

Abstract
In its “grand challenge” format in chemistry, “synthesis” as an activity sets out a goal that is substantially beyond current theoretical and technological capabilities. In pursuit of this goal, scientists are forced across uncharted territory, where they must answer unscripted questions and solve unscripted problems, creating new theories and new technologies in ways that would not be created by hypothesis-directed research. Thus, synthesis drives discovery and paradigm changes in ways that analysis cannot. Described here are the products that have arisen so far through the pursuit of one grand challenge in synthetic biology: Recreate the genetics, catalysis, evolution, and adaptation that we value in life, but using genetic and catalytic biopolymers different from those that have been delivered to us by natural history on Earth. The outcomes in technology include new diagnostic tools that have helped personalize the care of hundreds of thousands of patients worldwide. In science, the effort has generated a fundamentally different view of DNA, RNA, and how they work.
One challenge in synthetic biology is to recreate certain properties of life (e.g., evolution) using unnatural genetic and catalytic biopolymers. Many efforts have centered on artificial genetic systems (e.g., AEGIS).
On the occasion of the 90th birthday of Albert Eschenmoser, a master of synthesis.
Many have noted that the phrase “synthetic biology” has had no consistent meaning among the communities that have used it over the past 40 years (Brent 2004). This inconsistency is reflected in the literature. To some, synthetic biology means simply “synthesizing a lot of DNA,” perhaps even entire genomes (Ellington 2016; Glass 2016). To others, synthetic biology is a new name for the much older field of metabolic engineering, but on a grander scale than modestly constructing a microbe that manufactures a single natural product using a single heterologously expressed gene (Lechner et al. 2016). To others, “synthetic biology” is the redirecting of information in living systems, perhaps to create a microbial platform for further engineering (Gaj et al. 2016). To these can be added concepts not represented in this series, such as the construction of devices that use natural nucleic acids and proteins as biobricks (Smolke 2009; Win et al. 2009), perhaps to test a theory about how those parts work together naturally (Prehoda et al. 2000; Dueber et al. 2004). Others use “synthetic biology” as suggested by Eric Kool, to mean the use of unnatural molecules in the context of natural biological systems (Rawls 2000). In its original definition, synthetic biology meant the creation of artificial life (Leduc 1912).
We have ourselves long held the view that synthesis is not a field, but rather an activity, an activity that derives value not only from products that it creates, but also from what is learned as the syntheses are attempted (Sismour and Benner 2005a). Indeed, the heroes of synthesis in classical organic chemistry often chose targets that had no product value at all (Woodward 1968; Kishi 1989). Instead they chose targets that would present a “grand challenge,” a molecule whose synthesis was beyond current capabilities, whose pursuit would therefore challenge molecular theory.
Here, synthesis does something that “hypothesis-directed research” cannot. When scientists control the hypotheses that they test, they often strategically limit their activities to safe hypotheses that are likely to be true. If they do not, then their funding agencies will. Thus, “hypothesis-based research” tends to not challenge core convictions.
In contrast, synthesis in pursuit of a “grand challenge” forces scientists across uncharted grounds, where they must ask and answer unscripted questions. Thus, a well-selected synthetic grand challenge tests all of the theories and assumptions that go into any strategic synthetic plan. Many of these are unstated; the scientists involved might not even realize that they are making them. As a result, synthesis can drive discovery and paradigm change in ways that hypothesis cannot.
For example, in his classic defense of grand challenge synthesis, Woodward (1968) discussed his choice, with Albert Eschenmoser, of vitamin B12 as a synthetic target. In 1965 (and in some sense still), B12 was the most complicated nonpolymeric natural product known. The product of the synthesis itself had no commercial value; fermentation was already generating B12 for pennies per unit. However, the effort led to the discovery of the intimate relation between molecular reactivity and molecular orbital structure. That discovery, captured as the Woodward–Hoffman rules (1970), was later recognized by a Nobel Prize (Fukui 1982; Hoffmann 1982).
The celebrated total synthesis of the genome of a bacterium by Venter and his coworkers (Gibson et al. 2010) was, of course, nothing more (and nothing less) than the total synthesis of a natural product, one that happens to be involved in microbial inheritance. Venter reportedly said that he undertook this challenge because someone told him it “could not be done.” This “someone” was certainly not a synthetic organic chemist. The credo of chemistry, for at least 50 years, has held that if a molecular structure can be drawn, and if the arrangement of atoms that it represents is an energy minimum, then the molecule can be synthesized given enough effort and money. That credo rests on many successful syntheses; some examples include tetrodotoxin (which is barely an energy minimum) (Kishi et al. 1972) and palytoxin (whose complexity is almost boring) (Fig. 1) (Kishi 1989).
Figure 1.

The “grand challenge” target. Vitamin B12 (left) was synthesized to drive chemical theory, not because it was a multistep tour de force or because the product itself was valuable. Tetrodotoxin (middle) is barely stable, but was nevertheless synthesized (Kishi et al. 1972). Palytoxin (right) is not a biopolymer, although its structure reveals a biosynthetic pathway that involves repetitive assembly of building blocks; chemists could synthesize this also (Kishi 1989).
Despite that credo, chemists in the early 1980s fully understood that “structure theory” in chemistry had broad deficiencies. In particular, that theory could not tell us “what” molecules to synthesize to create a desired molecular behavior. This was especially true if the grand challenge goal related to artificial life (Leduc 1912). Chemical theory could not then, and still cannot, meet the following easy-to-express challenge: “Draw me structures of some molecules that, if synthesized, will together have the properties that we value in living systems.”
Indeed, even simpler tasks were (in 1980) and remain (today) beyond the power of chemical theory. Despite advanced computers, advanced theory (Pople 1999), and decades of effort, we still cannot predict the solubility of salts in water, the packing of organic crystals (Dunitz and Bernstein 1995), or the freezing point of water. Yet these “simple” processes are the elements of the molecular interactions that generate the molecular behaviors that we value in biology. We cannot create medicines by direct design. We cannot create molecular electronic devices by direct design. We cannot engineer self-assembling materials by direct design. We get these today only by trial, error, intuition, and experiment.
However, DNA has long appeared to be special. At first glance, the Watson–Crick model for the double helix did appear to allow design of molecules that bound to other molecules in water (a problematic solvent). Indeed, such design was being performed almost routinely by molecular biologists who had no training in the fraternity of organic chemistry. An entire industry (“antisense therapeutics”) (Miller and T’so 1988) was based on the notion that the Watson–Crick model of DNA–DNA binding could be easily transferred to other backbones, in particular, to nonionic backbones. Millions of dollars were spent to show that this transfer was not easy.
In contrast with models for other molecular systems in chemistry, the Watson–Crick model is almost trivially simple. First, the model holds that molecular recognition depends entirely on two very simple rules of complementarity: size complementarity (big purines pair with small pyrimidines) and hydrogen-bonding complementarity (hydrogen bond donors pair with hydrogen bond acceptors) (Fig. 2). High school students (ourselves included) were taught “how genetics works” using paper cutouts to illustrate these rules. Generations of future molecular biologists came to view the structure of DNA as “obviously correct.”
Figure 2.
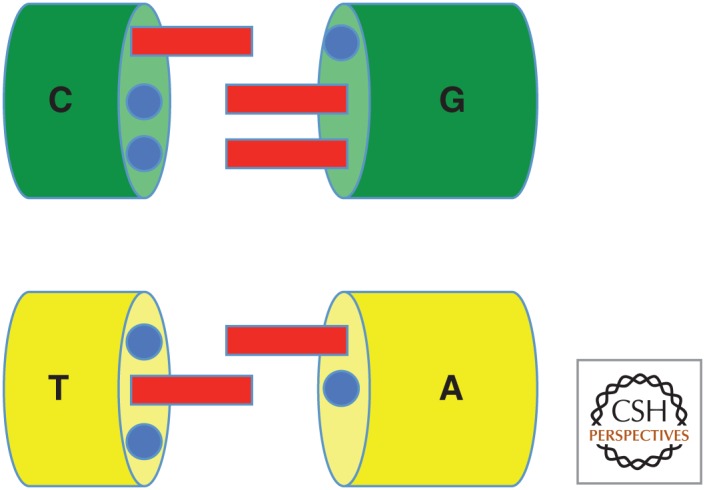
How genetics works. The cartoon that “explains everything”—paper cutouts that taught generations of schoolchildren that molecular genetics were simple chemistry.
Yet, even a newly minted Ph.D. in 1980 could see the multiple absurdities in this model. One hardly expects to get good molecular recognition from complementary hydrogen bonding in water; water as a solvent is “nothing but” competing hydrogen bonds. Further, the size complementarity required for Watson–Crick pairing is, at first glance, not likely to be enforced by the flexible backbone. Indeed, as Kool et al. (2000), Romesberg and collaborators (Malyshev et al. 2009, 2014), Hirao and collaborators. (Kimoto et al. 2011), Heuberger and Switzer (2008), and others (Doi et al. 2008) later showed, the backbone can easily adjust itself to accommodate geometries other than edge-on contacts (Fig. 3). Further, as “obvious” as nucleobase stacking seems (pennies stacked in a roll is a common analogy) (Bowman and Williams 2011), simple aromatic solids (benzene is an archetype) do not stack like pennies in a roll (Fig. 3).
Figure 3.

Some of the features of “noncartoon” natural DNA that make the Watson–Crick model less self-evident. (A) Unlike the “pennies in a roll” model for nucleobase stacking, the aromatic rings of benzene do not stack in a crystal (Cox et al. 1958). (B) Some synthetic nucleobase pairs that contribute to duplexes similar to the T:A pair, showing the flexibility of the backbone (Geyer et al. 2003; Gao et al. 2004; Heuberger and Switzer 2008; Winnacker and Kool 2013). (C) Adenine is missing a hydrogen-bonding group, creating many problems when using DNA to assemble nanostructures or to construct large DNA assemblies; diaminopurine forms a stronger pair. (D) Deamination of cytosine (shown), adenine, and guanine requires continuous repair. (E) G quartets are an example of a noncanonical structure that can dominate Watson–Crickery.
Once one begins down this path of reasoning, horrors pile on top of horrors. Adenine is “missing” a hydrogen-bonding group, perhaps because of how it emerged in prebiotic processes (Fig. 3) (Orgel 2004); the resulting instability of the A:T pair creates unending problems in biotechnology (Wei et al. 2012). Cytosine suffers spontaneous deamination in water, requiring constant repair in our genomes; so do adenine and guanine, at slower rates (Shapiro 1987). DNA sequences with consecutive dGs are so pathological that, at some point, commercial supply houses hesitated to make them (Mizusawa et al. 1986). And things get worse with RNA sequences that have consecutive Gs (Davis 2004).
These realizations helped make the 1980s a good time to expand synthesis past natural bioproducts to include unnatural bioproducts. Here, the “grand challenge” centered on questions “why?” and “why not?” Why did DNA have these molecular perplexities? What other molecular systems can “do” genetics? Could we synthesize alternative genetic molecules that perform better? The grand challenge question could even be put fancifully: If we encountered an alien species capable of Darwinian evolution, would their “DNA” be DNA or some other molecular system?
This article discusses the synthesis of alternative Watson–Crick systems (Benner et al. 1998; Benner 2004). As it turns out, the alternative molecular recognition systems do themselves have value as products. However, the major value of the synthetic effort to create unnatural genetic systems came from what was learned as the challenge was undertaken, under terms that did not allow failure to be an option (Bostick 2010).
REARRANGING THE SYNTHESIS OF HYDROGEN-BONDING UNITS—GENERATIONS OF ARTIFICIAL WATSON–CRICKERY
Even 30 years ago, one could easily draw nucleobase pairs that retained the Watson–Crick pairing “concept,” but had hydrogen-bonding units rearranged to give, at first glance, new Watson–Crick pairs. As shown in Figure 4, this rearrangement could readily generate 12 hypothetical nucleobases that might form a total of six orthogonal Watson–Crick pairs.
Figure 4.

Two rules of complementarity guide base pairing in DNA and RNA (collectively xeno nucleic acid [XNA]). The rules are (1) size complementarity (large purines pair with small pyrimidines), and (2) hydrogen-bonding complementarity (hydrogen bond donors, D, pair with hydrogen bond acceptors, A). Rearranging the nucleobase D and A groups gives artificially expanded genetic information systems (AEGIS). Chemical issues in the “first-generation” AEGIS (left pairs) are indicated in magenta. These motivated the synthesis of a second-generation AEGIS (right pairs). Electron density presented to the minor groove (green lobes) is recognized by polymerases (Benner et al. 1998).
The structures in Figure 4 came to be called “artificially expanded genetic information systems” (AEGIS). This term covers pairs that fully retain the Watson–Crick pairing concept; it distinguishes this concept from strategies from the Kool laboratory (in which interstrand hydrogen bonding is dispensed with entirely), the Hirao laboratory (in which size complementarity is key) (Hirao et al. 2002; Hirao 2006; Kimoto et al. 2009, 2013), and the Romesberg and Schultz laboratories (in which combinatorics defined the scope of polymerase–DNA interactions in a particularly interesting way) (McMinn et al. 1999; Malyshev et al. 2009).
We were not the first to hypothesize that an expanded genetic alphabet might be obtained by shuffling hydrogen-bonding units. Alex Rich, some 20 years earlier, had recognized that isoguanine (a natural product) and isocytosine might possibly form a third pair (Rich 1962) (the first-generation S:B pair in Fig. 1). Independently, Geoffrey Zubay (1988) proposed another alternative pair (Fig. 5), not recognizing that his hypothetical structure for the small component of the new pair lacked the aromatic planar geometry that the Watson–Crick model suggested was necessary for nucleobase stacking. Interestingly, even the writers of the movie E.T. the Extra-Terrestrial understood the possibility of an expanded genetic alphabet; E.T. has DNA built from six nucleotides, “inosine and a pyrimidine we cannot identify” (Mathison 1982).
Figure 5.

Different ring systems having different properties can implement the same Watson–Crick hydrogen-bonding pattern. (A) Zubay’s ring system formally implements a hydrogen bond donor–acceptor–donor pattern, but without the planar aromatic system that the Watson–Crick model implies (Zubay 1988); to get both, one must make a C-glycoside (Benner et al. 1987). (B) In nature, uridine and pseudouridine implement the same hydrogen-bonding pattern, the first as an N-glycoside, the second as a C-glycoside. (C) Obtaining a heterocycle to implement the puDDA hydrogen-bonding pattern was especially challenging, as various C-glycosides are easy to oxidize or easily epimerized. (D) Various different 5,6-ring systems implement the pu(DDA) hydrogen-bonding pattern, with different amounts of a minor tautomeric form, which implements a different pu(DAD) hydrogen-bonding pattern complementary to thymidine.
However, synthesis as an activity was necessary to determine whether nucleobase pairing was as simple as the Watson–Crick model implied. In this undertaking, it soon became clear that more than one heterocyclic system would support, or “implement,” any particular hydrogen-bonding pattern. For example, among natural nucleobases, uridine and pseudouridine both present a hydrogen bond acceptor–donor–acceptor hydrogen-bonding pattern (Fig. 5). Those seeking to meet the grand challenge of creating an artificial genetic system needed to decide which heterocyclic system to synthesize to implement each of the orthogonal hydrogen-bonding patterns.
In many cases, the first heterocyclic system prepared to implement each of the four additional hydrogen-bonding patterns turned out not to be the best heterocycle to support genetics. Several first-generation AEGIS components suffered from chemical defects, indicated in magenta in Figure 4. For example, the pyrazine heterocycles used first to implement the pyADD and pyDDA hydrogen-bonding patterns epimerized rapidly (Fig. 5) (Voegel et al. 1993a,b; Voegel and Benner 1994, 1996a,b; von Krosigk and Benner 2004). The purine ring system used to implement the puDDA hydrogen-bonding patterns had a substantial amount of a minor tautomer that created nucleobase-pairing ambiguity (Sepiol et al. 1976; Sismour et al. 2004). In a long process documented in the literature, second-generation implementations of various bonding patterns were then synthesized to fix problems in the new DNA (Benner 2009).
These second-generation improvements are summarized in Figure 4. The effort produced much new knowledge in heterocyclic chemistry. Within the framework of the Watson–Crick pair, a rather comprehensive view of what heterocycles can support genetics has now emerged. We can now even make a good guess about the “pyrimidine that we cannot identify” in E.T.’s DNA. We also know that if E.T. indeed had inosine in his/her genome, (s)he would have had difficulty surviving to the point where his/her species could attempt interplanetary travel.
NUCLEOBASE PAIRING IS CONSTRUCTIVELY AS SIMPLE AS THE WATSON–CRICK MODEL SUGGESTS
Once synthesis delivered heterocycles that adequately implemented the four extra hydrogen-bonding pairing patterns, it was relatively easy to adapt the then-emerging phosphoramidite-based solid-phase-synthesis chemistry (the key enabling technology for synthetic biology) to create DNA containing AEGIS components. These supported experiments to determine how AEGIS pairing contributes to overall duplex stability.
Remarkably, these studies found the Watson–Crick concept to be quite robust with respect to the shuffling of hydrogen-bonding units. In a study that looked at some 300 duplexes, pairs joined by three hydrogen bonds generally contributed more to duplex stability than pairs joined by just two; pairs joined by just one hydrogen bond contributed no stability to a duplex in competition with alternative interactions with bulk solvent (Geyer et al. 2003).
A set of second-order rules could also be discerned. For example, an uncompensated C=O group was not particularly disfavored in a pair (Fig. 6). However, an uncompensated –NH2 group was. Further, adding a proton and a positive charge to a nucleobase to create formal Watson–Crick complementarity was generally accepted. In contrast, losing a proton to put a negative charge on the nucleobase destabilized a formally Watson–Crick complement. These observations were used to develop a “self-avoiding molecular recognition system” (Hoshika et al. 2010), another unnatural DNA that is gaining in multiplex diagnostic systems and isothermal DNA amplification architectures (Sharma et al. 2014; Glushakova et al. 2015; Yang et al. 2015).
Figure 6.

Some second-order Watson–Crick pairing rules obtained via artificially expanded genetic information systems (AEGIS) development (Geyer et al. 2003). A negative charge in the nucleobase stack destabilizing (left). An uncompensated C–NH2 unit is destabilizing (center). An uncompensated C=O unit is acceptable (right).
The flexibility of the DNA backbone, and by implication its inability to enforce size complementarity, was also evident in these studies. For example, pairing of two small nucleobase analogs could stabilize the duplex if they were joined by three hydrogen bonds (Fig. 3B). Indeed, a small:small pair joined by three hydrogen bonds stabilized the duplex as much as a small:large pair joined by just two hydrogen bonds (Geyer et al. 2003). The observation that these pairs might compete with standard small:large pairs remains an important constraint on the design of artificial genetic systems.
These biophysical studies were followed by crystallographic studies that showed that AEGIS components did in fact pair with Watson–Crick geometry. For example, introduction of a single Z:P pair into the stem of an RNA riboswitch marginally increased the stability of that stem; a crystal structure showed essentially no geometric perturbation (Fig. 7, right) (Hernandez et al. 2015). In DNA, the crystal structure of a single Z:P pair likewise shows no substantial deviation from Watson–Crick geometry (Zhang et al. 2015) (Fig. 7, left). Indeed, duplexes with four or six Z:P pairs retain their overall Watson–Crick geometry (Fig. 7, center) (Georgiadis et al. 2015).
Figure 7.

Three crystal structures with Z:P artificially expanded genetic information systems (AEGIS) pairs. An isolated pair in a short, A-form DNA duplex crystallized with the aid of a selenium substitution (left) (Zhang et al. 2015). A 16-mer duplex with six consecutive Z:P pairs (center) (Georgiadis et al. 2015). A single Z:P pair in an RNA riboswitch in four views A, B, C, and D each rotated 90° (right) (Hernandez et al. 2015).
THE USE OF ORTHOGONAL AEGIS BINDING IN DIAGNOSTICS
Even as “why?” and “why not?” questions were being pursued in a grand challenge effort for discovery, not for technology, the intrinsic ability of AEGIS to fit within the canonical Watson–Crick structure was proving to be important for many applications. For example, Mickey Urdea and Thomas Horn at Chiron (Emeryville, CA) sought to create a branched DNA (b-DNA) assay kit to measure viral loads in patients infected with HIV, hepatitis B, and hepatitis C viruses (Bushnell et al. 1999). Viral load measurements are critical to determining when the management of a viral infection starts to fail because of mutation of the virus to create resistance to a drug being administered. Here, instead of amplifying the nucleic acid target, the b-DNA assay uses AEGIS components to assemble a signaling nanostructure (Fig. 8).
Figure 8.

The branched DNA assay (Bushnell et al. 1999). When artificially expanded genetic information systems (AEGIS) nucleotides (in this case, first-generation S and B) are placed in the amplifier nanostructure, the noise is dramatically decreased (Elbeik et al. 2004a,b).
Standard Watson–Crick pairing was used to capture the viral sequence on the solid support. Then, in a “sandwich” format, standard Watson–Crick pairing allowed the immobilized viral sequence to capture a second DNA molecule. The second DNA molecule then captured a b-DNA molecule, which, in turn, captured multiple signaling molecules. The b-DNA assay did not amplify the target viral nucleic acid sequence, creating a downstream contamination problem. Thus, the b-DNA assay became an alternative to a polymerase chain reaction (PCR) that required less skill to perform and less expertise to interpret.
Unfortunately, the b-DNA architecture with only natural nucleic acids failed to detect target nucleic acid sequence with low noise. Natural biological samples (e.g., blood) contain many nucleic acids, some of which partially complement “any” DNA sequences that might be used to assemble the signaling nanostructure. These can interact with the signaling molecules, perhaps mismatched, perhaps via concatenation, to immobilize signaling molecules on the surface of the support even in the absence of the target virus molecule.
This background noise was mitigated by putting AEGIS components (first-generation S and B) into the sequences that self-assembled to give the signaling nanostructure. AEGIS oligonucleotides cannot complement any natural oligonucleotides. Therefore, they cannot form structures that generate background noise. This allowed the b-DNA assay to become FDA approved with a level of detection at 30 molecules and serve millions of patients (Elbeik et al. 2004a,b). This represents the first examples of using DNA to construct commercially useful nanostructures.
The nonenzymatic hybridization of oligonucleotides containing AEGIS components showed that the simple Watson–Crick model could be generalized to increase the number of orthogonal pairs. In parallel, synthesis going on at the same time was showing the importance of the repeating charge in the backbone to molecular recognition (Steinbeck and Richert 1998; Benner and Hutter 2002). Here, various neutral backbone analogs of DNA, including polyamide nucleic acid (PNA) (Nielsen et al. 1991), were made and studied. The outcome of these studies was to show that to support Darwinian evolution, a linear genetic biopolymer was likely to require a repeating backbone charge. This “polyelectrolyte theory of the gene” (Benner and Hutter 2002) allowed the physical properties of the polymer to be largely independent of sequence, a property that is unusual in all molecular systems. Indeed, the repeating backbone charge is the reason why DNA molecules with expected properties are so easy to design.
At the same time, synthesis was showing the delicate and unpredictable impact of changing the structure of the sugar ring (Schneider and Benner 1990; Freier and Altmann 1997; Wilds et al. 2002). Here, a wide diversity of DNA analogs with different backbone carbohydrates (Eschenmoser 1999; Declercq et al. 2002; Wilds et al. 2002; Horhota et al. 2005) were synthesized and studied. Some had prebiotic significance (Krishnamurthy 2015).
Discussion of alternative backbone units for DNA and RNA is regrettably beyond the scope of this article. Nevertheless, the point was analogous: By the activity of synthesis, nucleobase pairing, at the core of genetics, was found to be more malleable than the phosphates and carbohydrates backbone units, the “uprights” in the “ladder” that the standard model had relegated as largely incidental to the performance of DNA in genetics.
CREATING A MOLECULAR BIOLOGY TO SUPPORT AEGIS
But could AEGIS components do more than just bind to other AEGIS components? Could an AEGIS DNA strand also dynamically participate in the enzymatic synthesis of its complement, a key property that we value in natural biomolecules?
Here, the grandness of the challenge arose from natural history. To serve in a genetic system, a polymerase must take instruction only from the template with extraordinary fidelity. Achieving this was no small trick. Polymerases have four natural substrates: A(template):T-triphosphate, T(template):A-triphosphate, G(template):C-triphosphate, and C(template):G-triphosphate. Thus, any direct contact between a polymerase and the nucleobase is “dangerous.” For example, contact in the major groove might cause a polymerase to prefer some nucleotides over others, disregarding the instructions from the template, as natural nucleobases differ greatly in the moieties that they present to the major groove.
The minor groove is different in this respect. As noted by Joyce and Steitz (1994), all of the four standard nucleobases present electron density to the minor groove. This density is delivered by the exocyclic C=O moieties of C and T and by the N3 nitrogens of A and G (Fig. 4). Crystallographic analysis of many polymerases identifies side chains that contact this electron density in both the template and primer in primer–template–enzyme complexes.
For just one AEGIS pair, both components present analogous electron density to the minor groove. Z has an exocyclic C=O moiety; its partner P carries electron density on its N3. In the three other second-generation pairs, the small component presents a hydrogen bond donating moiety (-NH2) to the minor groove. Thus, the Joyce–Steitz “minor groove scanning hypothesis” suggested that polymerases would readily synthesize duplex DNA containing Z:P pairs from templates and triphosphates, whereas relatively few would synthesize the S:B, K:X, and V:J pairs; those pairs would be synthesized best by polymerases that had been mutated.
This proved to be the case empirically. Nearly all polymerases examined over the past decade do a reasonable, sometimes acceptable, and occasionally an excellent job replicating Z:P pairs. In contrast, the S:B (Sismour and Benner 2005b) and K:X (Sismour et al. 2004) pairs, which lack this electron density, are often best replicated by polymerases that are first mutated.
To create polymerase variants that accept AEGIS components, a directed evolution tool developed by Tawfik and Griffiths (1998) and adapted to polymerases by Holliger (Ghadessy et al. 2001) proved to be useful. Called compartmentalized self-replication (CSR) (Fig. 9), bacteria containing plasmids encoding polymerase variants are placed in water droplets emulsified in oil. The water droplets carry buffer, primers, and triphosphates necessary for PCR. The bacteria biosynthesize the encoded variant polymerase, which is released with its encoding gene to the extracellular PCR mixture in the first heat step. If the variant can replicate its own gene, then that gene is amplified and the amplified gene presented to subsequent cycles of laboratory evolution. If, however, the variant cannot replicate its own gene, it does not survive. By placing AEGIS components strategically into a CSR experiment, polymerases that copy them with increased efficiency are evolved in the laboratory.
Figure 9.

Schematic of colony self-replication (CSR) (Ghadessy et al. 2001) for the selection of polymerases that amplify AEGIS pairs, here in a nested polymerase chain reaction (PCR) architecture. A library of genes encoding active (blue) and inactive (red) polymerases is cloned into bacterial cells, which are dispersed (one cell per droplet) into an emulsion where the extracellular buffer contains PCR primers and triphosphates. The initial heat cycle places the polymerases in contact with the PCR primers and triphosphates. The droplets keep the polymerase variant associated with its own encoding gene; if the gene is to be amplified, it must be amplified by its encoded polymerase. After the target-specific primers are consumed, the nested PCR is “carried” by external primers containing AEGIS components. The genes encoding polymerases that are able to copy AEGIS nucleotides (the blue arcs) are enriched in the product pool. The products are then recloned, and the selection is repeated.
Polymerases contain too many important amino acid residues to expect good results to emerge via random variation. Evidence of this comes, for example, from the fact that a library of 108 polymerase variants having 4–6 amino-acid replacements does not generate any that can be recovered in a laboratory selection experiment that has useful activity (Laos et al. 2014).
However, the natural history of polymerase evolution can rationally improve polymerase libraries. Sites that display unusual evolutionary behavior (such as heterotachy, homoplasy, and parallelism) (Fig. 10) are more productively altered in a nonrandom library (Chen et al. 2010). As a consequence, many polymerase systems are now available that replicate many different six-letter AEGIS alphabets (Sismour et al. 2004, 2005; Yang et al. 2010; Laos et al. 2014; Sefah et al. 2014).
Figure 10.

The pattern of amino acid replacement in natural history. Represented here by a phylogenetic tree for an individual site, the pattern can be used to guide protein engineering and directed evolution experiments. (A) Amino acid replacement at sites with low-level of replacements typically leads to inactivation of the polymerase. (B) Homoplasy in the form of parallel evolution indicating purifying selection removes variation that moves beyond a small set of amino acids; sites displaying this variation may be replaced in a controlled way. (C) Substantial variation indicates a site experiencing little purifying selective pressure; replacement at this site is unlikely to have little interesting impact on polymerase behavior. (D) Heterotachy, in which different branches have different rates of amino acid replacement, indicates changing functional constraints at a site; these sites are the foci of the most successfully designed protein engineering libraries (Chen et al. 2010).
USING AEGIS MOLECULAR BIOLOGY
With the availability of polymerases that replicate AEGIS pairs, the orthogonality of AEGIS pairing seen in the b-DNA assay can be exploited in useful processes and products that include AEGIS PCR. Consider, for example, the multiplexed PCR problem. In most cases, PCR amplicons can be extracted from a complex biological mixture by adding a single pair of primers. However, as the number of primer pairs is increased to target more and more amplicons, the primers interact with each other, find off-target sites in a complex genomic environment to bind, and create spurious amplicons.
AEGIS proved able to manage this problem in a nested PCR format (Fig. 11). Here, the PCR is initiated with low concentrations of chimeric primers with 3′-ends complementary to the target (natural) sequence, and a 5′-AEGIS tag. The initial rounds of PCR create amplicons carrying complementary AEGIS sequences at their ends. Therefore, after the small amounts of chimeric primers are consumed, the PCR is carried by large amounts of “external” AEGIS primers complementary to these tags. Even though they are present at high concentration, the AEGIS external primers cannot find any natural DNA to bind off-target, even in very complex biological mixtures. Therefore, AEGIS nested PCR is very clean, even in a multiplexed form (Fig. 11) (Yang et al. 2010).
Figure 11.

Although synthesized in a “grand challenge” to ask “what if?” and “why not?” questions, artificially expanded genetic information systems (AEGIS) components turned out to have practical value. Here, AEGIS orthogonality creates very clean multiplexed polymerase chain reaction (PCR) (right) in a nested PCR architecture (left). With AEGIS nucleotides in the external primers, PCR is clean, even in complex biological mixtures compared with the same PCR but with external primers having only standard nucleotides (Yang et al. 2010).
AEGIS orthogonality gained further use with the invention of “conversion” (Yang et al. 2013). Conversion copies a standard DNA molecule in a solution that lacks, for example, dCTP. Instead, the solution contains dZTP. With the correct polymerase, the correct buffer components, and the correct pH, the polymerase is forced to put in Z opposite G in the template (Yang et al. 2013). This creates a GAZT product, which cannot complement any natural xeno nucleic acid (XNA) sequence in any biological sample, no matter how complex. This allows clean and uniform capture of AEGIS DNA.
The use of conversion is shown in Figure 12, in a single assay that targets 22 different arboviral RNA sequences that might be present in a single mosquito carcass (Glushakova et al. 2015). Self-avoiding primers (Hoshika et al. 2010) are used with AEGIS external primers to cleanly create amplicons. Then, primers are extended with conversion to create an AEGIS tag that is the only oligonucleotide in the mixture complementary to a CTPA (computed tomography pulmonary angiogram) probe carried by a Luminex bead. This gives high and uniform detection of the amplicons arising from whatever RNA virus might be present. These combinations are now being used in assays to detect many infectious disease agents, from NiV (Nipah virus) and SARS (severe acute respiratory syndrome) virus to MERS (Middle East respiratory syndrome) virus and Zika virus (Benner et al. 2015; Yang et al. 2015).
Figure 12.

Artificially expanded genetic information systems (AEGIS) conversion supports an assay that allows 22 mosquito-borne viruses to be sought in a single mosquito carcass (for details, see Glushakova et al. 2015).
USING AEGIS INSTEAD OF NATURAL NUCLEOTIDES
This kind of synthetic biology shows that technology need not be constrained by the structure of the biopolymers that prebiotic chemistry (and four billion years of subsequent historical accident examined by natural selection) on Earth has delivered to us. Peter Schultz and others have made the parallel point with respect to the protein lexicon (Chatterjee et al. 2013).
The two can be joined. More nucleotide “letters” in a genetic alphabet should allow the writing of more amino acid “words” in the protein “lexicon.” Indeed, using the first-generation AEGIS S and B “letters” to support the codon:anticodon interaction in mRNA and tRNA molecules allowed an AEGIS mRNA molecule to encode a 21st amino acid in in vitro translation (Bain et al. 1992). One outcome of this synthesis was a deeper understanding of the role played by release factors in preventing frame shifting during translation termination. Parallel experiments using the second-generation Z:P pair, in which the AEGIS tRNA was charged by flexizymes (Morimoto et al. 2011) did the same. Flexizymes are RNA catalysts that charge tRNA molecules with amino acids; the inspiration for their design was the RNA enzymes that presumably charged tRNA in the RNA world that invented ribosomal translation.
AEGIS IN LARGE-SCALE DNA SYNTHESIS
AEGIS can also be used for another goal discussed in this volume: the synthesis of large DNA constructs from short fragments. Here, AEGIS components are placed in the overhangs of synthetic fragments that are mixed in a multicomponent ligation to create a large DNA molecule (Fig. 13). Because AEGIS nucleotides do not pair with standard nucleotides, these overhangs cannot form hairpins or other undesired secondary structures, either within their fragment or between fragments; they therefore are free to hybridize as designed. After ligation, conversion in the reverse direction replaces the AEGIS nucleotides by standard nucleotides, completing the synthesis of a large DNA construct. This was shown in a “one-pot” synthesis of a gene encoding kanamycin resistance (Merritt et al. 2014).
Figure 13.

Assembly of large DNA constructs using artificially expanded genetic information systems (AEGIS) pairs to assemble the strands without their forming hairpins or other undesired structures. Here, the added information density of a six-letter polymer diminishes assembly ambiguity. After the assembly is complete, AEGIS nucleotides are converted to standard nucleotides, giving an entirely natural construct (Merritt et al. 2014). This allows shorter fragments to be used, which, in turn, recognizes that the longer the synthetic fragment, the greater the chance of error.
AEGIS AS A PLATFORM FOR EVOLUTION
Natural DNA and RNA (collectively XNA) can perform functions beyond genetics (Ellington and Szostak 1992; Bartel and Szostak 1993; Breaker and Joyce 1994; Schneider et al. 1995; Kraemer et al. 2011). RNA catalysis may have supported the first forms of life on Earth; a current model for natural history holds that an earlier episode of life on Earth (the “RNA world”) used RNA as its only genetically encoded catalytic component (Benner et al. 1989). Indeed, the design of flexizymes to charge AEGIS tRNA with unnatural amino acids was based on the RNA-world model (Morimoto et al. 2011).
Adding replicable nucleobases should increase the binding and catalytic potential of the XNA libraries. However, adding nucleobases also expands the “sequence space” accessible to a biopolymer, from 4n species in a library n nucleotides in length to 6n in an XNA species with six building blocks, and 12n if the AEGIS is completed. It remains open whether AEGIS helps or hurts the search for functional nucleic acids, as the success of that search depends on the “density” of functional behavior in that space. Here, we might be advised to follow the Perrin or Silverman strategy of simply functionalizing the four standard nucleotides (Hollenstein et al. 2009a,b; Zhou et al. 2016).
The availability of polymerases that replicate AEGIS nucleotides makes it possible to apply laboratory in vitro evolution (LIVE) experiments to address that question (Fig. 14). The approach follows in vitro selection experiments applied to four-letter nucleic acids by Szostak, Ellington, Gold, Joyce, and others (Ellington and Szostak 1992; Bartel and Szostak 1993; Breaker and Joyce 1994; Schneider et al. 1995; Kraemer et al. 2011). These experiments have shown that natural nucleic acids are relatively poor reservoirs of receptors, ligands, and catalysts, perhaps because of their limited number and functionality of their monomers.
Figure 14.

Schematic of the cell-targeted artificially expanded genetic information systems–laboratory in vitro evolution (AEGIS-LIVE) reported in Zhang et al. (2015). A GACTZP DNA library with a randomized region flanked by primer binding sites was incubated with the target cells. Unbound sequences were then washed away. Bound sequences were eluted from the cells, and the supernatant enriched in AEGIS DNA molecules (“survivors”) having affinity for the cells was collected. Counterselections were performed against untransformed liver cells. The survivors were polymerase chain reaction (PCR)-amplified using a fluorescein isothiocyanate (FITC)-labeled primer and a biotinylated primer. Single-stranded DNA was made from the PCR products using the biotin handle and entered into the next round of selection. In each round, survivor pools were monitored for the appearance of “bulk” binding. After this was seen, survivors were sequenced and resynthesized for study.
AEGIS-LIVE is in its infancy, now with just four examples. Nevertheless, it appears as if adding functionalized AEGIS nucleotides to a DNA library delivers better and more specific receptors and ligands. In one example (Zhang et al. 2015), a GACTZP DNA library was used in an AEGIS-LIVE experiment to find AEGIS molecules that bind to HepG2 liver cancer cells. A counterselection against untransformed liver cells (Hu1545V) was used to remove nonspecific binders (Fig. 14) (Zhang et al. 2015). Binding was seen in the bulk pools after 12 rounds of affirmative selection. Four rounds of negative selection were embedded in the process, which also included 200 cycles of PCR. Sequencing recovered 17 motifs that contributed from 0.14% to 26% of the total surviving population. These were resynthesized and their affinities for HepG2 cells were measured (specificity data are shown in Fig. 15).
Figure 15.

DNA aptamers from artificially expanded genetic information systems–laboratory in vitro evolution (AEGIS-LIVE) (Zhang et al. 2015). (A) Sequences (only randomized region), dissociation constants (Kd), and their percentage in the pool of binders after 12 rounds of positive selection and four rounds of negative selection (Z and P in red). Sequences are arranged in order of increasing Kd. (B) Binding and specificity of AEGIS aptamers, arranged from the weakest binding (top) to the tightest (bottom). Binding is measured by incubating cells with fluorescently tagged binding species in cell sorter to give fluorescence intensity per cell (x-axis, log scale; the y-axis indicates the number of cells having the indicated intensity). (Left) Binding to transformed cancer cells, the cells that served as the target in the aptamer selection process. (Right) Binding to untransformed liver cells, which were the counterselection cells. Red distribution at bottom of each panel is the binding displayed by the DNA library before selection.
Several features of the data are striking. First, AEGIS-LIVE delivered cell-binding molecules that had more than one Z and/or P. These include several that had Z and P nearby (ZnP and PnnZ, where “n” is any nucleotide), multiple Z and P units nearby (e.g., PnnZnP), and even one with an adjacent Z and P (PZ). This suggests that the AEGIS-LIVE experiment searched much of the substantially larger “sequence space” of the GACTZP system. The binders were also more specific (Fig. 15).
Binders also emerged from the same experiment that had neither Z nor P. However, these had systematically weaker affinity for the cells, all with Kdiss values that were >200 nm. Those with Z and P typically had Kdiss values that were < 50 nm. These indicate that the additional functionality presented on Z, or possibly the additional information density of a six-letter nucleic acid, helped improve the quality of a DNA library as a reservoir of functional molecules. The nitro group is a “universal binding moiety” (remembering the ability of nitrocellulose to bind many proteins). Increasing the information density in an oligonucleotide manages folding ambiguity, a major problem in DNA catalysts that have been studied in detail (Carrigan et al. 2004).
MOVING AEGIS INTO LIVING CELLS
These results showed that the rather simple Watson–Crick model not only supported an enlarged molecular recognition system and an enlarged molecular biology, but also a genetic system that has enlarged functional potential. Further, if combined with natural enzymes, AEGIS provides many of the properties that we value in a genetic system. AEGIS forms duplexes with sequence specificity, directs its own replication, can adapt under selective pressure, and can evolve. Further, AEGIS has value, not only for what it has taught us about the intimate connection between molecular structure and genetics, but also in human diagnostics, pathogen surveillance, and in a platform to create receptors, ligands, and catalysts “on demand.”
Thus, AEGIS fits closely the Kool definition for synthetic biology (unnatural parts working in the context of natural systems) (Rawls 2000). However, AEGIS has not been placed into living cells. Here, the much more “unnatural” pair described by Romesberg has been replicated for ∼15 hr in Escherichia coli (Malyshev et al. 2014), as a single exemplar. Unfortunately, this tour de force required that an engineered E. coli cell be fed presynthesized triphosphates presented in the growth medium.
Fortunately, efforts to meet the grander challenge, a robust bacterial system that replicates AEGIS-containing plasmids, are well underway. Mutants of kinases that add a single phosphate to a nucleoside to make the nucleoside monophosphate, and then further transform the monophosphate to the di- and triphosphates inside of living cells, have been prepared (Matsuura et al. 2016). Cells have been engineered to manage the unnatural functional groups that AEGIS carries. The construction of a cell that makes a fifth and sixth triphosphate is teaching us much about how cells regulate this key machinery required for life. Perhaps someday E.T. will be present on Earth, not by interstellar travel, but rather by the hands of the synthetic biologist.
CONCLUSIONS
“Synthetic biology” means different things in different communities. Most communities, however, remain focused on activities that involve only rearranging natural bioparts. This is certainly pragmatic. Natural DNA can be ordered inexpensively from Integrated DNA Technologies (IDT). Natural polymerases that manage natural DNA can be likewise obtained inexpensively. Moving beyond “the natural” requires that one reinvent much of molecular biology, including polymerases, restriction enzymes (Chen et al. 2011), and other tools that biotechnologists take for granted. Moving beyond the natural also requires the development of new synthetic pipelines to create triphosphates and phosphoramidites and new analytical chemistry tools, including AEGIS sequencing technology (Yang et al. 2013). Thus, there is little wonder that most synthetic biology uses natural biological parts.
However, moving synthetic biology beyond “the natural” is not without benefits. As with classical synthesis, the effort to recreate the properties that we value in life, but with unnatural products, has provided (and is providing) unexpected insights into how biomolecules work in terran systems, and how biomolecular structure is intimately connected to biomolecular behavior.
But moving synthetic biology beyond the natural also benefits technology. Natural DNA is the product of prebiotically constrained starting molecules whose molecular structures have been conserved despite their multiple defects (Fig. 3). Those who do not move beyond the natural are condemned to suffer (in perpetuity) from these defects, which lead to failed microarrays (Wei et al. 2012), PCR multiplexed messes (Yang et al. 2010), expensive diagnostics (Glushakova et al. 2015), laborious DNA construct synthesis (Merritt et al. 2014), poor quality aptamers (Sefah et al. 2014), slow DNAzymes (Zhang et al. 2015), ambiguously folded DNA nanostructures, and proteins with only 20 (or 21) different kinds of amino acids (Chatterjee et al. 2013).
In return for its added effort, the unnatural kind of synthetic biology solves these problems. Indeed, given the advantages of redesigned DNA that “fixes God’s mistakes,” it might soon be surprising to find anyone who uses the natural stuff anymore.
ACKNOWLEDGMENTS
We are indebted to the National Aeronautics and Space Administration (NASA) (Experimental Approaches to Potential Alien Molecular Biologies: A Two-Biopolymer Darwinian System, Grant No. NNX14AK37G and Expanded Alphabets for Constructing Evolutionary Machines, Grant No. NNX15AF46G), The Defense Threat Reduction Agency (DTRA) (Aptamers from Artificial Genetic Systems, Grant No. HDTRA1-13-1-0004), and the Templeton World Charity Foundation (TWCF) (Doing Bioinformation Differently: A Two-Biopolymer Synthetic Life Form without Encoding or Instruction, with Bidirectional Information Flow, Grant No. TWCF0092/AB57) for their support of this work. This notwithstanding, the opinions expressed herein are those of the authors, and not of the Federal Government, NASA, the Department of Defense, or the TWCF.
Footnotes
Editors: Daniel G. Gibson, Clyde A. Hutchison III, Hamilton O. Smith, and J. Craig Venter
Additional Perspectives on Synthetic Biology available at www.cshperspectives.org
REFERENCES
*Reference is also in this collection.
- Bain JD, Chamberlin AR, Switzer CY, Benner SA. 1992. Ribosome-mediated incorporation of non-standard amino acids into a peptide through expansion of the genetic code. Nature 356: 537–539. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Szostak JW. 1993. Isolation of new ribozymes from a large pool of random sequences. Science 261: 1411–1418. [DOI] [PubMed] [Google Scholar]
- Benner SA. 2004. Understanding nucleic acids using synthetic chemistry. Accounts Chem Res 37: 784–797. [DOI] [PubMed] [Google Scholar]
- Benner SA. 2009. The life, the universe and the scientific method. FfAME, Gainesville, FL. [Google Scholar]
- Benner SA, Hutter D. 2002. Phosphates, DNA, and the search for nonterrean life. A second generation model for genetic molecules. Bioorg Chem 30: 62–80. [DOI] [PubMed] [Google Scholar]
- Benner SA, Allemann RK, Ellington AD, Ge L, Glasfeld A, Leanz GF, Krauch T, Macpherson LJ, Moroney SE, Piccirilli JA. 1987. Natural selection, protein engineering and the last riboorganism. Rational model building in biochemistry. Cold Spring Harbor Symp Quant Biol 52: 53–63. [DOI] [PubMed] [Google Scholar]
- Benner SA, Ellington AD, Tauer A. 1989. Modern metabolism as a palimpsest of the RNA world. Proc Natl Acad Sci 86: 7054–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner SA, Battersby TR, Kodra J, Switzer CY, Moroney SE, Voegel J, MacPherson L, von Krosigk U, Hammer C, Richert C, et al. 1998. Redesigning nucleic acids. Pure Appl Chem 70: 263–266. [DOI] [PubMed] [Google Scholar]
- Benner SA, Kim HJ, Merritt KB, Yang Z, McLendon C, Hoshika S, Hutter D. 2015. Next-generation DNA in pathogen detection, surveillance, and CLIA-waivable diagnostics. Advances in Global Health through Sensing Technologies 2015, 10.1117/12.2183481. [DOI] [Google Scholar]
- Bostick J. 2010. Origin of Apollo 13 quote: “Failure is not an option,” www.thespacereview.com/article/357/1. [Google Scholar]
- Bowman JC, Williams LD. 2011. Nucleic acids. In Encyclopedia of astrobiology, pp. 1141–1147. Springer, Berlin. [Google Scholar]
- Breaker RR, Joyce GF. 1994. A DNA enzyme that cleaves RNA. Chem Biol 1: 223–229. [DOI] [PubMed] [Google Scholar]
- Brent RL. 2004. A partnership between biology and engineering. Nat Biotechnol 22: 1211–1214. [DOI] [PubMed] [Google Scholar]
- Bushnell S, Budde J, Catino T, Cole J, Derti A, Kelso R, Collins ML, Molino G, Sheridan P, Monahan J, et al. 1999. ProbeDesigner for the design of probesets for branched DNA (bDNA) signal amplification assays. Bioinformatics 15: 348–355. [DOI] [PubMed] [Google Scholar]
- Carrigan MA, Ricardo A, Ang DN, Benner SA. 2004. Quantitative analysis of a deoxyribonucleotide catalyst obtained via in vitro selection. A DNA ribonuclease. Biochemistry 43: 11446–11459. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG. 2013. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52: 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Gaucher EA, Leal NA, Hutter D, Havemann SA, Govindarajan S, Ortlund EA, Benner SA. 2010. Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc Natl Acad Sci 107: 1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Yang ZY, Yang MC, Alvarado JB, Wang GG, Benner SA. 2011. Recognition of an expanded genetic alphabet by type-II restriction endonucleases and their application to analyze polymerase fidelity. Nucleic Acids Res 39: 3949–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EG, Cruickshank DWJ, Smith JAS. 1958. The crystal structure of benzene at −3°C. Proc R Soc A 247: 1–21. [Google Scholar]
- Davis JT. 2004. G quartets 40 years later: From 5′-GMP to molecular biology and supramolecular chemistry. Angew Chem Int Ed Engl 43: 668–898. [DOI] [PubMed] [Google Scholar]
- Declercq R, Van Aerschot A, Read RJ, Herdewijn P, Van Meervelt L. 2002. Crystal structure of double helical hexitol nucleic acids. J Am Chem Soc 124: 928–933. [DOI] [PubMed] [Google Scholar]
- Doi Y, Chiba J, Morikawa T, Inouye M. 2008. Artificial DNA made exclusively of nonnatural C-nucleosides with four types of nonnatural bases. J Am Chem Soc 130: 8762–8768. [DOI] [PubMed] [Google Scholar]
- Dueber JE, Yeh BJ, Bhattacharyya RP, Lim WA. 2004. Rewiring cell signaling: The logic and plasticity of eukaryotic protein circuitry. Curr Opin Struct Biol 14: 690–699. [DOI] [PubMed] [Google Scholar]
- Dunitz JD, Bernstein J. 1995. Disappearing polymorphs. Acc Chem Res 28: 193–200. [Google Scholar]
- Elbeik T, Markowitz N, Nassos P, Kumar U, Beringer S, Haller B, Ng V. 2004a. Simultaneous runs of the Bayer VERSANT HIV-1 version 3.0 and HCV bDNA version 3.0 quantitative assays on the system 340 platform provide reliable quantitation and improved work flow. J Clin Microbiol 42: 3120–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbeik T, Surtihadi J, Destree M, Gorlin J, Holodniy M, Jortani SA, Kuramoto K, Ng V, Valdes R, Valsamakis A, et al. 2004b. Multicenter evaluation of the performance characteristics of the Bayer VERSANT HCV RNA 3.0 assay (bDNA). J Clin Microbiol 42: 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Ellington AD. 2016. Review on DNA synthesis and assembly approaches. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. 1992. Selection in vitro of single-stranded molecules that fold into specific ligand-binding structures. Nature 355: 850–852. [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. 1999. Chemical etiology of nucleic acid structure. Science 284: 2118–2124. [DOI] [PubMed] [Google Scholar]
- Freier SM, Altmann KH. 1997. The ups and downs of nucleic acid duplex stability. Structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res 25: 4429–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui K. 1982. Role of frontier orbitals in chemical reactions. Science 218: 747–754. [DOI] [PubMed] [Google Scholar]
- *.Gaj T, Sirk SJ, Shui SI, Liu J. 2016. Genome editing technologies: Principles and applications. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JM, Liu HB, Kool ET. 2004. Expanded-size bases in naturally sized DNA: Evaluation of steric effects in Watson–Crick pairing. J Am Chem Soc 126: 11826–11831. [DOI] [PubMed] [Google Scholar]
- Georgiadis MM, Singh I, Kellett WF, Hoshika S, Benner SA, Richards NGJ. 2015. Crystal structures of non-natural nucleobase pairs in A- and B-DNA. J Am Chem Soc 137: 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer CR, Battersby TR, Benner SA. 2003. Nucleobase pairing in expanded Watson–Crick like genetic information systems. The nucleobases. Structure 11: 1485–1498. [DOI] [PubMed] [Google Scholar]
- Ghadessy FJ, Ong JL, Holliger P. 2001. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci 98: 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, et al. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329: 52–56. [DOI] [PubMed] [Google Scholar]
- *.Glass J. 2016. Minimal cells. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakova LG, Bradley A, Bradley KM, Alto BW, Hoshika S, Hutter D, Sharma N, Yang Benner SA. 2015. High-throughput multiplexed xMAP Luminex array panel for detection of twenty two medically important mosquito-borne arboviruses based on innovations in synthetic biology. J Virol Meth 214: 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AR, Shao Y, Hoshika S, Yang Z, Shelke SA, Herrou J, Kim HJ, Kim MJ, Piccirilli JA, Benner SA. 2015. A crystal structure of a functional RNA molecule containing an artificial nucleobase pair. Angew Chem Int Ed Engl 54: 9853–9856. [DOI] [PubMed] [Google Scholar]
- Heuberger BD, Switzer C. 2008. An alternative nucleobase code: Characterization of purine–purine DNA double helices bearing guanine–isoguanine and diaminopurine 7-deazaxanthine base pairs. ChemBioChem 9: 2779–2783. [DOI] [PubMed] [Google Scholar]
- Hirao I. 2006. Unnatural base pair systems for DNA/RNA-based biotechnology. Curr Opin Chem Biol 10: 622–627. [DOI] [PubMed] [Google Scholar]
- Hirao I, Ohtsuki T, Fujiwara T, Mitsui T, Yokogawa T, Okuni T, Nakayama H, Takie K, Yabuki T, Kigawa T, et al. 2002. An unnatural base pair for incorporating amino acid analogs into proteins. Nat Biotechnol 20: 177–182. [DOI] [PubMed] [Google Scholar]
- Hoffmann R. 1982. Building bridges between inorganic and organic chemistry (Nobel Lecture). Angew Chem Int Ed Engl 21: 711–724. [Google Scholar]
- Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. 2009a. A DNAzyme with three protein-like functional groups. Enhancing catalytic efficiency of M2+-independent RNA cleavage. ChemBioChem 10: 1988–1992. [DOI] [PubMed] [Google Scholar]
- Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. 2009b. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+). Nucleic Acids Res 37: 1638–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horhota A, Zou KY, Ichida JK, Yu B, McLaughlin LW, Szostak JW, Chaput JC. 2005. Kinetic analysis of an efficient DNA-dependent TNA polymerase. J Amer Chem Soc 127: 7427–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshika S, Leal N, Chen F, Benner SA. 2010. Artificial genetic systems. Self-avoiding DNA in PCR and multiplexed PCR. Angew Chem Int Ed Engl 49: 5554–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce CM, Steitz TA. 1994. Function and structure relationships in DNA polymerases. Ann Rev Biochem 63: 777–822. [DOI] [PubMed] [Google Scholar]
- Kimoto M, Kawai R, Mitsui T, Yokoyama S, Hirao I. 2009. An unnatural base pair system for efficient PCR amplification and functionalization of DNA molecules. Nucleic Acids Res 37: e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimoto M, Cox RS, Hirao I. 2011. Unnatural base pair systems for sensing and diagnostic applications. Expert Rev Mol Diagn 11: 321–331. [DOI] [PubMed] [Google Scholar]
- Kimoto M, Yamashige R, Matsunaga K, Yokoyama S, Hirao I. 2013. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat Biotechnol 31: 453–457. [DOI] [PubMed] [Google Scholar]
- Kishi Y. 1989. Natural products synthesis. Palytoxin. Pure Appl Chem 61: 313–324. [Google Scholar]
- Kishi Y, Fujuyama T, Aratani M, Nakatsubo F, Goto T, Inoue S, Tanini H, Sugiura S, Kakoi H. 1972. Synthetic studies on tetrodotoxin and related compounds. IV: Stereospecific total synthesis of dl-tetrodotoxin. J Am Chem Soc 94: 9219–9221. [DOI] [PubMed] [Google Scholar]
- Kool ET, Morales JC, Guckian KM. 2000. Mimicking the structure and function of DNA. Insights into DNA stability and replication. Angew Chem Int Ed Engl 39: 990–1009. [DOI] [PubMed] [Google Scholar]
- Kraemer S, Vaught JD, Bock C, Gold L, Katilius E, Keeney TR, Kim N, Saccomano NA, Wilcox SK, Zichi D, et al. 2011. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 6: e26332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy R. 2015. On the emergence of RNA. Israel J Chem 55: 837–850. [Google Scholar]
- Laos R, Thomson JM, Benner SA. 2014. DNA polymerases engineered by directed evolution to incorporate non-standard nucleotides. Front Microbiol 5: 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Lechner A, Brunk E, Keasling JD. 2016. The need for integrated approaches in metabolic engineering. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc S. 1912. La biologie synthetique. Poinat, Paris. [Google Scholar]
- Malyshev DA, Seo YJ, Ordoukhanian P, Romesberg FE. 2009. PCR with an expanded genetic alphabet. J Am Chem Soc 131: 14620–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR Jr, Romesberg FE. 2014. A semi-synthetic organism with an expanded genetic alphabet. Nature 509: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathison M. 1982. E.T. the Extra-Terrestrial. Directed by Steven Spielberg, Universal Pictures.
- Matsuura MF, Shaw RW, Moses JD, Kim HJ, Kim MJ, Kim MS, Benner SA. 2016. Assays to detect the formation of triphosphates of unnatural nucleotides: Application to Escherichia coli nucleoside diphosphate kinase. ACS Synth Biol 5: 234–240. [DOI] [PubMed] [Google Scholar]
- McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. 1999. Efforts toward expansion of the genetic alphabet. DNA polymerase recognition of a highly stable, self-pairing hydrophobic base. J Am Chem Soc 121: 11585–11586. [Google Scholar]
- Merritt KB, Bradley KM, Hutter D, Benner SA. 2014. Whole genes from autonomous self-assembly of synthetic oligonucleotides incorporating artificial nucleotides. Beilstein J Org Chem 10: 2348–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Ts’o POP. 1988. Oligonucleotide inhibitors of gene-expression in living cells. New opportunities in drug design. Ann Rep Med Chem 23: 295–304. [Google Scholar]
- Mizusawa S, Nishimur S, Seela F. 1986. Improvement of the dideoxy chain termination method of DNA sequencing by use of deoxy-7-deazaguanosine triphosphate in place of dGTP. Nucleic Acids Res 14: 1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto J, Hayashi Y, Iwasaki K, Suga H. 2011. Flexizymes: Their evolutionary history and the origin of catalytic function. Acc Chem Res 44: 1359–1368. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Berg RH, Buchardt O. 1991. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254: 1497–1500. [DOI] [PubMed] [Google Scholar]
- Orgel LE. 2004. Prebiotic adenine revisited: Eutectics and photochemistry. Orig Life Evol Biosph 34:: 361–369. [DOI] [PubMed] [Google Scholar]
- Pople JA. 1999. Nobel lecture. Quantum mechanical models. Rev Mod Phys 71:: 1267–1274. [Google Scholar]
- Prehoda KE, Scott JA, Mullins RD, Lim WA. 2000. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290: 801–806. [DOI] [PubMed] [Google Scholar]
- Rawls RL. 2000. “Synthetic biology” makes its debut. Chem Eng News 78: 49–53. [Google Scholar]
- Rich A. 1962. On the problems of evolution and biochemical information transfer. In Horizons in biochemistry (ed. Kasha M, Pullmann B), pp. 103–126. Academic, New York. [Google Scholar]
- Schneider KC, Benner SA. 1990. Oligonucleotides containing flexible nucleoside analogs. J Am Chem Soc 112: 453–455. [Google Scholar]
- Schneider DJ, Feigon J, Hostomsky Z, Gold L. 1995. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochemistry 34: 9599–9610. [DOI] [PubMed] [Google Scholar]
- Sefah K, Yang Z, Bradley KM, Hoshika S, Jimenez E, Zhu G, Shanker S, Yu F, Tan WH, Benner SA. 2014. In vitro selection with artificial expanded genetic information systems (AEGIS). Proc Natl Acad Sci 111: 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepiol J, Kazimierczuk Z, Shugar DZ. 1976. Tautomerism of iso-guanosine and solvent-induced keto-enol equilibrium. Z Naturforsch 31: 361–370. [DOI] [PubMed] [Google Scholar]
- Shapiro R. 1987. Origins: A skeptic’s guide to the creation of life on Earth. Bantam Dell, New York. [Google Scholar]
- Sharma N, Hoshika S, Hutter D, Bradley KM, Benner SA. 2014. Recombinase-based isothermal amplification of nucleic acids with self-avoiding molecular recognition systems (SAMRS). ChemBioChem 15: 2268–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sismour AM, Benner SA. 2005a. Synthetic biology. Expert Opin Biol Ther 5: 1409–1414. [DOI] [PubMed] [Google Scholar]
- Sismour AM, Benner SA. 2005b. The use of thymidine analogs to improve the replication of an extra DNA base pair. A synthetic biological system. Nucleic Acids Res 33: 5640–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sismour AM, Lutz S, Park J-H, Lutz MJ, Boyer PL, Hughes SH, Benner SA. 2004. PCR amplification of DNA containing non-standard base pairs by variants of reverse transcriptase from human immunodeficiency virus-1. Nucleic Acids Res 32: 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolke CD. 2009. Building outside of the box: iGEM and the BioBricks Foundation. Nat Biotechnol 27: 1099–1102. [DOI] [PubMed] [Google Scholar]
- Steinbeck C, Richert C. 1998. The role of ionic backbones in RNA structure. An unusually stable non-Watson–Crick duplex of a nonionic analog in an apolar medium. J Am Chem Soc 120: 11576–11580. [Google Scholar]
- Stoker HS. 1988. General, organic and biological chemistry, p. 745 McGraw-Hill, New York. [Google Scholar]
- Tawfik DS, Griffiths AD. 1998. Man-made cell-like compartments for molecular evolution. Nat Biotechnol 16: 652–656. [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Benner SA. 1994. Non-standard hydrogen bonding in duplex oligonucleotides. The base pair between an acceptor–donor–donor pyrimidine analog and a donor–acceptor–acceptor purine analog. J Am Chem Soc 116: 6929–6930. [Google Scholar]
- Voegel JJ, Benner SA. 1996a. Synthesis and characterization of non-standard nucleosides and nucleotides bearing the acceptor–donor–donor pyrimidine analog 6-amino-3-methylpyrazin-2(1H)-one. Helv Chim Acta 79: 1863–1880. [Google Scholar]
- Voegel JJ, Benner SA. 1996b. Synthesis, molecular recognition and enzymology of oligonucleotides containing the non-standard base pair between 5-aza-7-deaza-isoguanine and 6-amino-3-methylpyrazin-2(1H)-one, a donor–acceptor–acceptor purine analog and an acceptor–donor–donor pyrimidine analog. Helv Chim Acta 79: 1881–1898. [Google Scholar]
- Voegel JJ, Altorfer MM, Benner SA. 1993a. The donor–acceptor–acceptor purine analog: Transformation of 5-aza-7-deaza-1H-isoguanine (=4 aminoimidazo-[1,2-a]-1,3,5-triazin-2(1H)-one) to 2′-deoxy-5-aza-7-deaza-isoguanosine using purine nucleoside phosphorylase. Helv Chim Acta 76: 2061–2069. [Google Scholar]
- Voegel JJ, von Krosigk U, Benner SA. 1993b. Synthesis and tautomeric equilibrium of 6-amino-5-benzyl-3-methylpyrazin-2-one. An acceptor–donor–donor nucleoside base analog. J Org Chem 58: 7542–7547. [Google Scholar]
- von Krosigk U, Benner SA. 2004. Expanding the genetic alphabet. Pyrazine nucleosides that support a donor–donor–acceptor hydrogen bonding pattern. Helv Chim Acta 87: 1299–1324. [Google Scholar]
- Wei T, Pearson MN, Armstrong K, Blohm D, Liu J. 2012. Analysis of crucial factors resulting in microarray hybridization failure. Mol Biosyst 8: 1325–1339. [DOI] [PubMed] [Google Scholar]
- Wilds CJ, Wawrzak Z, Krishnamurthy R, Eschenmoser A, Egli M. 2002. Crystal structure of a B-form DNA duplex containing (l)-α-threofuranosyl (3′→2′) nucleosides: A four-carbon sugar is easily accommodated into the backbone of DNA. J Am Chem Soc 124: 13716–13721. [DOI] [PubMed] [Google Scholar]
- Win MN, Liang JC, Smolke CD. 2009. Frameworks for programming biological function through RNA parts and devices. Chem Biol 16: 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnacker M, Kool ET. 2013. Artificial genetic sets composed of size-expanded base pairs. Angew Chem Int Ed Engl 52: 12498–12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward RB. 1968. Recent advances in the chemistry of natural products. Pure Appl Chem 17: 519–547. [DOI] [PubMed] [Google Scholar]
- Woodward RB, Hoffmann R. 1970. The conservation of orbital symmetry. Springer, Heidelberg. [Google Scholar]
- Yang Z, Chen F, Chamberlin SG, Benner SA. 2010. Expanded genetic alphabets in the polymerase chain reaction. Angew Chem Int Ed Engl 49: 177–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Durante M, Glushakova LG, Sharma N, Leal NA, Bradley KM, Chen F, Benner SA. 2013. Conversion strategy using an expanded genetic alphabet to assay nucleic acids. Anal Chem 85: 4705–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, McLendon C, Hutter D, Bradley KM, Frye C, Benner SA. 2015. Helicase dependent isothermal amplification of DNA and RNA using self-avoiding molecular recognition systems. ChemBioChem 16: 1365– 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang Z, Sefah K, Bradley KM, Hoshika S, Zhu G, Cansiz S, Teng IT, Champanhac C, McLendon C, et al. 2015. Evolution of functional six-nucleotide DNA. J Am Chem Soc 137: 6734–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Avins JL, Klauser PC, Brandsen BM, Lee Y, Silverman SK. 2016. DNA-catalyzed amide hydrolysis. J Am Chem Soc 138: 2106–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubay G. 1988. The roots of modern biochemistry (ed. Kleinkauf H, von Döehren H, Jaenicke L), pp. 911–916. Walter de Gruyter, Berlin. [Google Scholar]