these to have different chemical shift profiles.
Figure 7.
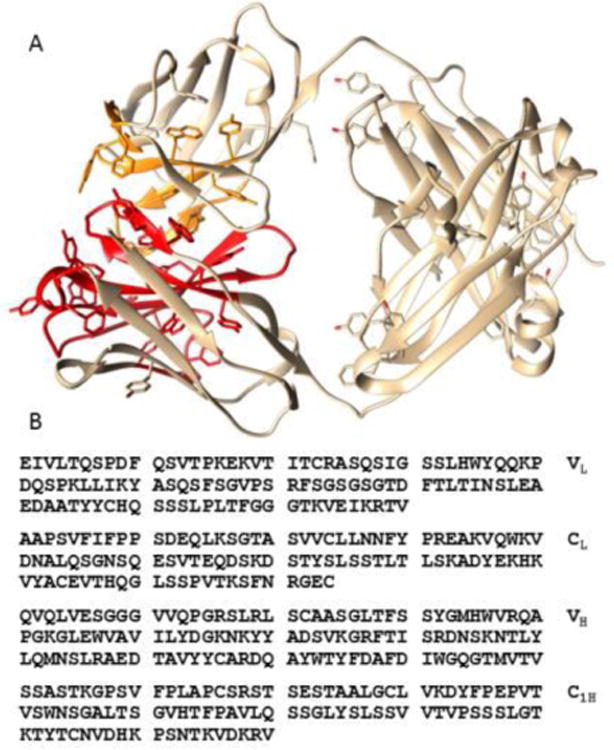
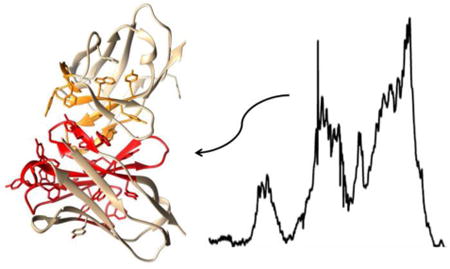
Amino acid sequence of Fab of mAbB (B) and structure of homologous region from canakinumab (A) (4G5Z). Aromatic amino acid rich regions of VH are shown in red. Aromatic amino acid rich regions of VL are shown in orange. Molecular graphics were produced with UCSF Chimera supported by NIGMS P41-GM1033111
The 7.0 ppm spectral region coincides with predictions for aromatic proton resonances found in tyrosine, phenylalanine, or tryptophan. The 2.0 ppm peak coincides with methylene protons in proline, lysine, arginine, glutamic acid and glutamine, and the peaks in the 4.0 ppm region coincide with predictions for proton resonances found in glycine, serine, and threonine. The most distinctive feature, of the sharp component spectrum in Figure 6F, may be the increased intensity of peaks in the aromatic region near 7.0 ppm. Examining a whole antibody crystal structure (1HZH)19 with moderately high sequence identity to mAbB (79%) one finds a region involving the first heavy chain domain (VH) and a contact region on the first light chain domain (VL) that is particularly rich in aromatic amino acids. This persists in a crystal structure of the Fab fragment of the therapeutic antibody, canakinumab20, which is 87% identical to mAbB in VH -C1H and 98% identical in the light chain. This Fab fragment is depicted in figure 7A. These regions in mAbB correspond approximately to 25-70 and 90-115 on the heavy chain and 31-36, 49-56 and 86-100 on the light chain. Analysis of amino acid composition of this region shows more than 20% aromatic residues compared to 9% for the more structured Fc region of the heavy chain and 10.3% for the entire heavy chain. Some pH induced unfolding specifically in this region could account for the shift in intensity in the aromatic region of the sharp component spectrum. There are also regions predicted to be disordered before and after the heavy chain regions mentioned above21. These regions are rich in serine, glycine, and hydrophilic amino acids. They may account for the increases in the 2.0 and 4.0 ppm regions if they participate in the unfolding.
A suggestion that the VH-VL part of the full antibody may prematurely begin to unfold as pH is lowered is consistent with some thermodynamic data. The variable domain of the light chain and the variable domain of the heavy chain have been suggested to act as a cooperative unit, and that this unit is typically less stable than a cooperative unit formed by the constant parts of the heavy chain and light chain22. While a structural interpretation of changes in resonance intensities at particular chemical shift values must remain open to question in the absence of experiments directed at specific assignments, such an analysis can be useful in directing future research.
Supplementary Material
Acknowledgments
This work benefitted from instrumentation provided in part by the Georgia Research Alliance and the NIH through a shared instrumentation grant, S10 RR027097.
Footnotes
Associated Content:Supporting Information. An illustration of additional suppression of excipient using a pulsed field gradient deconvolution is provided. In addition, MATLAB scripts for the extraction of R1ρ or diffusion decay constants, clustering of these constants, and deconvolution of spectra based on these constants are provided.
Notes. The authors declare no competing financial interest.
References
- 1.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 2.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Nature Reviews Drug Discovery. 2012;11:527–540. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soergel F, Lerch H, Lauber T. Biodrugs. 2010;24:347–357. doi: 10.2165/11585100-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Drug Discovery Today. 2014;19:95–102. doi: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubin Y, Gingras G, Sauve S. Analytical Chemistry. 2008;80:2623–2627. doi: 10.1021/ac7026222. [DOI] [PubMed] [Google Scholar]
- 6.Arbogast LW, Brinson RG, Marino JP. Analytical Chemistry. 2015;87:3556–3561. doi: 10.1021/ac504804m. [DOI] [PubMed] [Google Scholar]
- 7.Poppe L, Jordan JB, Rogers G, Schnier PD. Analytical Chemistry. 2015;87:5539–5545. doi: 10.1021/acs.analchem.5b00950. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann B, Eichmuller C, Steinhauser O, Konrat R. In: Nuclear Magnetic Resonance of Biological Macromolecules. James TL, editor. Part C. 2005. pp. 142–+. [Google Scholar]
- 9.Poppe L, Jordan JB, Lawson K, Jerums M, Apostol I, Schnier PD. Analytical Chemistry. 2013;85:9623–9629. doi: 10.1021/ac401867f. [DOI] [PubMed] [Google Scholar]
- 10.Kutyshenko VP, Cortijo M. Protein Science. 2000;9:1540–1547. doi: 10.1110/ps.9.8.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer B, Peters T. Angewandte Chemie-International Edition. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 12.Altieri AS, Hinton DP, Byrd RA. Journal of the American Chemical Society. 1995;117:7566–7567. [Google Scholar]
- 13.Vlasov AP, Kravchuk ZI, Martsev SP. Biochemistry-Moscow. 1996;61:155–171. [PubMed] [Google Scholar]
- 14.Rowe ES. Biochemistry. 1976;15:905–916. doi: 10.1021/bi00649a028. [DOI] [PubMed] [Google Scholar]
- 15.Cerasoli E, Ravi J, Garfagnini T, Gnaniah S, le Pevelen D, Tranter GE. Analytical and Bioanalytical Chemistry. 2014;406:6577–6586. doi: 10.1007/s00216-014-7970-x. [DOI] [PubMed] [Google Scholar]
- 16.Vermeer AWP, Norde W. Biophysical Journal. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Mabs. 2015;7:84–95. doi: 10.4161/19420862.2014.985494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Analytical Chemistry. 2009;81:2644–2651. doi: 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saphire EO, Parren P, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 20.Blech M, Peter D, Fischer P, Bauer MMT, Hafner M, Zeeb M, Nar H. Journal of Molecular Biology. 2013;425:94–111. doi: 10.1016/j.jmb.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 21.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothlisberger D, Honegger A, Pluckthun A. Journal of Molecular Biology. 2005;347:773–789. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.