

In this review, Tuzlak et al. summarize the consequences of impaired apoptotic cell death in vertebrate development and tissue homeostasis and discuss possible explanations for the overall limited impact of apoptosis deficiency on these processes.
Keywords: BCL2 family, BH3-only proteins, cell death, development, tissue homeostasis
Abstract
“Programmed cell death or ‘apoptosis’ is critical for organogenesis during embryonic development and tissue homeostasis in the adult. Its deregulation can contribute to a broad range of human pathologies, including neurodegeneration, cancer, or autoimmunity…” These or similar phrases have become generic opening statements in many reviews and textbooks describing the physiological relevance of apoptotic cell death. However, while the role in disease has been documented beyond doubt, facilitating innovative drug discovery, we wonder whether the former is really true. What goes wrong in vertebrate development or in adult tissue when the main route to apoptotic cell death, controlled by the BCL2 family, is impaired? Such scenarios have been mimicked by deletion of one or more prodeath genes within the BCL2 family, and gene targeting studies in mice exploring the consequences have been manifold. Many of these studies were geared toward understanding the role of BCL2 family proteins and mitochondrial apoptosis in disease, whereas fewer focused in detail on their role during normal development or tissue homeostasis, perhaps also due to an irritating lack of phenotype. Looking at these studies, the relevance of classical programmed cell death by apoptosis for development appears rather limited. Together, these many studies suggest either highly selective and context-dependent contributions of mitochondrial apoptosis or significant redundancy with alternative cell death mechanisms, as summarized and discussed here.
BCL2, the founding member of the BCL2 family, was identified 30 years ago upon characterization of a frequently observed t14,18 chromosomal translocation in follicular lymphoma (Cleary et al. 1986; Tsujimoto and Croce 1986). After realizing that the presumed oncogenic potential of this gene lies within its anti-apoptotic property (Vaux et al. 1988), a number of related proteins were identified in protein–protein interaction screens, by sequence similarity, and/or by structural homology in humans as well as different vertebrate model organisms and viral pathogens. Some of these proteins exert inherent prosurvival function by blocking apoptosis, and some of them may even be converted into killers by limited proteolysis or upon alternative splicing of the encoding RNA or act exclusively as inducers of cell death (Tait and Green 2013; Czabotar et al. 2014). Orthologs of these proteins found in nematodes regulate apoptosis during embryonic development and upon exogenous stress in the germline, but molecular details do differ substantially from vertebrates, while, in insects, more distant BCL2 relatives seem to have no role in apoptosis regulation at all. In fact, it seems that “intrinsic” or “mitochondrial” BCL2-regulated apoptosis has been specially tuned to fit the needs of vertebrates (Bender et al. 2012). For more detailed information on the history of the discovery of the extended BCL2 family, we refer the interested reader to a recent comprehensive “time-line” article by Delbridge et al. (2016).
Next to anti-apoptotic BCL2, BCLXL, BCLW, BCLB, MCL1, and A1/BFL1, cell death-inducing family members include apical “BH3-only” proteins (e.g., BIM, PUMA, or BID) that act as preformed sentinels or are induced transcriptionally in response to stress as well as BAX-like proteins that control mitochondrial outer membrane (MOM) integrity. Once activated, BAX and related BAK can promote MOM permeabilization (MOMP) by pore formation, much like some bacterial toxins (Garcia-Saez et al. 2010) or phage-derived holins do (Pang et al. 2011), causing the release of “danger signals” from the intermitochondrial membrane space, including cytochrome c and inhibitor of apoptosis protein (IAP) antagonists such as SMAC/DIABLO (Tait and Green 2013). This leads to the activation and amplification of a proteolytic cascade, initiated and executed by endopeptidases of the caspase family. Along this route, caspases orchestrate largely immunologically silent cell death (and may even induce tolerance), contrasting their role in other, proinflammatory cell death paradigms (Martin et al. 2012). Although not all apoptotic cell death needs to be immunologically silent, it is fair to say that apoptosis occurring during vertebrate development indeed is. This is most likely also true in tissue homeostasis in the adult, where apoptosis aims to maintain steady state in the absence of injury or other types of stress (Martin et al. 2012).
Numerous studies have explored the role of anti-apoptotic BCL2 proteins in mouse development, and the consequences of loss of function of these proteins—highlighting tissue and cell type-specific prosurvival function of these proteins—have been reviewed recently elsewhere (Sochalska et al. 2015). Clearly, these studies pinpoint that probably all cell types, even platelets, rely on apoptosis inhibition by different BCL2 family proteins during their development or in the adult. But what about the other side of the coin? What can we learn about physiology (rather than pathophysiology) when we block apoptosis? This review summarizes the consequences of impaired apoptotic cell death in vertebrate development and tissue homeostasis, largely based on mouse studies, and discusses possible explanations for the overall limited impact of apoptosis deficiency on these processes. We close with some possibly provocative thoughts about putative redundancies among the different cell death machineries described today (Fig. 1) and lines of research that might address these possibilities.
Figure 1.

Signaling cascades implicated in developmental cell death and tissue homeostasis. Prevention of classic apoptosis pathways by genetic deletion of Bax and Bak (intrinsic) or impaired death receptor (DR) signaling (extrinsic) does not lead to major developmental abnormalities during embryogenesis or in surviving adults. Hence, additional cell death pathways have been implicated in the removal of excess cells during development. These pathways may involve caspases (green boxes) or not (blue boxes). Caspase-dependent cell death modalities involve apoptosis, ultimately engaging effector caspases 3, 6, and 7 and immunologically silent cell death that ultimately leads to corpse removal by phagocytes. Pyroptosis—a pathway activated by Caspases 1 and 11 in innate immune cells, mainly macrophages—leads to the cleavage of Gasdermin D and possibly other members of this family that ultimately triggers plasma membrane rupture. Lysosomal cell death involves lysosomal membrane permeabilization (LMP) and can be induced by several triggers, such as Ca2+ overload, H2O2, extensive DNA damage, or possibly even DR signaling, leading to the release of cathepsins. These enzymes were reported to trigger either a necrotic, apoptotic, or apoptosis-like response, e.g., by involving the BCL2 family protein BID. Accordingly, caspase-dependent as well as caspase-independent forms of lysosomal cell death have been reported. Entosis is a cell death that is triggered by signals from neighboring cells expressing E-cadherins or α-catenin, and those cells are engulfed, followed by phagosome-to-lysosome fusion, leading to cell degradation. Necroptosis is characterized by the activation of RIPK3 and phosphorylation of MLKL. It requires engagement of cell surface receptors, such as toll-like receptor 4 (TLR4), TNFR family members, or the interferon receptor (IFN). Engagement of these or other intracellular receptors, including DAI or TLR3, under conditions where Caspase 8 is inhibited triggers necroptosis. Caspase 8 can cleave and thereby inhibit RIPK1/RIPK3, which are needed to activate MLKL's pore-forming potential, leading to plasma membrane rupture during necroptosis. Programmed necrosis is induced by excess of Ca2+, increased reactive oxygen species (ROS) levels, or heavy metals, causing persistent mitochondrial membrane permeabilization through the mitochondrial permeability transition (MPT) pore that spans both the inner and outer mitochondrial membrane. Finally, ferroptosis is triggered by deprivation of cysteine, leading to glutathione (GSH) depletion and inhibition of the detoxifying enzyme GPX4, thereby causing lipid peroxidation and plasma membrane rupture by so-far undefined mechanisms.
Mitochondrial apoptosis: initiation, execution, and the enigmatic apoptotic pore
Intense research over the past decades has shaped a reasonably sharp picture of how prodeath BCL2 proteins initiate and execute mitochondrial apoptosis via MOMP (Fig. 2). The early “rheostat model” proposed that the ratio of proapoptotic and anti-apoptotic proteins in a given cell known at that time—e.g., BCL2:BAX (or other combinations)—defines cellular destiny (Oltvai et al. 1993; Sedlak et al. 1995). This model had to be dismissed soon due to the discovery that BH3-only proteins can promote apoptosis by direct interaction with BCL2 prosurvival proteins, displacing BAX (Yang et al. 1995; Wang et al. 1996). The “neutralization” model of mitochondrial apoptosis hence suggested that BAX/BAK are constitutively active when all BCL2-like proteins present in a given cell are neutralized by direct engagement with BH3-only proteins that display different affinities for the former (Chen et al. 2005; Willis et al. 2005, 2007). This model had some competition based on findings that some BH3-only proteins, such as truncated BID (tBID) (Wang et al. 1996) or BIM, can trigger BAX/BAK conformational changes and activation by direct, albeit transient, interaction (Cheng et al. 2001; Letai et al. 2002; Kuwana et al. 2005). The ensuing “direct activation” model (Kim et al. 2006; Brunelle and Letai 2009) built on these parallel observations and gained much support by additional studies making use of full-length recombinant proteins to study permeabilization of artificial lipid vesicles in the absence or presence of mitochondria-specific lipids (in particular cardiolipin) but also whole mitochondria. These observations formed the basis of the “embedded together” model (Lovell et al. 2008), which incorporated the role of the mitochondrial membrane in the regulation of the interactions between Bcl-2 proteins in an advanced version of the “direct activation” model (Fig. 2; Bogner et al. 2010). Furthermore, the generation of “BH3 domain swap mutants,” where BH3 domains of different origin (e.g., those derived from BAD, PUMA, or NOXA) were grafted into the mouse Bim gene locus, provided long-sought-after in vivo evidence for this model (Merino et al. 2009). Previous nuclear magnetic resonance (NMR) (Walensky et al. 2006; Gavathiotis et al. 2008) and crystallography-based structural studies also suggested that a conformational change in BAX (and BAK) triggered by direct BH3-only protein binding is crucial to generate the minimum functional unit of these proteins (Czabotar et al. 2013; Brouwer et al. 2014), referred to as the “BH3 in groove” symmetric homodimer (involving helices α2–α5). Dimer formation in the MOM leads to the exposure of additional contact sites, likely involving helices 6 and 9 and allowing formation of higher-order oligomers between individual dimers that can span the MOM like a clamp (Bleicken et al. 2014). This culminates in the progressive assembly of a toroidal pore (a mix of proteins and lipids) that was visualized only recently in elegant studies using superresolution microscopy and stimulated emission depletion (STED) nanoscopy (Grosse et al. 2016; Salvador-Gallego et al. 2016). Of note, an autoinhibited BAX dimer has been described recently to be present in the cytosol (Garner et al. 2016). This dimer conformation needs to be broken up to allow BAX activation, translocation, and accumulation in the MOM. However, this process is antagonized in healthy cells by continuous BCL2/BCLX-driven retrotranslocation of heterodimers back into the cytosol (Edlich et al. 2011; Todt et al. 2015), while retention at the MOM is facilitated by membrane-resident VDAC2 and BAK (Ma et al. 2014), adding additional layers of complexity to apoptosis regulation (Fig. 2).
Figure 2.

Models for the activation of BAX/BAK and apoptotic pore formation at the MOM. (A) The neutralization model proposes that BAX and BAK are sequestered in their active form by BCL2 prosurvival proteins and that apoptotic stimuli lead to the recruitment of BH3-only proteins by either post-translational stabilization or transcriptional induction. These BH3-only proteins then bind to BCL2-like proteins, thereby releasing BAX/BAK, which are intrinsically active. (B) The “direct activation” model implies that BAX or BAK needs to be directly engaged by “activator BH3-only” proteins in order to impose a conformational change needed for dimerization and subsequent oligomerization. In order to inhibit the initiation of apoptosis, BCL2 proteins sequester activator BH3-only proteins to prevent BAX/BAK activation. Binding of BCL2 to sensitizer BH3-only proteins (e.g., BAD, HRK, BMF) is needed to free activator BH3-only proteins (BIM, BID, and PUMA) to activate BAX or BAK. (C) The embedded together model implies that BCL2-like proteins sequester BH3-only proteins at the MOM, thereby inhibiting their interaction with and direct activation of BAK/BAX. In the MOM, BCL2-like proteins themselves also prevent dimerization and pore formation of BH3-only protein-activated/embedded BAX/BAK monomers. Additionally, the retrotranslocation concept proposes that BAX (and also BAK with slower kinetics) is in an equilibrium and is extracted from the MOM in a BCL2/BCLX-dependent manner and shuttled back into the cytoplasm, where it oscillates between an autoinhibitory dimer and a monomeric state. Only the latter is able to translocate to the mitochondria, where it needs to accumulate in order to form an active dimer that can then form the apoptotic pore upon oligomerization.
However, to the delight of some but dismay of others, the requirement of BH3-only proteins for direct BAX/BAK activation needs to be reconsidered (Garcia Saez and Villunger 2016). First of all, in an attempt to reconcile all of these models, it became evident that BCL2 proteins can engage BH3-only proteins as well as activated BAX/BAK with high affinity (Llambi et al. 2011). Along that line, a recent study combining structural biology with mouse genetics provided the first evidence that impaired binding of BCLXL to BAK due to mutation of Glu75 of BAK (a mutant generated randomly during ENU mutagenesis in mice not affecting its structure or binding to direct activator BH3-only proteins and killing potential in vitro) is sufficient to trigger spontaneous cell death upon MCL1 inhibition in mouse embryonic fibroblasts (MEFs). Importantly, loss of BAK–BCLXL interaction triggered a number of in vivo phenotypes associated usually with loss of BclxL in vivo, including T-cell loss and reduced platelet life span (Lee et al. 2016). This study also provides some evidence that BAK can act as an activator of BAX in some settings, an interesting idea proposed earlier (Ma et al. 2014). Overall, this suggests that sequestration of BAK by BCLXL is much more critical for keeping cells alive than preventing BAK from becoming directly activated by BH3-only proteins. These findings are consistent with other studies proposing a unifying or hierarchical model of BCL2 protein function (Llambi et al. 2011; Chen et al. 2015) and also a recent report by Dai et al. (2015), who observed that drug sensitivity of hematopoietic cancers correlates well with the oligomeric state and the amount of BAK present in a cell rather than with the ability of BAK to engage direct activators, such as BIM or PUMA. Perhaps all of these interactions between BH3-only proteins and prosurvival BCL2 proteins serve only one higher purpose: facilitating direct interactions of BAX with BAK.
Furthermore, BAX or BAK can spontaneously accumulate at and permeabilize the MOM in the absence of any of the proteins previously reported to possess direct activation potential (i.e., BID, BIM, and PUMA as well as unrelated p53 and Rb) when all prosurvival proteins present are effectively neutralized. Of note, literally all known BH3-only proteins can be rendered to become direct activators at least in vitro, depending on experimental conditions (Du et al. 2011; Chen et al. 2015). Hence, it is even more surprising that this autoactivation also happens in the absence of any known BH3-only protein ever reported to be involved in mitochondrial apoptosis or autophagy (O'Neill et al. 2016). While these studies do not completely exclude an accelerating or modulatory role of direct interaction of some BH3-only proteins with BAX or BAK in a given cellular- or stimulus-dependent context, it challenges the universal validity of the “direct activation/embedded together” models. It also points to the existence of so-far undefined membrane components, possibly even of lipid origin, that suffice to promote BAX or BAK dimerization and subsequent pore formation. As such, interrogation of protein–protein and lipid–protein interactions in the context of the BAX/BAK pore can be expected to yield new insights into the molecular biology of MOMP.
Along this line, despite the rather absolute apoptosis resistance phenotype of cells lacking BAX and BAK (see below), we need to keep in mind that the putative BAX-like family member BOK might be a mediator of mitochondrial apoptosis, as it can exert pore-forming properties in certain contexts. Work over the last few years has indicated that BOK functions may partly overlap with those of BAX/BAK but that there might also be unique roles to this still poorly understood family member. Regarding its proapoptotic functions, it remains to be clarified whether BOK can directly form pores in the MOM and, if confirmative, of what nature these pores are. For example, is the intermediate of the elusive BOK pore a dimer? BOK-induced membrane activity so far has been shown only in artificial membranes (liposomes), and proteasomal degradation of BOK, regulated by members of the ERAD pathway, was also put forward recently as a major means of controlling (proapoptotic) BOK activity (Llambi et al. 2016), yet, in several tissues, BOK is found readily expressed (Ke et al. 2012). However, more in-depth biophysical analyses, preferably within a lipid membrane environment (as done for BAX) (for example, see Bleicken et al. 2014), are necessary to determine the protein–protein interactions, composition, size, and stability of pores. This would also allow one to determine whether members of the BCL-2 family can cooperate with BOK (e.g., tBID, BAX, and BAK) or inhibit its activity (e.g., MCL-1 and BFL-1/A1). Furthermore, solving the three-dimensional structure in the membrane-bound conformation is a prerequisite to advance our understanding of the molecular events regulating BOK activation.
What have we learned about developmental cell death from the analysis of BH3-only knockout mouse mutants?
BH3-only proteins are considered to be stress-induced proteins and stimulus-dependent initiators of mitochondrial apoptosis that depend on the presence of BAX or BAK to execute their function (Wei et al. 2001). Despite remaining issues on how these proteins ultimately activate apoptosis, it is generally accepted that some BH3-only proteins are more potent killers than others: These include BIM, BID, and PUMA, which can bind to all prosurvival homologs with comparable affinity, while NOXA, BAD, BMF, DP5/HRK, and BIK/BLK, on the other hand, are believed to act mainly as auxiliary prodeath proteins by helping to sequester BCL2 and the like; however, among those, NOXA recently has been assigned direct BAX/BAK-activating potential (Chen et al. 2015). Since such properties have also been assigned to BMF before (Du et al. 2011), a strict discrimination of two groups of BH3-only proteins appears difficult.
Substantial redundancy between BH3-only proteins has been observed in in vitro analyses and gene targeting studies in mice exploring the consequences of loss of one or a combination of BH3-only proteins (Table 1). Therefore, it became evident that Bim is probably the most critical member of the family, as single deletion of this gene has the most pronounced effects of all BH3-only protein genes studied in isolation (Pinon et al. 2008). Most strikingly, ablation of Bim causes the loss of approximately half of the progeny before embryonic day 10.5 (E10.5) in mouse development (Bouillet et al. 1999). The basis for this phenomenon and its partial penetrance is still unclear but may be related to the observation that FGF4-driven receptor tyrosine kinase signaling in trophoblast stem (TS) cells limits BIM protein levels, thereby setting the balance between TS cell proliferation and differentiation in early embryogenesis (Yang et al. 2006).
Table 1.
Phenotypic characteristics of BH3-only protein loss-of-function mutants

Adult Bim−/− animals show the most profound developmental defects within the hematopoietic system, displaying severe lymphoid hyperplasia. Increased secretion of immunoglobulins (Igs), including autoantibodies, by accumulating plasma cells leads to fatal kidney disease in ∼50% of 1-yr-old Bim−/− mice (Bouillet et al. 1999). These effects were found ameliorated upon backcrossing onto a C57BL/6 genetic background, but cell death-resistance phenotypes of leukocytes and lymphadenopathy persisted. Importantly, loss of only one allele can prevent lymphopenia and polycystic kidney disease observed in mice lacking Bcl-2, while loss of both alleles are needed to prevent melanocytes from dying (Bouillet et al. 2001). Thus, BIM seems to be a crucial player in regulating apoptosis during kidney development, in hair follicle stem cells, and in the hematopoietic system. Concerning the latter, BIM is necessary for not only the deletion of autoreactive B and T lymphocytes during development (Bouillet et al. 2002; Enders et al. 2003; Villunger et al. 2004) but also the termination of T-cell- and B-cell-mediated immune responses (Hildeman et al. 2002; Pellegrini et al. 2003), affinity maturation (Fischer et al. 2007), or the survival of granulocytes (Villunger et al. 2003b). Moreover, it also acts as a gatekeeper, preventing cancer in response to oncogenic stress, as exemplified in the accelerated onset of c-MYC-driven lymphomas in the absence of a single allele of Bim (Egle et al. 2004; Frenzel et al. 2010).
Although, BIM-deficient cells show profound protection from a number of apoptotic stimuli, such as cytokine withdrawal and ionomycin or steroid treatment, they still remain sensitive to Fas ligation, etoposide treatment, or γ irradiation (Bouillet et al. 1999). These observations already suggested that apoptosis following DNA damage is regulated by other BH3-only proteins. Shortly thereafter, the p53 up-regulated modulators of apoptosis, PUMA (Nakano and Vousden 2001; Yu et al. 2001) and NOXA/PMAIP (Oda et al. 2000), two BH3-only proteins induced by p53, were first described. NOXA seems to be less critical in this process, since its deletion does not impair apoptosis upon DNA damage in most cell types (Villunger et al. 2003a) but protected gastrointestinal epithelial cells from the consequence of high-dose γ irradiation in vivo (Shibue et al. 2003). Of note, keratinocytes and fibroblasts are significantly protected from UV-mediated cell death in its absence (Naik et al. 2007). Interestingly, NOXA might also play a role in immunity, as Noxa−/− mice were reported to show defects in the selection of high-affinity antigen receptors upon activation of T and B cells (Wensveen et al. 2010, 2012) and in emergency hematopoiesis (Wensveen et al. 2011). Loss of Puma, on the other hand, protects hematopoietic (stem) cells (Labi et al. 2010; Michalak et al. 2010), hepatocytes, and gastrointestinal epithelium (Qiu et al. 2008, 2011) effectively from genotoxic stress and γ irradiation (Happo et al. 2010), while combined loss of Puma and Noxa further increases radio resistance in some cell types, such as mouse oocytes (Michalak et al. 2008; Kerr et al. 2012). Interestingly, only the combined deletion of Puma, Noxa, and Bim provides protection to an extent comparable with p53 deficiency, at least in MYC-driven lymphoma cells (Happo et al. 2010), even though BIM is not a direct target of p53. The latter suggests the existence of a cross-talk between the DNA damage response and BIM, most likely involving activation of JNK signaling (Pinon et al. 2008).
Notably, PUMA-deficient lymphocytes are also refractory to certain p53-independent apoptotic stimuli such as dexamethasone or growth factor deprivation (Jeffers et al. 2003; Villunger et al. 2003a), and compound mutants lacking BIM and PUMA reveal synergistic functions of these proteins in cell death induction (Erlacher et al. 2006). However, no additional developmental defects were noted in Bim−/−Puma−/− mice when compared with Bim mutant mice (Erlacher et al. 2006). with the exception of a more pronounced lymphadenopathy and more severely compromised deletion of autoreactive T cells (Gray et al. 2012). More recently, it has been shown that Puma−/− mice display an increase in adipocyte size and increased serum leptin levels. Accordingly, food intake was reduced during a high-fat diet; however, no differences in body mass and glucose metabolism were observed, probably due to reduced physical activity compared with control mice (Litwak et al. 2016). However, it is difficult to put the observed phenotype in a developmental context, as the used mouse model had PUMA deleted in all somatic cells. Of note, PUMA has also been associated with glucose-induced cell death of pancreatic islets (McKenzie et al. 2010), maintaining the interest for this protein in diabetes- and obesity-associated research.
BID is a particular BH3-only protein because it is the only member of the subfamily that is intrinsically structured and needs to be cleaved by caspases, most commonly by caspase 8, for activation (Li et al. 1998; Luo et al. 1998). As such it connects the extrinsic and intrinsic apoptosis signaling pathways in certain cell types that rely on efficient neutralization of XIAP to generate above-threshold caspase activity upon death receptor (DR) ligation (Jost et al. 2009). As a consequence, hepatocytes or pancreatic β cells from BID-deficient mice are insensitive to Fas ligation in vivo (Yin et al. 1999). Neurons from Bid−/− mice were reported to better resist ischemic cell death that can involve caspase 8-activating ligands of the TNF family (Plesnila et al. 2001). However, the latter has been a matter of debate (Engel et al. 2010). Furthermore, BID has been reported to be a p53 target, and Bid−/− MEFs were suggested to be more resistant to DNA-damaging agents (Sax et al. 2002). Along this line, it is interesting to note that BID can be phosphorylated by ATM and has been reported to affect S-phase checkpoint stringency and mitochondrial reactive oxygen species (ROS) production (Kamer et al. 2005; Zinkel et al. 2005; Kaufmann et al. 2007), yet the role of BID in the DNA damage response remains disputed (Kaufmann et al. 2007). The combined deletion of BIM and BID did not cause any developmental abnormalities or phenotypes that exceed those observed in BIM mutant mice, with the notable exception that double-knockout (DKO) mice were protected from TNF-driven fulminant hepatitis more than single-mutant mice, placing BID and BIM downstream from TNFR signaling in hepatocytes (Kaufmann et al. 2009).
In an attempt to phenocopy BAX/BAK mutant mice (discussed below) in support of the direct activation model of BH3-only protein function, mice lacking BID, BIM, and PUMA were generated (Ren et al. 2010). However, ultimately, these triple-mutant (triple-knockout [TKO]) mice did not mimic combined loss of BAX and BAK, as indicated, e.g., by the fact that the rate of perinatal lethality and certain developmental phenotypes, such as persisting interdigital webbing and vaginal atresia, were clearly less pronounced and less penetrant than in BAX/BAK DKO mice (Ren et al. 2010). Furthermore, a follow-up publication by the very same group showed that TKO MEFs are less resistant to apoptosis induction than BAX/BAK DKO MEFs, and deletion of NOXA on top of BID, BIM, and PUMA (QKO) was required to generate comparable death resistance phenotypes in response to some but not all stimuli tested (Chen et al. 2015). The latter suggests that any of the remaining BH3-only proteins in TKO mice may become an activator of cell death in the right context.
Deletion of any of the other BH3-only proteins on their own failed to reveal key roles in development, and only minor roles in cell death signaling were observed. The synergistic potential of these “less potent” BH3-only proteins can be observed best when looking at loss-of-function studies on a BIM-deficient background. While BMF-deficient mice show only mild phenotypes, including B-cell-restricted lymphadenopathy and reduced sensitivity of lymphoid cells to glucocorticoids or histone deacetylase inhibitors (Labi et al. 2008; Grespi et al. 2010), the combined loss of BMF and BIM leads to pronounced developmental abnormalities, including webbed feet, vaginal atresia, and malocclusion of the incisors (Hubner et al. 2010; Labi et al. 2014). Additionally, increased lymphadenopathy, higher serum Ig levels, and a higher incidence of glomerulonephritis and malignant disease, leading to premature lethality, were observed when compared with BIM mutant mice (Labi et al. 2014; Woess et al. 2015). BMF also supports BIM in promoting mammary epithelial cell death during organ morphogenesis (Mailleux et al. 2007; Schmelzle et al. 2007) and the late stage of involution after the cessation of lactation, a classic example of developmental apoptosis (Sakamoto et al. 2016; Schuler et al. 2016).
Another prominent example for cell type-specific redundancy of BH3-only proteins is BIK, the first protein to be identified in this class (Boyd et al. 1995). BIK has been shown to reside at the endoplasmic reticulum (ER), where it might be involved in the transfer of Ca2+ to mitochondria. Although expressed at significant levels in several tissues, no overt phenotype or cell death deficit could be observed in mice lacking Bik (Coultas et al. 2004). However, BIK codeletion with BIM causes infertility in males due to perturbed spermatogenesis, resembling the phenotype of BAX-deficient mice, although the block in spermatogenesis occurs somewhat later in their development (Coultas et al. 2005). Notably, no additive cell death effects were observed when BIK and NOXA were codeleted (Happo et al. 2012).
Despite intensive research since its early discovery, the cell death function of the BH3-only protein BAD (Yang et al. 1995) remains somewhat elusive. BAD deficiency itself does not cause any aberrations in embryonic development, although Bad−/− mice were reported to show impaired B-cell function and develop diffuse large B-cell lymphoma with increased incidence (Ranger et al. 2003). Subsequent reports suggested a role in platelet life span (Kelly et al. 2010) and as regulator of glucose-induced insulin secretion and gluconeogenesis upon phosphorylation of its BH3 domain, precluding its binding to BCL2 (Danial et al. 2003, 2008; Gimenez-Cassina et al. 2014). Findings that BAD-deficient mice are less sensitive in a model of systemic TNF-mediated hepatitis (Yan et al. 2013) could not be confirmed in an independent study (Ottina et al. 2015). Of note, responses to TNF or glucose are strongly influenced by the genetic background of the mouse model used and, when poorly controlled for, can contribute to sometimes inconsistent findings (Leiter 2002; Vanden Berghe et al. 2015). Combined deletion of BAD plus BIM (Kelly et al. 2010) or BMF (Baumgartner et al. 2013), respectively, did not result in any particular developmental phenotypes and revealed only mildly increased cell death resistance upon cytokine withdrawal, B-cell-specific lymphadenopathy, and increased susceptibility to spontaneous as well as radiation-induced tumorigenesis.
Finally, deletion of Dp5/Hrk, a BH3-only family member that is expressed mainly in the central and peripheral nervous system (Imaizumi et al. 1997), does not impair neuronal development, although sensory neurons derived from these mice are less sensitive to nerve growth factor withdrawal ex vivo (Coultas et al. 2007). HRK-deficient mice were also reported to be protected from high-fat-diet-induced loss of glucose tolerance and displayed increased pancreatic β-cell mass (Cunha et al. 2012); however, this phenotype awaits independent confirmation.
In summary, these studies point toward a large degree of redundancy among the BH3-only proteins in developmental cell death and substantial flexibility regarding their use and requirement in stress-induced apoptosis. Based on the reported phenotypes in single and compound BH3-only protein mouse mutants, the general relevance of mitochondrial cell death for organ development in the embryo and tissue homeostasis in the adult must be strongly questioned. Certainly, it remains possible that the combined deletion of all BH3-only proteins listed above may phenocopy BAX/BAK mutant mice, but, given the assumption that not all of these proteins have exclusive roles in apoptosis but also intersect, e.g., with the regulation of mitochondrial dynamics or the autophagy machinery, data interpretation will remain difficult.
Defining the role of mitochondrial cell death in development by loss-of-function analysis in mice lacking BAK, BAX, BOK, or combinations thereof
BAX and BAK loss-of-function mutants have been generated early on but failed to reveal major developmental defects (Table 2). BAX mutant mice showed mild cell death resistance of neurons to trophic factor deprivation ex vivo, an increase in neurons and lymphocytes, and, paradoxically, male sterility due to increased spermatogonia death, halting spermatogenesis (Knudson et al. 1995; Deckwerth et al. 1996; White et al. 1998; Fan et al. 2001). BAK mutant mice showed no discernable defects in development or mitochondrial or DR-mediated apoptosis. In striking contrast, mice lacking both genes showed significant developmental defects and a severe impairment in cell death in response to a broad range of apoptotic triggers. Most BAX/BAK DKO mice died perinatally, and only <10% of mice survived to adulthood (Lindsten et al. 2000). Of note, clear developmental phenotypes associated with defective developmental apoptosis, such as a persistent interdigitating mesenchyme and an imperforate vaginal canal, were observed. Furthermore, these mice also showed neurological deficits, as seen by circling behavior and frequent seizures upon stress. Unresponsiveness to auditory stimulation indicated deafness, and pups frequently died after a few days, often showing suckling defects and social isolation from littermates (Lindsten et al. 2000). The neurological defects were associated with excess numbers of undifferentiated cells in the periventricular zone, and follow-up studies documented massive lymphadenopathy in surviving or fetal liver-reconstituted mice (Lindsten et al. 2000; Rathmell et al. 2002), increased neuronal progenitor cell number in situ (Lindsten et al. 2003), and persisting fetal ocular vasculature and inner retinal cells, including rod photoreceptors, which normally die if they fail to migrate to the outer retina (Hahn et al. 2002, 2003). However, formation of other organs—including the kidney, heart, liver, lung, pancreas, or bladder—appeared to develop and function normally in the surviving mice (Lindsten et al. 2000; Lindsten and Thompson 2006). This raises the possibility that the development of these organs may not be linked to developmental apoptosis or that alternative means to induce apoptosis or other forms of developmental cell death are engaged (Fig. 1).
Table 2.
Phenotypic characteristics of Bax/Bak/Bok loss-of-function mutants
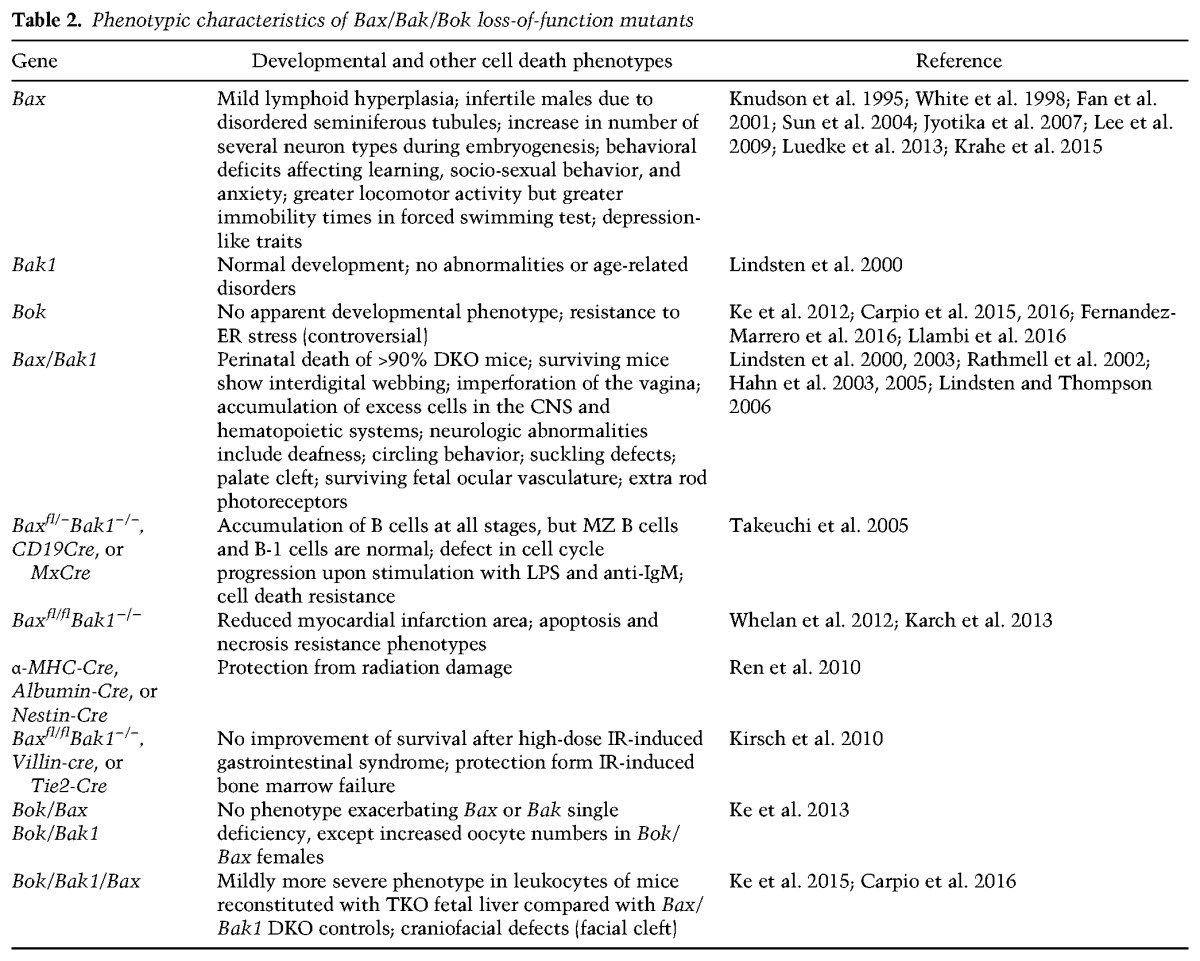
One such cell death mediator might be BOK; however, apart from overexpression data, physiological evidence for a proapoptotic role of BOK is still meager. No developmental abnormalities or spontaneous phenotypes were reported for the first Bok gene-deficient mouse model generated, and hematopoietic cells derived from these mice were normally sensitive to classical apoptotic stresses (Ke et al. 2012). Along that line, Bok−/−Bax−/− and Bok−/−Bak−/− doubly deficient mice displayed no exacerbated phenotype over Bax or Bak single-deficient mice, respectively, with the exception of increased oocyte numbers in aged Bok−/−Bax−/− females (Ke et al. 2013). The latter finding may point toward a role of BOK in oocyte apoptosis that warrants further investigation. Recently, Ke et al. (2015) also reported a more pronounced phenotype in lymphocytes in the peripheral blood of mice reconstituted with Bok−/−Bax−/−Bak−/− TKO hematopoietic stem cells compared with Bax−/−Bak−/− DKO controls, indicating that BOK may have redundant functions with BAX and BAK in this compartment. This is an interesting observation, taking into account the low BOK expression in mouse hematopoietic cells (Ke et al. 2012). Maybe not surprising, a comparison of DKO and TKO lymphocytes failed to reveal any differences in response to various apoptosis inducers, given that DKO cells were already fully protected. It is conceivable that more pronounced effects may be seen during development or in the adult organism in TKO tissues that are normally high in BOK expression, and a thorough analysis of embryogenesis in TKO mice or conditional deletion of all three genes in a given tissue is eagerly awaited.
Mitochondrial apoptosis and its role in early embryogenesis
Despite the fact that loss of individual BH3-only proteins or combinations thereof points toward rate-limiting roles in tissue homeostasis in adults, best documented in the hematopoietic system, the phenotypes observed are, at second thought, rather minor (Table 1). The same holds true when considering that some Bax−/−Bak−/− mice can survive to adulthood (Lindsten and Thompson 2006), and chimeric mice carrying a reconstituted hematopoietic system lacking BAX/BAK and BOK develop “only” gradual lymphadenopathy with signs of autoimmunity but not cancer or inflammatory phenotypes due to excess leukocytes (Ke et al. 2015). Based on this and the fact that most BAX/BAK-deficient mice die perinatally only by largely unexplored causes, we need to conclude that cell death observed early in embryogenesis does not necessarily involve mitochondria or, alternatively, may be mediated by alternative factors that can promote MOMP. It is worth mentioning here that, in most vertebrates, cell death is not observed prior to gastrulation (Penaloza et al. 2006). However, in mammals, such as mice, cell death occurs already at the morula stage during compaction as well as during blastocoel formation and differentiation of the inner cell mass (ICM) in the early blastocyst (Zakeri et al. 2005; Busso et al. 2010). Why, then, is development not perturbed in the absence of BAX/BAK? One is tempted to argue here that an excess of cells of the ICM can be tolerated easily at that early stage, as we also introduce extra embryonic stem cells into the blastocyst in the process of generating gene-modified mice. Perhaps cell death in the blastocyst stage embryo is simply not critical for development or may not follow classic routes. Excess cells in BAX/BAK DKO blastocysts have not yet been demonstrated but might be cleared by other means; for example, cell cannibalization via entosis (Krishna and Overholtzer 2016) or nonprofessional phagocytosis by neighboring cells upon exposure of “eat me” signals (Arandjelovic and Ravichandran 2015). Certainly, a failure to clear dying cells from embryoid bodies—e.g., observed in the context of ATG5 or Beclin-1 deficiency—blocks cavitation (Qu et al. 2007), suggesting that lack of cell death and accumulation of cells at that stage should do the same. Clearly, this process does not seem perturbed in the absence of BAX/BAK. However, aficionados of alternative modes of cell death, such as necroptosis, may find it intriguing that general caspase inhibition by z-VAD-FMK or selective inhibition of caspase 8 halts the development of the preimplantation embryo (Zakeri et al. 2005; Busso et al. 2010). In fact, caspase inhibition even enhances rates of cell death normally observed in the ICM in vitro. However, whether this death occurs by necroptosis is yet unclear (Fig. 1).
Taken together, it seems that timed removal of excess cells of the ICM in the early preimplantation development of the embryo is either not mediated by BAX/BAK, compensated by alternative types of death, or simply well tolerated and balanced by other means (e.g., reduced proliferation rates or phagocytic clearance mechanisms). Interestingly, lack of Bax/Bak appears to delay the differentiation of embryonic stem cells upon retinoic acid treatment or embryoid body or teratoma formation by blocking CD95-mediated cell death of less differentiated cells (Wang et al. 2015). The notion that limited caspase activity—most likely generated by caspase 8 temporarily activating caspase 3 (Fujita et al. 2008)—seems required for proper blastocyst formation in vitro is certainly interesting but somewhat confounded by the fact that, in the absence of Casp8, embryogenesis halts only around E10.5 due to defective angiogenesis (Varfolomeev et al. 1998), while Casp3-deficient mice can come to term (Honarpour et al. 2000; Zheng et al. 2000), meaning that preimplantation development is not affected by lack of caspase activity. Whether BAX/BAK can generate such caspase activity in the absence of caspase 8 needs to be analyzed, but it is worth mentioning that caspase 6 can also cleave RIPK1 (van Raam et al. 2013) and could hence put a halt to necroptosis in the early embryo when caspase 8 is absent.
Following similar lines of thought, tube formation in the embryo is crucial to the normal development of many organs, including the mammary glands, kidneys, and lungs, the latter two developing normally in BAX/BAK DKO mice. Tube formation can be achieved by the closure of an epithelial sheet layer (e.g., neural tube and primitive gut) or by lumen formation involving cell clearance in a compact cell mass, also known as cavitation (female reproductive tract and exocrine glands). In mouse embryos, the first cavitation event happens shortly prior to gastrulation, converting the solid embryonic ectoderm into a hollow egg cylinder, thereby generating the proamniotic cavity, surrounded by a single epithelial cell layer (Coucouvanis and Martin 1995). The basic principles observed at that stage most likely reflect what is happening in organogenesis later many times. The cell death observed at that stage shows classic hallmarks of apoptosis (DNA laddering and pyknotic nuclei) and is induced by cues generated from the visceral endoderm, while the columnar epithelium is rescued from death by signals generated from its interaction of the basement membrane (i.e., integrin signaling) (Coucouvanis and Martin 1995; Murray and Edgar 2000). This type of cell death is hence highly reminiscent of what is often referred to in the cell death field as “anoikis,” which is induced upon loss of contact with the extracellular matrix or expulsion of cells from the epithelium (e.g., in the gastrointestinal tract). This death is blocked by BCL2 overexpression or loss of certain BH3-only proteins (Schmelzle et al. 2007) and hence is supposedly dependent on BAX/BAK. Again, this process must progress normally in their absence in the early embryo, as it occurs during lumen formation in many other organs during embryogenesis, e.g., in the kidneys, lungs, or heart. It deserves mentioning here that caspase 8 has been implicated in anoikis and lumen formation before (Frisch and Screaton 2001). Although this has been deemed controversial, one may speculate that caspase 8 might well play a critical role in development, in particular when BAX and BAK are missing. Whether the surviving DKO mice have impaired duct formation in exocrine glands, such as the mammary gland, remains unexplored.
Mitochondrial apoptosis as a regulator of tissue homeostasis in the adult
Tissue-specific codeletion of Bax and Bak, usually achieved by conditional deletion of Bax on a Bak−/− background, raises the question of whether mitochondrial apoptosis is critical for homeostasis in the adult. For example, Bax/Bak codeletion in the gastrointestinal tract using Villin-Cre does not cause spontaneous tissue hyperplasia, documenting that cell death of gastrointestinal epithelial cells can happen in the absence of mitochondrial apoptosis, and tissue homeostasis is effectively maintained in its absence (Kirsch et al. 2010). This is also supported by the fact that surviving DKO mice have not been reported to develop hyperplasia in the gastrointestinal tract. It remains possible, though, that shedding of cells from the tips of villi and their excretion together with feces are sufficient to maintain gut homeostasis.
Also, in tissues with a low proliferative index, like the brain (Ren et al. 2010), heart, and liver (Whelan et al. 2012; Karch et al. 2013), BAX and BAK seem dispensable for homeostasis, as their codeletion does not seem to trigger a hyperplastic phenotype in these organs (Table 2). This may be less of a surprise, as the promoters driving Cre expression to delete Bax on a Bak-deficient background usually stem from genes that associate with terminal differentiation. As the mitotic index and rate of post-mitotic cell death in these organs are known to be low, the observation time in these studies may not have been long enough to reveal developing organ deficits. Alternatively, mechanisms other than mitochondrial apoptosis may compensate to maintain organ homeostasis. It will certainly be of interest to monitor such conditional mouse mutants long term or expose them to challenges that associate with tissue regeneration that needs to be balanced by cell death induction; e.g., upon recovery from partial hepatectomy.
Taken together, all of these findings challenge textbook dogma about the importance of apoptosis in embryonic development or tissue homeostasis. As deletion of BAX and BAK effectively protects many cell types from death induced by diverse apoptosis-inducing agents and signaling cues but only causes modest changes in adult tissue homeostasis and embryogenesis, alternative explanations are needed.
As quickly touched on before, such an explanation might be redundancy with BOK. However, expression of Bok mRNA in tissue from BAX and BAK single or compound knockout mice as well as BOK protein in tissue from BAX or BAK single-knockout mice appears, on the whole, unaffected, arguing against compensatory effects, as do studies combining BAX or BAK deficiency with loss of BOK (Ke et al. 2012, 2013). The fact that mice reconstituted with Bax/Bak/Bok TKO hematopoiesis showed increased lymphocyte numbers in peripheral blood and a higher infiltration rate of lymphocytes into the gut may also be explained by loss of other processes, as published data also point toward nonapoptotic roles of BOK, such as in trophoblast proliferation (Ray et al. 2010). Even protective roles have been reported for BOK. It has been shown that Bok cells can be more sensitive to the Golgi/ER-impacting drug brefeldin A, presenting with a defective IRE1α-mediated stress response and enhanced apoptosis (Echeverry et al. 2013). Likewise, D'Orsi et al. (2016) reported Bok-deficient neurons to be more sensitive to oxygen/glucose deprivation-induced or seizure-induced neuronal injury. In essence, these findings point toward nonapoptotic or even protective roles of BOK at the ER. Certainly, it will be very interesting to see whether the Bax/Bak/Bok triple deficiency affects embryogenesis more severely than loss of BAK and BAX in tissues that do express discernable levels of BOK, such as the ovaries, uterus, or gastrointestinal tract (Ke et al. 2012). However, whether putative changes are related to impaired cell death due to lack of BOK needs to be thoroughly validated.
Redundancies between cell death modalities as a means to control developmental cell death?
Assuming that the analysis of the above-mentioned TKO mice will not provide a universally satisfying answer to the question of whether mitochondrial apoptosis is strictly essential and uniquely required for embryogenesis and organ homeostasis, the follow-up question would be: What cell death mechanisms could contribute to the development of or compensate for Bax/Bak/Bok deficiency in tissue homeostasis? While it was easy to speculate why cells die when caspase activation is inhibited in the presence of MOMP, this will require entirely different thinking, looking at developmental cell death in the absence of BAX/BAK/BOK. In the first case, it is quite easy to accept that energy depletion, breakdown of membrane gradients, and solute influx in cells undergoing MOMP in a quantitative manner ultimately trigger cellular demise even in the absence of caspase activation, with features of programmed necrosis (Miyazaki et al. 2001; Tait and Green 2008). This was also confirmed by observations made in embryos surviving loss of Apaf-1, Caspase 9, or Caspase 3 and managing to come to term (Honarpour et al. 2000; Zheng et al. 2000). These mice have cleared tissues usually removed by apoptotic mechanisms during development. Consistently, necrosis-like morphology of interdigital cells was observed in embryonic limb buds that were allowed to develop in culture in the presence of pharmacological caspase inhibitors (Chautan et al. 1999). This is in line with the notion that none of the reported mouse strains lacking caspases that are required for apoptosis exhibits soft-tissue syndactyly (Wang and Lenardo 2000). This suggests that, in some cell types and tissues, programmed necrosis can compensate for impaired mitochondrial apoptosis or that caspases can be engaged in a noncanonical manner during development, as suggested by observations that lymphocytes lacking Apaf-1 or Caspase 9 can still undergo BCL2-regulated apoptosis (Marsden et al. 2002).
Although necrotic cell death can clearly be programmed in response to certain types of stress, such as ischemia/reperfusion injury in the kidney or heart, Ca2+ overload, or excitotoxicity in neurons, where necrosis plus secondary inflammatory cytokine-driven apoptosis frequently contributes to tissue damage (Linkermann et al. 2014), we do not have strong evidence that programmed necrosis contributes to organ development. This may be simply due to a lack of understanding of the underlying molecular machinery (Karch and Molkentin 2015). The only gene confirmed so far to be rate-limiting in this process is cyclophilin D (CypD), a peptidylprolyl isomerase present in the mitochondrial matrix, which controls mitochondrial permeability transition (MPT). However, although mice lacking CypD show reduced programmed necrosis in ischemia models, embryogenesis is not perturbed (Schinzel et al. 2005).
Clearly, defects in MPT pore (MTP) opening do not impair apoptosis. Recently, a structural contribution of BAX and BAK that differs from their pore-forming potential driving MOMP has been proposed to be required for programmed necrosis upon MTP opening, e.g., in response to H2O2, Ca2+ overload, or heavy-metal poisoning (Tischner et al. 2012; Karch et al. 2013). Most strikingly, MEFs and liver mitochondria isolated from Bax/Bak DKO mice showed clear deficits in MTP activity and codeletion of Bax/Bak in the heart (αMHC-Cre) protected from coronary artery ligation-induced myocardial infarction, an insult usually associated with rapid ATP depletion converting apoptotic death into necrosis involving MTP (Karch et al. 2013). Interestingly, however, conditional deletion of Bax on a Bak/CypD DKO background yielded comparable results and not a higher degree of protection, as anticipated (Whelan et al. 2012). Hence, these studies provide evidence that BAX/BAK contribute to MTP-driven programmed necrosis in some settings. Importantly, this activity does not require their oligomerization potential but rather their mitochondrial fusion potential, as fragmented mitochondria, like those found in BAX/BAK-deficient cells, have a deficit in MTP opening. Hence, the MTP-opening defect could be ameliorated by addition of an inhibitor of mitochondrial fission (Whelan et al. 2012). The investigators also conclude that BAX/BAK may actually form the outer constituent of the elusive MTP complex that is needed to promote solute entry and organ swelling during necrosis. However, assuming that this is indeed the case and that BAX/BAK can control apoptosis plus programmed necrosis at the same time, the rather mild phenotypes observed in BAX/BAK-deficient embryos would represent consequences of impairment of both cell death pathways, excluding a major role for MTP and programmed necrosis in embryogenesis.
Whether other proteolytic enzymes, such as cytosolic calpains or lysosomal cathepsins, may be needed to trigger death in the absence of BAX/BAK is more difficult to assess, as these enzymes are members of large families, some of which are essential for embryogenesis (Repnik and Turk 2010; Smith and Schnellmann 2012). Calcium-regulated calpains have been implicated repeatedly in cell death associated with neurodegenerative disorders, such as amyotrophic lateral sclerosis or multiple sclerosis, but not necessarily developmental cell death (Smith and Schnellmann 2012), although m-calpain mutant embryos fail to implant (Dutt et al. 2006). In contrast, induction of lysosomal cathepsins and their subsequent release have been reported to precede interdigital cell death in avian limb buds, and up-regulation of cathepsin D has also been observed in the limb buds of mouse embryos. Simultaneous pharmacological inhibition of caspases and cathepsins delayed mesodermal cell death in chicken limbs more potently than caspase inhibition alone (Zuzarte-Luis et al. 2007). Furthermore, a critical role of cathepsins B and L has been reported in mammary gland remodeling during involution (Kreuzaler et al. 2011), although its physiological role remains disputed (Sakamoto et al. 2016). Regardless, these observations again support the idea that mitochondrial apoptosis is not solely responsible for developmentally programmed cell death in vertebrates but works in conjunction with, or can be compensated for by, other cell killing mechanisms.
Based on available data today, components of the DR pathway of apoptosis are equally unlikely to compensate for mitochondrial cell death during embryonic development, although unrestrained necroptosis causes embryonic lethality in animals lacking FADD, FLIP, or Caspase 8. This excess cell death that destroys the vasculature of the embryo, leading to developmental arrest around E10.5 (Varfolomeev et al. 1998; Yeh et al. 1998, 2000), can be fully rescued by codeletion of RIPK3 or MLKL, the key driver and effector of necroptosis, respectively. FADD/MLKL-deficient and Caspase 8/MLKL-deficient mice, which lack literally all DR-mediated apoptosis as well as programmed necroptosis are born at a normal Mendelian frequency and show no developmental abnormalities, as do mice lacking FADD plus RIPK3 or Caspase 8 plus RIPK3 (Kaiser et al. 2011; Oberst et al. 2011; Alvarez-Diaz et al. 2016; Zhang et al. 2016). Tissue homeostasis also appears unaffected in these mice, with one notable exception, the hematopoietic compartment, which gradually expands and provokes fatal autoimmunity in aged mice (Alvarez-Diaz et al. 2016; Zhang et al. 2016). As such, all of these mutants mimic the pathology noted in lpr mutant mice that lack functional FAS/CD95 but develop normally otherwise (Watanabe-Fukunaga et al. 1992). Synergy between mitochondrial and DR-mediated apoptosis has been reported before as being critical for immune response termination (Hughes et al. 2008; Weant et al. 2008). Whether they cooperate during development needs to be investigated.
The generation of BAX/BAK DKO mice that lack MLKL or RIPK3 may reveal unexpected insight regarding the potential redundancy between these cell death pathways in embryogenesis. However, as MLKL-deficient (Murphy et al. 2013) or RIPK3-deficient (Newton et al. 2004) mice do not show developmental phenotypes, one would need to speculate that BAX/BAK can compensate well for their absence in cell death initiation during embryogenesis. However, this is unlikely, as BCL2 overexpression or loss of BAX/BAK fails to block MLKL-driven cell death, and activation of BAX/BAK does not trigger necrosome formation (Pasparakis and Vandenabeele 2015). Whether BAX/BAK-deficient embryos show increased rates of inflammation due to nonapoptotic cell death—e.g., via necrosis that may then activate MLKL-driven necroptosis—remains to be sorted out. Clearly, simultaneous deletion of RIPK3 mediated necroptosis, requiring MLKL for execution, and CypD, which is required for regulated necrosis by the MTP, does not impair embryogenesis and tissue homeostasis (Linkermann et al. 2013). Whether simultaneous inhibition of several of these cell death pathways—e.g., in a BAX/BAK/MLKL/CYPD/CASP8 compound mutant mouse—will halt embryogenesis in its tracks needs to be investigated in the future by those who can afford the involved mouse work. But what about other pore-forming complexes that can kill cells, such as Caspase 1/11-activated Gasdermin D (Ding et al. 2016; Liu et al. 2016)? Is this killing machinery really exclusive for innate immune cells? What about ferroptosis? This cell death process is regulated by the antioxidant enzyme glutathione peroxidase 4 (GPX4), which depends on glutathione to prevent toxic iron-dependent lipid peroxidation and cell death (Yang and Stockwell 2016). Of note, ferroptosis inhibition can ameliorate developmental defects noted in the absence of MDM2 when the canonical effector functions of p53 are impaired (Jiang et al. 2015). Hence, under certain conditions, this type of cell death can become activated during embryogenesis.
However, in times of CRISPR/CAS9-mediated gene editing of mouse embryos, those with the necessary resources at hand may find these intriguing possibilities to explore. Regardless of the phenotype of such “multi-CRISPR” embryos, establishing a direct link to absence of cell death of any kind will be challenging. Clearly, signals emanating from dying cells serve as a cue to control proliferation, migration, or differentiation during development, and separating cause from consequence and phenotype will be difficult. Do we need cell death execution at all to clear superfluous cells? Perhaps the exposure of certain engulfment signals suffices to clear cells during organogenesis. In the end, we may learn yet again that nature finds its way.
Conclusions and perspectives
Given that work on BCL2 family proteins has been overwhelming in its quantity in the past decades and that apoptosis has made it into every cell biology and embryology textbook as a key regulator of development and tissue homeostasis, it is surprising how little we actually do know about its actual relevance for these processes. One is tempted to say that (cell) death is not an essential part of life after all, which might sound comforting but ultimately must be wrong. We all know it. Of course, this may in part be due to the fact that cell death events during vertebrate and in particular mammalian development are difficult to follow and study. Regardless, keeping in mind that there are still huge gaps in our knowledge of what controls cavitation and duct and organ formation during embryonic development and how homeostasis is maintained in the adult in the absence of mitochondrial apoptosis, particularly outside the hematopoietic system, we can anticipate that research along these lines will still be rewarding. We may find not only novel regulators of these developmental processes but also valuable new insights for human disease.
Acknowledgments
We apologize to those scientists who made valuable contributions to the field over the years but were not cited here due to space constrains. We thank members of the Villunger and Kaufmann laboratories as well as Ana Garcia-Saez for helpful discussion. Our work on BCL2 family members is supported by grants from the Austrian Science Fund (grant I1298; FOR-2036), the Molecular Cell Biology and Oncology Doctoral College (W1101), and the Swiss National Science Foundation (grant 310030E150805, FOR-2036, to T.K.). S.T. is supported by a doctoral fellowship from the Austrian Academy of Science (ÖAW).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.289298.116.
References
- Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O'Reilly LA, et al. 2016. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45: 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandjelovic S, Ravichandran KS. 2015. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16: 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner F, Woess C, Pedit V, Tzankov A, Labi V, Villunger A. 2013. Minor cell-death defects but reduced tumor latency in mice lacking the BH3-only proteins Bad and Bmf. Oncogene 32: 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, Pellettieri J, Sanchez Alvarado A, Salvesen GS, Green DR. 2012. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci 109: 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, Garcia-Saez AJ, Bordignon E. 2014. Structural model of active Bax at the membrane. Mol Cell 56: 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner C, Leber B, Andrews DW. 2010. Apoptosis: embedded in membranes. Curr Opin Cell Biol 22: 845–851. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F, Adams JM, Strasser A. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Cory S, Zhang L-C, Strasser A, Adams JM. 2001. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev Cell 1: 645–653. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415: 922–926. [DOI] [PubMed] [Google Scholar]
- Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ, et al. 1995. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene 11: 1921–1928. [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE. 2014. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946. [DOI] [PubMed] [Google Scholar]
- Brunelle JK, Letai A. 2009. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 122: 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso D, Dominguez C, Perez-Acle T, Moreno RD. 2010. Life-giving caspases: revealing new roles during mouse embryo preimplantation development. Int J Dev Biol 54: 857–865. [DOI] [PubMed] [Google Scholar]
- Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG. 2015. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci 112: 7201–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG. 2016. Reply to Fernandez-Marrero, et al.: role of BOK at the intersection of endoplasmic reticulum stress and apoptosis regulation. Proc Natl Acad Sci 113: E494–E495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. 1999. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol 9: 967–970. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17: 393–403. [DOI] [PubMed] [Google Scholar]
- Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang YF, Ren DC, Kim HJ, Takeda S, Reyna DE, Chan PM, et al. 2015. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 17: 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. 2001. BCL-2, BCL-xL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705–711. [DOI] [PubMed] [Google Scholar]
- Cleary ML, Smith SD, Sklar J. 1986. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47: 19–28. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR. 1995. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 83: 279–287. [DOI] [PubMed] [Google Scholar]
- Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM, Strasser A. 2004. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol 24: 1570–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas L, Bouillet P, Loveland KL, Meachem S, Perlman H, Adams JM, Strasser A. 2005. Concomitant loss of proapoptotic BH3-only Bcl-2 antagonists Bik and Bim arrests spermatogenesis. EMBO J 24: 3963–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas L, Terzano S, Thomas T, Voss A, Reid K, Stanley EG, Scott CL, Bouillet P, Bartlett P, Ham J, et al. 2007. Hrk/DP5 contributes to the apoptosis of select neuronal populations but is dispensable for haematopoietic cell apoptosis. J Cell Sci 120: 2044–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha J, Boim M, Ferreira V, Naves M. 2012. Effect of high glucose concentration on the osteoblast function. Nephrol Dial Transpl 27: 494. [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. 2014. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63. [DOI] [PubMed] [Google Scholar]
- Dai HM, Ding HS, Meng XW, Peterson KL, Schneider PA, Karp JE, Kaufmann SH. 2015. Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells. Genes Dev 29: 2140–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. 2003. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424: 952–956. [DOI] [PubMed] [Google Scholar]
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, Datta SR, Pitter KL, Bird GH, Wikstrom JD, et al. 2008. Dual role of proapoptotic BAD in insulin secretion and β cell survival. Nat Med 14: 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM Jr, Snider WD, Korsmeyer SJ. 1996. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17: 401–411. [DOI] [PubMed] [Google Scholar]
- Delbridge AR, Grabow S, Strasser A, Vaux DL. 2016. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 16: 99–109. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116. [DOI] [PubMed] [Google Scholar]
- D'Orsi B, Engel T, Pfeiffer S, Nandi S, Kaufmann T, Henshall DC, Prehn JH. 2016. Bok is not pro-apoptotic but suppresses poly ADP-ribose polymerase-dependent cell death pathways and protects against excitotoxic and seizure-induced neuronal injury. J Neurosci 36: 4564–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. 2011. BH3-domains other than Bim and Bid can directly activate BAX/BAK. J Biol Chem 286: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt P, Croall DE, Arthur JS, Veyra TD, Williams K, Elce JS, Greer PA. 2006. m-Calpain is required for preimplantation embryonic development in mice. BMC Dev Biol 6: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, Kaufmann T. 2013. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ 20: 785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ. 2011. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell 145: 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. 2004. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci 101: 6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. 2003. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med 198: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Caballero-Caballero A, Schindler CK, Plesnila N, Strasser A, Prehn JHM, Henshall DC. 2010. BH3-only protein Bid is dispensable for seizure-induced neuronal death and the associated nuclear accumulation of apoptosis-inducing factor. J Neurochem 115: 92–101. [DOI] [PubMed] [Google Scholar]
- Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, Villunger A. 2006. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 203: 2939–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Favero M, Vogel MW. 2001. Elimination of Bax expression in mice increases cerebellar purkinje cell numbers but not the number of granule cells. J Comp Neurol 436: 82–91. [PubMed] [Google Scholar]
- Fernandez-Marrero Y, Ke F, Echeverry N, Bouillet P, Bachmann D, Strasser A, Kaufmann T. 2016. Is BOK required for apoptosis induced by endoplasmic reticulum stress? Proc Natl Acad Sci 113: E492–E493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SF, Bouillet P, O'Donnell K, Light A, Tarlinton DM, Strasser A. 2007. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood 110: 3978–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H, Tzankov A, Villunger A. 2010. Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood 115: 995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Screaton RA. 2001. Anoikis mechanisms. Curr Opin Cell Biol 13: 555–562. [DOI] [PubMed] [Google Scholar]
- Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA, Zwaka TP. 2008. Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2: 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Saez AJ, Villunger A. 2016. MOMP in the absence of BH3-only proteins. Genes Dev 30: 878–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Saez AJ, Fuertes G, Suckale J, Salgado J. 2010. Permeabilization of the outer mitochondrial membrane by Bcl-2 proteins. Adv Exp Med Biol 677: 91–105. [DOI] [PubMed] [Google Scholar]
- Garner TP, Reyna DE, Priyadarshi A, Chen HC, Li S, Wu Y, Ganesan YT, Malashkevich VN, Almo SS, Cheng EH, et al. 2016. An autoinhibited dimeric form of BAX regulates the BAX activation pathway. Mol Cell 63: 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. 2008. BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Garcia-Haro L, Choi CS, Osundiji MA, Lane EA, Huang H, Yildirim MA, Szlyk B, Fisher JK, Polak K, et al. 2014. Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab 19: 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DH, Kupresanin F, Berzins SP, Herold MJ, O'Reilly LA, Bouillet P, Strasser A. 2012. The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity 37: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grespi F, Soratroi C, Krumschnabel G, Sohm B, Ploner C, Geley S, Hengst L, Hacker G, Villunger A. 2010. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ 17: 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S. 2016. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35: 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn P, Lindsten T, Tolentino M, Bennett J, Milam AH, Thompson C, Dunaief J. 2002. Bax and bak are essential for developmental apoptosis of ocular fetal vasculature and INL photoreceptors. Invest Ophthalmol Vis Sci 43: U177. [DOI] [PubMed] [Google Scholar]
- Hahn P, Lindsten T, Ying GS, Bennett J, Milam AH, Thompson CB, Dunaief JL. 2003. Proapoptotic Bcl-2 family members, Bax and Bak, are essential for developmental photoreceptor apoptosis. Invest Ophthalmol Vis Sci 44: 3598–3605. [DOI] [PubMed] [Google Scholar]
- Hahn P, Lindsten T, Tolentino M, Thompson CB, Bennett J, Dunaief JL. 2005. Persistent fetal ocular vasculature in mice deficient in bax and bak. Arch Ophthalmol 123: 797–802. [DOI] [PubMed] [Google Scholar]
- Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, Dewson G, Michalak EM, Vandenberg CJ, Smyth GK, et al. 2010. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 116: 5256–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happo L, Phipson B, Smyth GK, Strasser A, Scott CL. 2012. Neither loss of Bik alone, nor combined loss of Bik and Noxa, accelerate murine lymphoma development or render lymphoma cells resistant to DNA damaging drugs. Cell Death Dis 3: e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. 2002. Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member, Bim. Immunity 16: 759–767. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J. 2000. Adult Apaf-1-deficient mice exhibit male infertility. Dev Biol 218: 248–258. [DOI] [PubMed] [Google Scholar]
- Hubner A, Cavanagh-Kyros J, Rincon M, Flavell RA, Davis RJ. 2010. Functional cooperation of the proapoptotic Bcl2 family proteins Bmf and Bim in vivo. Mol Cell Biol 30: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. 2008. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity 28: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi K, Tsuda M, Imai Y, Wanaka A, Takagi T, Tohyama M. 1997. Molecular cloning of a novel polypeptide, DP5, induced during programmed neuronal death. J Biol Chem 272: 18842–18848. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328. [DOI] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, et al. 2009. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460: 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyotika J, McCutcheon J, Laroche J, Blaustein JD, Forger NG. 2007. Deletion of the Bax gene disrupts sexual behavior and modestly impairs motor function in mice. Dev Neurobiol 67: 1511–1519. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471: 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC, Gross A. 2005. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122: 593–603. [DOI] [PubMed] [Google Scholar]
- Karch J, Molkentin JD. 2015. Regulated necrotic cell death: the passive aggressive side of Bax and Bak. Circ Res 116: 1800–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EHY, Robbins J, et al. 2013. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2: e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, Johnstone RW, Dixit VM, Strasser A. 2007. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell 129: 423–433. [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S, Cretney E, Smyth MJ, Silke J, Hakem R, et al. 2009. Fatal hepatitis mediated by tumor necrosis factor TNFα requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity 30: 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, Kaufmann T. 2012. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ 19: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, Voss AK. 2013. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis 4: e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Grabow S, Kelly GL, Lin A, O'Reilly LA, Strasser A. 2015. Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis 6: e1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PN, White MJ, Goschnick MW, Fairfax KA, Tarlinton DM, Kinkel SA, Bouillet P, Adams JM, Kile BT, Strasser A. 2010. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ 17: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, Bouillet P, Mills A, Scott CL, Findlay JK, et al. 2012. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell 48: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJD, Cheng EHY. 2006. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8: 1348–1358. [DOI] [PubMed] [Google Scholar]
- Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, Jeffords LB, Sodha P, Mercer KL, Cohen R, et al. 2010. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327: 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. 1995. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99. [DOI] [PubMed] [Google Scholar]
- Krahe TE, Medina AE, Lantz CL, Filgueiras CC. 2015. Hyperactivity and depression-like traits in Bax KO mice. Brain Res 1625: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. 2011. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 13: 303–309. [DOI] [PubMed] [Google Scholar]
- Krishna S, Overholtzer M. 2016. Mechanisms and consequences of entosis. Cell Mol Life Sci 73: 2379–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17: 525–535. [DOI] [PubMed] [Google Scholar]
- Labi V, Erlacher M, Kiessling S, Manzl C, Frenzel A, O'Reilly L, Strasser A, Villunger A. 2008. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates γ irradiation-induced thymic lymphoma development. J Exp Med 205: 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. 2010. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev 24: 1602–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, Tzankov A, Villunger A. 2014. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood 123: 2652–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Kim WR, Sun W, Jung MW. 2009. Role of dentate gyrus in aligning internal spatial map to external landmark. Learn Mem 16: 530–536. [DOI] [PubMed] [Google Scholar]
- Lee EF, Grabow S, Chappaz S, Dewson G, Hockings C, Kluck RM, Debrincat MA, Gray DH, Witkowski MT, Evangelista M, et al. 2016. Physiological restraint of Bak by Bcl-xL is essential for cell survival. Genes Dev 30: 1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH. 2002. Mice with targeted gene disruptions or gene insertions for diabetes research: problems, pitfalls, and potential solutions. Diabetologia 45: 296–308. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik M, Walensky L, Sorcinelli M, Weiler S, Korsmeyer S. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu C-J, Yuan J. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Thompson CB. 2006. Cell death in the absence of Bax and Bak. Cell Death Differ 13: 1272–1276. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. 2000. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 6: 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. 2003. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci 23: 11112–11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, et al. 2013. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci 110: 12024–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Stockwell BR, Krautwald S, Anders HJ. 2014. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 14: 759–767. [DOI] [PubMed] [Google Scholar]
- Litwak SA, Loh K, Stanley WJ, Pappas EG, Wali JA, Selck C, Strasser A, Thomas HE, Gurzov EN. 2016. p53-upregulated-modulator-of-apoptosis (PUMA) deficiency affects food intake but does not impact on body weight or glucose homeostasis in diet-induced obesity. Sci Rep 6: 23802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, et al. 2016. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084. [DOI] [PubMed] [Google Scholar]
- Luedke AC, Boucher PO, Niel L, Holmes MM. 2013. Altered anxiety and defensive behaviors in Bax knockout mice. Behav Brain Res 239: 115–120. [DOI] [PubMed] [Google Scholar]
- Luo X, Budlhardjo I, Zou H, Slaughter C, Wang X. 1998. Bid, a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490. [DOI] [PubMed] [Google Scholar]
- Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S, Stroud DA, Menard M, Kluck RM, Ryan MT, Dewson G. 2014. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ 21: 1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. 2007. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell 12: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, et al. 2002. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Henry CM, Cullen SP. 2012. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell 46: 387–397. [DOI] [PubMed] [Google Scholar]
- McKenzie MD, Carrington EM, Kaufmann T, Strasser A, Huang DC, Kay TW, Allison J, Thomas HE. 2008. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic β-cells. Diabetes 57: 1284–1292. [DOI] [PubMed] [Google Scholar]
- McKenzie MD, Jamieson E, Jansen ES, Scott CL, Huang DCS, Bouillet P, Allison J, Kay TWH, Strasser A, Thomas HE. 2010. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes 59: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, et al. 2009. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 186: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak EM, Villunger A, Adams JM, Strasser A. 2008. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 15: 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, Strasser A. 2010. Apoptosis-promoted tumorigenesis: γ-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev 24: 1608–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Yoshida H, Sasaki M, Hara H, Kimura G, Mak TW, Nomoto K. 2001. Caspase-independent cell death and mitochondrial disruptions observed in the Apaf1-deficient cells. J Biochem 129: 963–969. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. 2013. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453. [DOI] [PubMed] [Google Scholar]
- Murray P, Edgar D. 2000. Regulation of programmed cell death by basement membranes in embryonic development. J Cell Biol 150: 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik E, Michalak EM, Villunger A, Adams JM, Strasser A. 2007. Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol 176: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM. 2004. Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24: 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. 2011. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. 2000. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619. [DOI] [PubMed] [Google Scholar]
- O'Neill KL, Huang K, Zhang J, Chen Y, Luo X. 2016. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev 30: 973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottina E, Sochalska M, Sgonc R, Villunger A. 2015. The BH3-only protein Bad is dispensable for TNF-mediated cell death. Cell Death Dis 6: e1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X, Moussa SH, Targy NM, Bose JL, George NM, Gries C, Lopez H, Zhang L, Bayles KW, Young R, et al. 2011. Active Bax and Bak are functional holins. Genes Dev 25: 2278–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P. 2015. Necroptosis and its role in inflammation. Nature 517: 311–320. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Belz G, Bouillet P, Strasser A. 2003. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci 100: 14175–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaloza C, Lin L, Lockshin RA, Zakeri Z. 2006. Cell death in development: shaping the embryo. Histochem Cell Biol 126: 149–158. [DOI] [PubMed] [Google Scholar]
- Pinon JD, Labi V, Egle A, Villunger A. 2008. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene 27: S41–S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, et al. 2001. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci 98: 15318–15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, Zhang L, Yu J. 2008. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2: 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Wang X, Leibowitz B, Yang W, Zhang L, Yu J. 2011. PUMA-mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology 54: 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. 2007. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128: 931–946. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME, Korsmeyer SJ. 2003. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci 100: 9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB. 2002. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol 3: 932–939. [DOI] [PubMed] [Google Scholar]
- Ray JE, Garcia J, Jurisicova A, Caniggia I. 2010. Mtd/Bok takes a swing: proapoptotic Mtd/Bok regulates trophoblast cell proliferation during human placental development and in preeclampsia. Cell Death Differ 17: 846–859. [DOI] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. 2010. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330: 1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U, Turk B. 2010. Lysosomal-mitochondrial cross-talk during cell death. Mitochondrion 10: 662–669. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Wehde BL, Yoo KH, Kim T, Rajbhandari N, Shin HY, Triplett AA, Radler PD, Schuler F, Villunger A, et al. 2016. Janus kinase 1 is essential for inflammatory cytokine signaling and mammary gland remodeling. Mol Cell Biol 36: 1673–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, Garcia-Saez AJ. 2016. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. 2002. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4: 842–849. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. 2005. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci 102: 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS. 2007. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci 104: 3787–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler F, Baumgartner F, Klepsch V, Chamson M, Muller-Holzner E, Watson CJ, Oh S, Hennighausen L, Tymoszuk P, Doppler W, et al. 2016. The BH3-only protein BIM contributes to late-stage involution in the mouse mammary gland. Cell Death Differ 23: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. 1995. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci 92: 7834–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N. 2003. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev 17: 2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Schnellmann RG. 2012. Calpains, mitochondria, and apoptosis. Cardiovasc Res 96: 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochalska M, Tuzlak S, Egle A, Villunger A. 2015. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: implications for targeted therapy. FEBS J 282: 834–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Winseck A, Vinsant S, Park OH, Kim H, Oppenheim RW. 2004. Programmed cell death of adult-generated hippocampal neurons is mediated by the proapoptotic gene Bax. J Neurosci 24: 11205–11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, Green DR. 2008. Caspase-independent cell death: leaving the set without the final cut. Oncogene 27: 6452–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. 2013. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. 2005. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci 102: 11272–11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischner D, Manzl C, Soratroi C, Villunger A, Krumschnabel G. 2012. Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 17: 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, Ichim G, Tait SWG, Frank S, Langer HF, et al. 2015. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J 34: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Croce CM. 1986. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci 83: 5214–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Hulpiau P, Martens L, Vandenbroucke RE, Van Wonterghem E, Perry SW, Bruggeman I, Divert T, Choi SM, Vuylsteke M, et al. 2015. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity 43: 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Raam BJ, Ehrnhoefer DE, Hayden MR, Salvesen GS. 2013. Intrinsic cleavage of receptor-interacting protein kinase-1 by caspase-6. Cell Death Differ 20: 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. 2003a. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038. [DOI] [PubMed] [Google Scholar]
- Villunger A, Scott C, Bouillet P, Strasser A. 2003b. Essential role for the BH3-only protein Bim but redundant roles for Bax, Bcl-2, and Bcl-w in the control of granulocyte survival. Blood 101: 2393–2400. [DOI] [PubMed] [Google Scholar]
- Villunger A, Marsden VS, Zhan Y, Erlacher M, Lew AM, Bouillet P, Berzins S, Godfrey DI, Heath WR, Strasser A. 2004. Negative selection of semimature CD4+8−HSA+ thymocytes requires the BH3-only protein Bim but is independent of death receptor signaling. Proc Natl Acad Sci 101: 7052–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. 2006. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24: 199–210. [DOI] [PubMed] [Google Scholar]
- Wang J, Lenardo MJ. 2000. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 113: 753–757. [DOI] [PubMed] [Google Scholar]
- Wang K, Yin X-M, Chao DT, Milliman CL, Korsmeyer SJ. 1996. BID: a novel BH3 domain-only death agonist. Genes Dev 10: 2859–2869. [DOI] [PubMed] [Google Scholar]
- Wang ES, Reyes NA, Melton C, Huskey NE, Momcilovic O, Goga A, Blelloch R, Oakes SA. 2015. Fas-activated mitochondrial apoptosis culls stalled embryonic stem cells to promote differentiation. Curr Biol 25: 3110–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356: 314–317. [DOI] [PubMed] [Google Scholar]
- Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. 2008. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity 28: 218–230. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA, Eldering E. 2010. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity 32: 754–765. [DOI] [PubMed] [Google Scholar]
- Wensveen FM, Alves NL, Derks IA, Reedquist KA, Eldering E. 2011. Apoptosis induced by overall metabolic stress converges on the Bcl-2 family proteins Noxa and Mcl-1. Apoptosis 16: 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Derks IA, van Gisbergen KP, de Bruin AM, Meijers JC, Yigittop H, Nolte MA, Eldering E, van Lier RA. 2012. BH3-only protein Noxa regulates apoptosis in activated B cells and controls high-affinity antibody formation. Blood 119: 1440–1449. [DOI] [PubMed] [Google Scholar]
- Wensveen FM, Geest CR, Libregts SF, Derks IA, Ekert PG, Labi V, Villunger A, Nolte MA, Eldering E. 2013a. BH3-only protein Noxa contributes to apoptotic control of stress-erythropoiesis. Apoptosis 18: 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, Ten Brinke A, de Vries N, Cekinovic D, Jonjic S, et al. 2013b. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol 190: 1180–1191. [DOI] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. 2012. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci 109: 6566–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. 1998. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci 18: 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315: 856–859. [DOI] [PubMed] [Google Scholar]
- Woess C, Tuzlak S, Labi V, Drach M, Bertele D, Schneider P, Villunger A. 2015. Combined loss of the BH3-only proteins Bim and Bmf restores B-cell development and function in TACI-Ig transgenic mice. Cell Death Differ 22: 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J, Lin A. 2013. Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell 152: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. 2016. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26: 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. 1995. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285–291. [DOI] [PubMed] [Google Scholar]
- Yang WT, Klaman LD, Chen BB, Araki T, Harada H, Thomas SM, George EL, Neel BG. 2006. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 10: 317–327. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954–1958. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, et al. 2000. Requirement for casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12: 633–642. [DOI] [PubMed] [Google Scholar]
- Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. 1999. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7: 673–682. [DOI] [PubMed] [Google Scholar]
- Zakeri Z, Lockshin RA, Criado-Rodriguez LM, Martinez AC. 2005. A generalized caspase inhibitor disrupts early mammalian development. Int J Dev Biol 49: 43–47. [DOI] [PubMed] [Google Scholar]
- Zhang X, Fan C, Zhang H, Zhao Q, Liu Y, Xu C, Xie Q, Wu X, Yu X, Zhang J, et al. 2016. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep 16: 3247–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. 2000. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med 6: 1241–1247. [DOI] [PubMed] [Google Scholar]
- Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ. 2005. A role for proapoptotic BID in the DNA-damage response. Cell 122: 579–591. [DOI] [PubMed] [Google Scholar]
- Zuzarte-Luis V, Montero JA, Kawakami Y, Izpisua-Belmonte JC, Hurle JM. 2007. Lysosomal cathepsins in embryonic programmed cell death. Dev Biol 301: 205–217. [DOI] [PubMed] [Google Scholar]