

Balmus et al. report that Paxx/Xlf double-knockout mice display embryonic lethality associated with genomic instability, cell death in the central nervous system, and an almost complete block in lymphogenesis, phenotypes that closely resemble those of Xrcc4−/− and Lig4−/− mice.
Keywords: ATM, development, NHEJ, PAXX, synthetic lethality, XLF
Abstract
PAXX was identified recently as a novel nonhomologous end-joining DNA repair factor in human cells. To characterize its physiological roles, we generated Paxx-deficient mice. Like Xlf−/− mice, Paxx−/− mice are viable, grow normally, and are fertile but show mild radiosensitivity. Strikingly, while Paxx loss is epistatic with Ku80, Lig4, and Atm deficiency, Paxx/Xlf double-knockout mice display embryonic lethality associated with genomic instability, cell death in the central nervous system, and an almost complete block in lymphogenesis, phenotypes that closely resemble those of Xrcc4−/− and Lig4−/− mice. Thus, combined loss of Paxx and Xlf is synthetic-lethal in mammals.
DNA double-strand breaks (DSBs) are extremely toxic lesions that must be repaired for an organism to pass on its genetic material intact to the next generation (Jackson and Bartek 2009). Cells have evolved two principal DSB repair pathways to address this challenge: homologous recombination (HR) and nonhomologous end-joining (NHEJ). HR is restricted to S and G2 phases of the cell cycle because it requires a sister chromatid as the template for repair. In contrast, NHEJ is the dominant DSB repair pathway throughout interphase in mammalian cells, although it is restrained during DNA replication (Beucher et al. 2009; Karanam et al. 2012). NHEJ is initiated by Ku, a ring-shaped heterodimeric protein complex consisting of Ku70 and Ku80 subunits that specifically recognizes DSB ends (Grundy et al. 2014). Ku forms a platform for the downstream recruitment of core NHEJ factors such as DNA ligase IV (LIG4); its stable binding partner, XRCC4; and XLF, a protein structurally related to XRCC4 (Ochi et al. 2014).
Most DSBs in vertebrate cells are generated by agents such as ionizing radiation (IR) or molecules that directly damage DNA by chemically reacting with it or through processing of other DNA lesions during DNA replication or mitosis. Additionally, some DSBs are induced deliberately by enzymatic cleavage in certain cell types at various stages of development; for example, to generate immune receptor diversity in B and T lymphocytes during variable, diversity, and joining [V(D)J] recombination (Jackson and Bartek 2009). NHEJ plays a central role in V(D)J recombination, a fact highlighted by the severe immunodeficiency found in some human patients and mice with NHEJ defects (Woodbine et al. 2014).
Mouse models of NHEJ deficiency show both overlapping and unique features. Ku-deficient mice are subviable and fertile but have profound growth defects, increased neuronal cell death, and immunodeficiency (Nussenzweig et al. 1996; Zhu et al. 1996; Ouyang et al. 1997; Gu et al. 2000). Loss of Xrcc4 or Lig4 results in a more severe phenotype with embryonic growth defects, blocked lymphogenesis, and late embryonic lethality associated with a large increase in cell death in the developing central nervous system (CNS) (Barnes et al. 1998; Frank et al. 1998; Gao et al. 1998). In contrast, Xlf−/− mice have a relatively mild phenotype with no growth defects, neuronal cell death, or overt immunodeficiency despite XLF being a core NHEJ factor (Ahnesorg et al. 2006; Buck et al. 2006; Li et al. 2008). Furthermore, Xlf−/− B cells perform V(D)J recombination at almost wild-type levels, which explains the lack of significant immunodeficiency in these mice and suggests that compensatory mechanisms can mitigate loss of XLF in developing lymphocytes. One of these mechanisms comprises the ATM–H2AX–53BP1 axis of DSB repair, as combined loss of any one of these factors with XLF deficiency causes profound defects in V(D)J recombination and lymphocyte development even though loss of any of these proteins individually is not significantly detrimental to these processes (Zha et al. 2011; Liu et al. 2012; Oksenych et al. 2012).
Recently, we and others identified a third XRCC4-like NHEJ protein, called PAXX (Craxton et al. 2015; Ochi et al. 2015; Xing et al. 2015). PAXX is required for cellular resistance to DSB-inducing agents and is rapidly recruited to DNA damage sites, where it stabilizes NHEJ factors on chromatin and promotes DNA repair by NHEJ. In vitro, PAXX can stimulate DNA end ligation in the presence of LIG4, XRCC4, and Ku, with most if not all of these functions requiring its direct binding to Ku. However, it is still unknown whether PAXX functions similarly to other core NHEJ factors in physiological settings or how PAXX loss might impact at the organism level in terms of growth and development.
In this study, we describe the generation and characterization of PAXX-deficient mice. Like Xlf−/− mice, Paxx−/− mice are viable, grow normally, and are fertile. However, these mice are radiosensitive and show a mild reduction in splenic lymphocyte numbers. Strikingly, combined loss of Paxx and Xlf is synthetic-lethal, as the majority of double-knockout embryos dies before birth with significant growth defects, increased genomic instability and subsequent cell death in the developing CNS, and an almost complete block in lymphocyte development, phenotypes that are strongly reminiscent of Xrcc4−/− or Lig4−/− mice. Thus, PAXX and XLF share a redundant function that is critical for DNA repair during mammalian development.
Results and Discussion
Generation of Paxx−/− mice
To characterize PAXX function in mice, we disrupted the Paxx locus in C57BL/6NTac zygotes using CRISPR–Cas9 (Wright et al. 2016) by injection of wild-type Cas9 mRNA together with two small guide RNAs (sgRNAs) and an oligonucleotide template to collapse the entire Paxx genomic region, resulting in a 1.6-kb deletion (Fig. 1A). For ease of mutant selection when using Cas9, Paxx sgRNAs were coinjected with an sgRNA to disrupt exon 1 of the tyrosinase gene (Tyr) to yield an albino phenotype in successfully targeted mice (Supplemental Fig. S1A,B). We identified four Paxx F0 founders, all of which showed the albino phenotype (Supplemental Fig. S2A,B). Upon confirmation of Paxx locus deletion by PCR and sequencing (Supplemental Fig. S2C), we backcrossed founders to the C57BL/6NTac original background for three generations to segregate the Tyr deletion and any off-target mutations. Resulting mice showed complete absence of PAXX protein in tissues such as thymus, lung, and brain that normally highly express Paxx (Fig. 1A).
Figure 1.
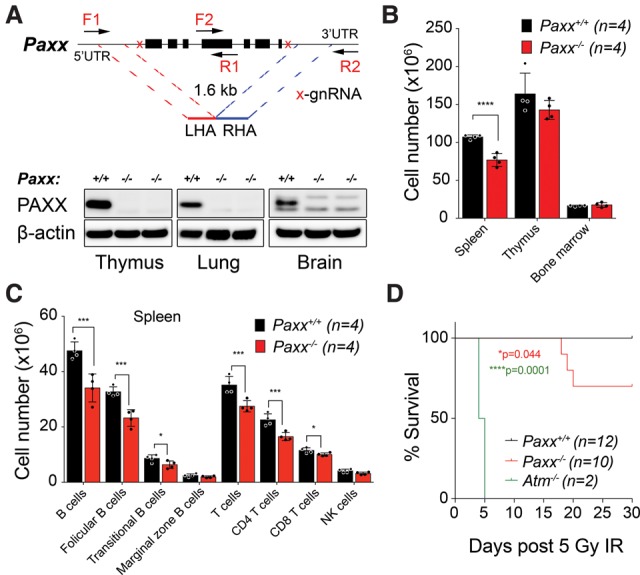
Paxx−/− mice are viable and show increased sensitivity to IR. (A) Schematic representation of the CRISPR design deployed to delete the entire coding sequence of the Paxx locus on mouse chromosome 2. Representative Western blot showing PAXX protein level in wild-type versus Paxx knockout thymi, lungs, and brains. β-Actin was the loading control. (B) Paxx−/− mice show a significant reduction of total cell numbers in the spleen ([****] P < 0.0001, Student's t test; 6-wk-old female mice; mean ± SD) but not in the thymus or bone marrow when compared with Paxx+/+ mice. (C) B-cell and T-cell distribution in the spleens of Paxx+/+ as compared with Paxx−/− littermate mice. (***) P < 0.001; (*) P < 0.05, Student's t test. Mean ± SD. Leukocytes were identified as CD45+. B cells were defined as CD19+ with the subsets on the basis of CD21 and CD23 expression. (Follicular B) CD23 + CD21low/neg; (marginal zone B) CD23low/neg CD21high; (transitional B) CD23low CD21low. NK cells were defined as NK1.1+ CD3−, and T cells were defined as CD3+ NK1.1− with T-cell subsets on the basis of surface CD4 or CD8 expression. (D) Mice (12–14 wk old) of the indicated genotypes were exposed to 5 Gy of whole-body IR. Kaplan-Meyer χ2 statistical analysis showed no lethality in Paxx+/+ mice but increased sensitivity to IR in Paxx−/− mice. Atm−/− mice were used as positive controls.
Paxx−/− mice were born at the expected Mendelian frequencies, were fertile, and displayed no developmental or hematological abnormalities (Supplemental Fig. S2D,E; Supplemental Table S1). Paxx−/− mice also showed no increased tumor predisposition up to 400 d (data not shown) and no significant increase in micronucleus formation (Supplemental Fig. S3A), a sensitive method for detecting genomic instability in vivo (Balmus et al. 2015).
Paxx−/− mice are radiosensitive but show no overt immune phenotype
In light of the immune defects of NHEJ-deficient mouse models, we examined whether Paxx−/− mice displayed any overt immunodeficiency by analyzing leukocyte numbers. We detected a modest reduction in cell counts in the spleens of Paxx−/− mice compared with wild-type littermates, but this was not observed in the thymus or bone marrow (Fig. 1B). Splenic cell reduction was due to decreased B-cell and T-cell populations, while the NK cell lineage was not significantly affected (Fig. 1C). T-cell development in the thymus (as measured by CD4 and CD8 expression) was essentially normal in Paxx−/− mice (Supplemental Fig. S3B), although they did exhibit a small increase in the number and percentage of mature B cells within the bone marrow (Supplemental Fig. S3C; data not shown). To test whether Paxx−/− mice were capable of mounting a proper immune response, we immunized Paxx−/− and wild-type littermates with purified fragment C of tetanus toxin and found that immune responses as measured by total immunoglobulin (Ig), IgG1, and IgG2a antibody titers were similar in both (Supplemental Fig. S3D). Furthermore, class switch recombination (CSR) was normal in the absence of PAXX (Supplemental Fig. S3E). As PAXX is important for efficient DSB repair in human cells (Craxton et al. 2015; Ochi et al. 2015; Xing et al. 2015), we treated Paxx−/− mice with IR. Following whole-body IR treatment, we observed significantly increased lethality in Paxx−/− mice when compared with wild-type controls, although this was not as dramatic as that observed in Atm−/− mice (Fig. 1D; Barlow et al. 1996). Taken together, these data implied that although PAXX is important for effective DSB repair, it does not play an essential role in mammalian development and is unlikely to function as a strong tumor suppressor. In addition, Paxx−/− mice showed only a mild reduction in lymphocyte numbers and displayed no overt immunodeficiency phenotype. In this regard, Paxx−/− mice most closely resemble Xlf−/− mice (Li et al. 2008) rather than mice deficient in other core NHEJ factors.
Paxx loss is epistatic with Ku, Lig4, and Atm deficiency
Genetic crosses between NHEJ mutant mice and with other DNA repair-deficient mouse models have revealed both synthetic viability (e.g., Ku80−/− Lig4−/− mice) and synthetic lethality (e.g., Atm−/− Xlf−/− mice) relationships (Karanjawala et al. 2002; Zha et al. 2011). Such genetic crosses can highlight antagonistic roles and/or functional redundancies between DNA repair factors and can help position such factors in NHEJ and other events. We therefore crossed our Paxx−/− mice into Ku80-deficient, Lig4-deficient, Atm-deficient, or Xlf-deficient backgrounds.
Paxx−/− Ku80−/− mice were born at frequencies similar to Ku80−/− mice and did not show any overt phenotypes compared with Ku80−/− animals (Fig. 2A; Supplemental Table S2A), consistent with Paxx being epistatic with Ku80 in regard to NHEJ. Notably, this was similar to what we observed with Xlf−/− Ku80−/− mice, which also closely resemble Ku80−/− mice (Supplemental Fig. S4A,B). In contrast to Ku80−/− mice, Lig4−/− mice die during embryonic development in a manner associated with dramatically increased apoptosis in the CNS (Barnes et al. 1998; Frank et al. 1998; Gao et al. 1998). As Ku80 loss rescues the lethality of Lig4−/− mice (Karanjawala et al. 2002) and because PAXX functions in physical contact with Ku in NHEJ, we wondered whether PAXX loss would result in a similar rescue. However, multiple rounds of breeding did not produce any viable Paxx−/− Lig4−/− offspring, indicating that, unlike Ku80 loss, PAXX loss cannot rescue the embryonic lethality caused by LIG4 deficiency (Fig. 2B; Supplemental Table S2B). Next, we generated Paxx−/− Atm−/− mice to explore potential functional relationships between these two genes. Notably, in contrast to the relationship between Atm and Xlf (Zha et al. 2011), Paxx−/− Atm−/− mice displayed no additional phenotypes compared with Atm−/− mice (Fig. 2C; Supplemental Fig. S5; Supplemental Table S2C), indicating that Paxx and Atm are epistatic for immune functions such as CSR and during development.
Figure 2.
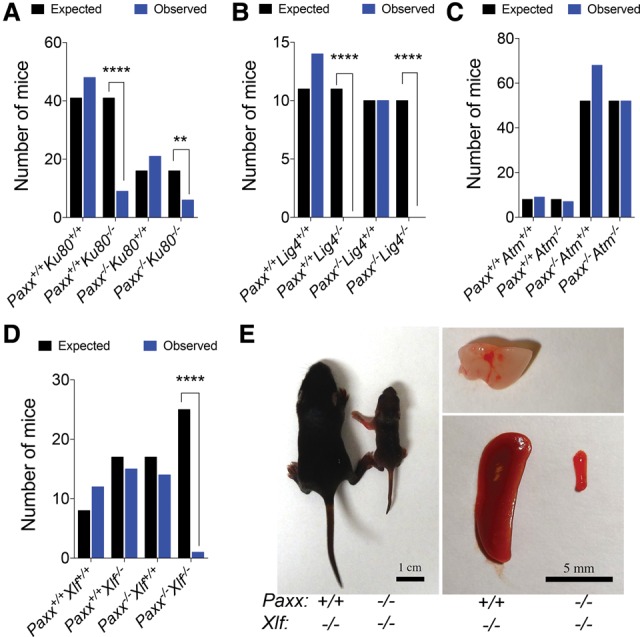
Paxx/Xlf deficiency leads to a synthetic-lethal phenotype. Mice were genotyped using ear snip biopsies, and expected versus observed numbers were used to calculate χ2. Bar graphs presenting selected genotype combinations are presented. (A) Paxx/Ku80-deficient mice are significantly underrepresented compared with wild-type mice (**P < 0.01) but are represented at ratios similar to that of Ku80 deficiency alone ([****] P < 0.0001). (B) Paxx deficiency cannot save the Lig4-associated lethality. Neither Lig4−/− nor Paxx−/− Lig4−/− mice are born. (****) P < 0.0001. (C) Paxx deficiency does not modify the phenotypes associated with Atm loss. Like Atm−/− mice, Paxx−/− Atm−/− mice are born at the expected Mendelian frequencies. (D) Unlike Paxx−/− or Xlf−/− mice, Paxx−/−Xlf−/− mice are not born at the expected frequencies. (****) P < 0.0001. (E) Photograph of a 10-d-old Paxx−/− Xlf−/− mouse as compared with a Paxx+/+ Xlf−/− littermate control. Isolated spleens and thymus of the represented mice are shown. In the case of the Paxx−/− Xlf−/− mouse no thymus was identified.
Paxx loss is synthetic-lethal with Xlf deficiency
We also crossed Paxx−/− mice with Xlf−/− mice to examine potential functional relationships between these genes. Strikingly, these double-knockout mice were dramatically underrepresented, with only one Paxx−/− Xlf−/− mouse born out of 25 expected (Fig. 2D; Supplemental Table S2D). Furthermore, this mouse was born smaller and, at 5 d of age, was clearly distinguishable from its littermate controls (Supplemental Fig. S6A). By 10 d of age, this Paxx−/− Xlf−/− mouse failed to thrive and, at necropsy, exhibited a microspleen and a complete absence of a thymus, similar to what is observed in Ku80−/− animals (Fig. 2E). In accord with these findings, there was a significant decrease in splenic cell counts relative to body weight, and, upon red blood cell lysis, no lymphocytes were recovered (Supplemental Fig. S6B,C). Taken together, these data indicated that while Paxx loss is epistatic with Ku80, Lig4, and Atm in terms of its developmental roles, it is synthetic-lethal in combination with Xlf loss.
To determine when Paxx−/− Xlf−/− mice die during embryonic development, we established timed matings and performed embryo dissections starting at embryonic day 9.5 (E9.5) when Paxx/Xlf double mutants showed no gross abnormalities (Supplemental Fig. S7A). By E10.5, although double mutants were recovered at the expected frequencies, more than half of the Paxx−/− Xlf−/− embryos were smaller than their littermate controls (Fig. 3A; Supplemental Fig. S7B). Nevertheless, the Paxx/Xlf double mutants were not developmentally delayed at this stage, showing somite numbers similar to those of wild-type, Paxx−/−, or Xlf−/− embryos (Fig. 3B), thus hinting at a possible cell fate abnormality. Similarly, at E14.5, double-mutant embryos were obtained at the expected Mendelian frequencies (Fig. 3C; Supplemental Table S3A) but were smaller than controls (Fig. 3D,E). By E18.5, Paxx−/− Xlf−/− double mutants were no longer obtained at the expected frequencies, and a significant proportion of them was found to have died (Fig. 3F; Supplemental Table S3B). A few were viable but showed reduced body weight, much smaller spleens, and a drastic involution of the thymus (Fig. 3G–I), thus highlighting a requirement for either PAXX or XLF in lymphatic organ development and for life for more than a few days postnatally.
Figure 3.
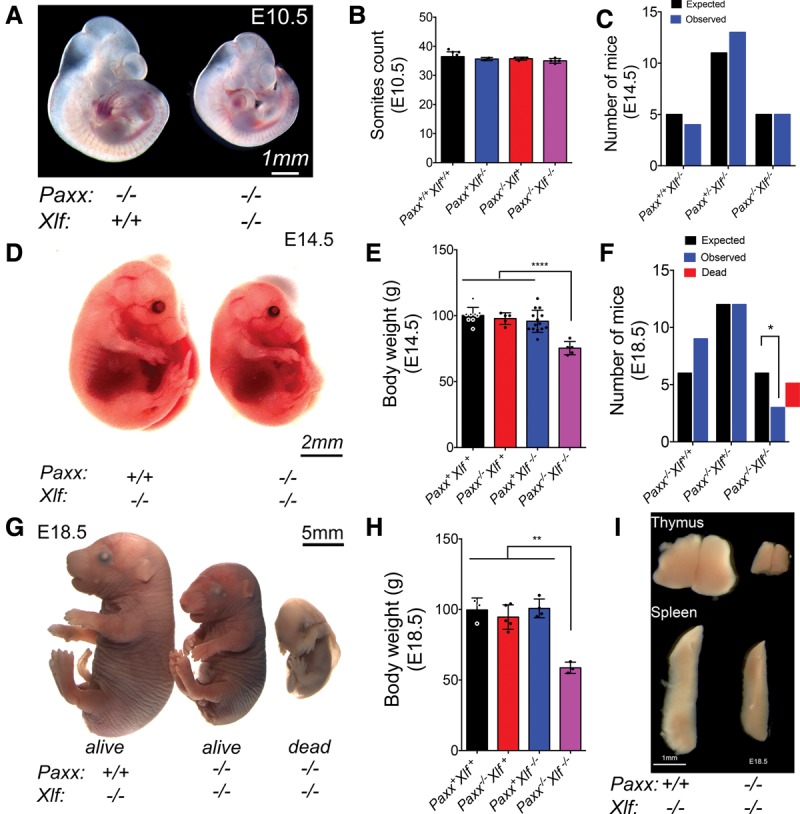
Embryonic lethality upon simultaneous loss of Paxx and Xlf. Embryos from timed matings were isolated at the indicated stage of embryonic development, imaged for morphological assessment, and genotyped by PCR from the yolk sacs or tail snips. (A) Representative images of Paxx−/− and Paxx−/− Xlf−/− E10.5 littermate embryos. (B) Bar graph of somite counts from E10.5 dissected litters. (C) Bar graph (mean ± SD) of expected versus observed E14.5 embryos. (D) Representative images of Paxx−/− and Paxx−/− Xlf−/− E14.5 littermate embryos. (E) Bar graph representing the percentage relative body weight as compared with control littermates at E14.5. (****) P < 0.0001, Student's t test. (F) Bar graph of expected versus observed E18.5 embryos. (*) P < 0.05, Student's t test. The red bar was added to represent the number of Paxx−/− Xlf−/− embryos found dead. (G) Representative images of Xlf−/− (left) and Paxx−/− Xlf−/− E18.5 littermate embryos. (H) Bar graph (mean ± SD) representing the percentage relative body weight as compared with control littermates at E18.5. (**) P < 0.01, Student's t test. (I) Representative images of thymi and spleens of Xlf−/− as compared with Paxx−/− Xlf−/− littermate E18.5 live embryos.
To determine the cause of embryonic death in Paxx−/− Xlf−/− mice, we looked for markers of genomic instability (phosphorylated histone H2AX; γH2AX) and apoptosis (cleaved caspase 3) in E10.5 and E14.5 embryos via immunohistochemistry. At both E10.5 and E14.5, we consistently observed an increase in γH2AX-positive cells in the CNS of the Paxx−/− Xlf−/− double-mutant mice as compared with littermate controls (Fig. 4A,B; Supplemental Fig. S7C). This was associated with increased apoptosis, as evidenced by the accumulation of cleaved caspase 3, especially in the cortical plate region of the cerebral cortex (Fig. 4B; Supplemental Fig. S7C,D). In contrast, significant increases in these markers were not detected in most other tissues (Supplemental Fig. S8A,B). Together, these findings highlighted a critical requirement for Paxx or Xlf for development of not only lymphatic organs but also the CNS. We thus conclude that lack of effective maintenance of genome stability during development results in increased cell death and embryonic lethality in the absence of both PAXX and XLF.
Figure 4.
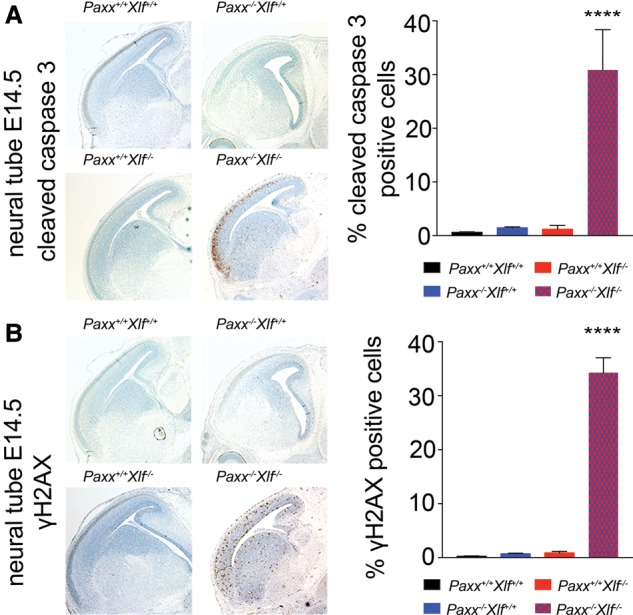
Increased genomic instability and apoptosis in the CNS of Paxx−/− Xlf−/− double-mutant embryos. Representative images (5× magnification) depicting immunohistochemical staining for cleaved caspase 3-positive apoptotic cells (A) and γH2AX-positive cells (B) in the neural tubes of E14.5 embryos. Bar graphs (mean ± SD) present the percentage of cleaved caspase 3-positive (A) and pan-nuclear γH2AX-positive (B) cells of the representative genotypes. More than 500 cells per embryo were counted. n ≥ 3 per genotype. Statistical analysis was performed using one-way ANOVA. (****) P < 0.0001, Dunnett's multiple comparisons test.
In this study, we described the generation of PAXX-deficient mice. We found that while Paxx−/− mice are radiosensitive, they are born at Mendelian ratios and display no overt immunodeficiency or developmental defects. In this regard, Paxx−/− mice are reminiscent of Xlf−/− mice, although the immunological phenotype of the latter is slightly more pronounced (Li et al. 2008). Furthermore, our data support a model in which PAXX functions in NHEJ in a manner connected to and dependent on Ku because Ku80−/− Paxx−/− double-mutant mice were essentially identical to Ku80−/− mice. Furthermore, we found that PAXX loss could not rescue the embryonic lethality phenotype of Lig4−/− mice. This was expected based on previous cell-based studies and biochemical analyses of PAXX, which showed that PAXX functions downstream from Ku in the NHEJ process, being recruited to Ku-bound DNA ends by direct binding of the PAXX C terminus to Ku (Ochi et al. 2015; Xing et al. 2015).
Perhaps surprisingly, we found that combined PAXX and XLF deficiency is lethal in mice. This was unexpected based on our previous results in human cells, where we found that depletion of XLF in PAXX−/− RPE-1 cells caused no additional radiosensitivity (Ochi et al. 2015). Interestingly, we were able to recapitulate this epistasis in mouse embryonic fibroblasts (MEFs) derived from our Paxx/Xlf single-knockout and double-knockout mice exposed to IR and the radiomimetic drug phleomycin (Supplemental Fig. S9A–C). Due to the extensive cell death in lymphoid progenitors in developing embryos, we were unable to isolate B cells from Paxx−/− Xlf−/− mice to examine their radiosensitivity in vitro. However, recent data using Paxx/Xlf single-knockout and double-knockout v-Abl transformed pro-B cells derived from wild-type mice demonstrated that, in this cell lineage as in the developing CNS, a synthetic relationship between PAXX and XLF exists in terms of cellular sensitivity to IR as well as in the process of V(D)J recombination (Kumar et al. 2016; Lescale et al. 2016b). Thus, there appears to be a tissue-specific requirement for PAXX and XLF: In some cells, both PAXX and XLF are required for NHEJ, but, in others, either one or the other suffices to a major degree. In this regard, we note that tissue-specific requirements for XLF in mice have been demonstrated previously (Li et al. 2008).
Since the generation of XLF-deficient mice was first reported (Li et al. 2008), it has been unclear why the phenotype of these mice is relatively mild compared with mice in which other core NHEJ factors are mutated given that XLF is important for DSB repair by NHEJ in cells (Ahnesorg et al. 2006; Buck et al. 2006). To help explain these findings, functional redundancy with other factors was suggested and, in due course, demonstrated by XLF loss being shown to display nonepistatic or synthetic-lethal relationships with loss of components of the ATM–H2AX–53BP1 pathway or with C-terminal mutants of the V(D)J recombinase protein RAG2 (Zha et al. 2011; Liu et al. 2012; Oksenych et al. 2012; Lescale et al. 2016a). Our data reveal that PAXX and XLF also share one or more redundant functions, as, unlike single mutants, mice lacking both proteins display severe growth defects, extensive cell death in the CNS, and an almost complete block to lymphocyte development similar to that of Xrcc4−/− or Lig4−/− mice. Interestingly, unlike the situation when combining XLF and ATM loss, combined loss of PAXX and ATM did not lead to a phenotype more severe than that of mice lacking ATM alone. Considering our findings, it will be of interest to further explore the overlapping and nonoverlapping functions of PAXX and XLF in NHEJ during development, define the basis for the apparent tissue-specific effects of their combined loss, and establish how PAXX might operate in the context of ATM-mediated DSB repair processes.
Materials and methods
Animals
Care and use of all mice used to generate data for this protocol were carried out in accordance with UK Home Office regulations, UK Animals (Scientific Procedures) Act of 2013. Paxx-deleted (C57BL/6NTac-Paxxem1Gb) mice were generated using CRISPR/Cas9. Ku80 (B6.129-Xrcc5tm1Nus/J; stock no: 004361), Lig4 (B6;129S6-Lig4tm1Fwa/Kvm; stock no: 006482), and Atm (129S6-Atmtm1Awb/J); stock no. 008671) knockout mice were imported from Jackson Laboratories. Xlf (129S6/SvEvTac-Nhej1tm1Fwa) knockout mice (Li et al. 2008) were imported from the laboratory of Dr. Shan Zha at Columbia University. The double-mutant combinations were maintained on the selected mixed backgrounds. For analysis of embryonic development, timed matings were performed. Noon of the day of vaginal plug detection was defined as E0.5.
IR treatment
For analysis of survival upon IR, mice were subjected to 5 Gy of γ irradiation using a sealed 137Cs source γ irradiator (CIS-IBL 437C). Subsequently, mice were monitored by daily weighing for up to 30 d or until reaching humane endpoint criteria based on the clinical disease scoring system detailed in the Supplemental Material.
Histology, immunohistochemistry, and immunoblotting
All major organs were isolated following euthanasia and then fixed in 10% formalin overnight. Embryos were dissected on cold PBS and fixed in 4% paraformaldehyde. On the second day, the fixed organs were transferred to 70% ethanol, placed in cassettes, and embedded in paraffin, and serial 5-µm sections were collected on Superfrost Plus slides (Thermo Fisher Scientific) using a Leica microdissection system (LMD7000). For immunohistochemistry apoptosis and DNA damage, quantitation was done using antibodies against cleaved caspase 3 (Abcam, ab13847) or γH2AX (clone JBW301; Millipore), respectively. For Western blotting, antibodies against PAXX (Abcam, ab126353) and β-actin (Sigma, A5316) were used. Histology images were obtained using an Aperio Scanscope (Aperio Technologies). Statistical analysis was performed using Prism 7 (Graphpad). Student's t tests were always two-tailed.
Supplementary Material
Acknowledgments
We thank Simon Clare for help with mouse immunizations, Josep Forment and Julia Coates for technical assistance with MEFs, and Emmanouil Metzakopian and Robert Weiss for critical reading of the manuscript. Research in S.P.J.’s laboratory is funded by Cancer Research UK (CRUK) program grant number C6/A11224, the European Research Council, and the European Community Seventh Framework Programme grant agreement number HEALTH-F2-2010-259893 (DDResponse). Core funding is provided by CRUK (C6946/A14492) and the Wellcome Trust (WT092096). S.P.J. receives his salary from the University of Cambridge, UK, supplemented by CRUK. L.D.’s laboratory is funded by the Institut Pasteur as well as the European Research Council (ERC) under starting grant agreement number 310917. D.J.A.’s laboratory is supported by CRUK and the Wellcome Trust. A.N.B. is supported by a CRUK Career Development Fellowship (C29215/A20772).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.290510.116.
Freely available online through the Genes & Development Open Access option.
References
- Ahnesorg P, Smith P, Jackson SP. 2006. XLF interacts with the XRCC4–DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124: 301–313. [DOI] [PubMed] [Google Scholar]
- Balmus G, Karp NA, Ng BL, Jackson SP, Adams DJ, Mcintyre RE. 2015. A high-throughput in vivo micronucleus assay for genome instability screening in mice. Nat Protoc 10: 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. 1998. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 8: 1395–1398. [DOI] [PubMed] [Google Scholar]
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Löbrich M. 2009. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 28: 3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche M-C, Sanal O, Plebani A, Stéphan J-L, Hufnagel M, le Deist F, et al. 2006. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124: 287–299. [DOI] [PubMed] [Google Scholar]
- Craxton A, Somers J, Munnur D, Jukes-Jones R, Cain K, Malewicz M. 2015. XLS (c9orf142) is a new component of mammalian DNA double-stranded break repair. Cell Death Differ 22: 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Davidson L, Kangaloo L, Alt FW. 1998. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396: 173–177. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95: 891–902. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Moulding HA, Caldecott KW, Rulten SL. 2014. One ring to bring them all—the role of Ku in mammalian non-homologous end joining. DNA Repair (Amst) 17: 30–38. [DOI] [PubMed] [Google Scholar]
- Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, Ferguson D, Hasty P, Chun J, Alt FW. 2000. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci 97: 2668–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, Lahav G. 2012. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanjawala ZE, Adachi N, Irvine RA, Oh EK, Shibata D, Schwarz K, Hsieh C-L, Lieber MR. 2002. The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair (Amst) 1: 1017–1026. [DOI] [PubMed] [Google Scholar]
- Kumar V, Alt FW, Frock RL. 2016. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc Natl Acad Sci 113: 10619–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescale C, Abramowski V, Bedora-Faure M, Murigneux V, Vera G, Roth DB, Revy P, de Villartay J-P, Deriano L. 2016a. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat Commun 7: 10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, Bacoccina L, Jarade A, Clouin C, Sivapalan R, et al. 2016b. Specific roles of XRCC4 paralogs PAXX and XLF during V(D)J recombination. Cell Rep 16: 2967–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Alt FW, Cheng H-L, Brush JW, Goff PH, Murphy MM, Franco S, Zhang Y, Zha S. 2008. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell 31: 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. 2012. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci 109: 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. 1996. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 382: 551–555. [DOI] [PubMed] [Google Scholar]
- Ochi T, Wu Q, Blundell TL. 2014. The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair (Amst) 17: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, et al. 2015. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347: 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Patel H, Su A, Guo C. 2012. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci 109: 2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H, Nussenzweig A, Kurimasa A, Soares VC, Li X, Cordon-Cardo C, Li WH, Cheong N, Nussenzweig M, Iliakis G, et al. 1997. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J Exp Med 186: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, Gennery AR, Jeggo PA. 2014. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 16C: 84–96. [DOI] [PubMed] [Google Scholar]
- Wright AV, Nuñez JK, Doudna JA. 2016. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell 164: 29–44. [DOI] [PubMed] [Google Scholar]
- Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, et al. 2015. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun 6: 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Guo C, Boboila C, Oksenych V, Cheng H-L, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, et al. 2011. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 469: 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. 1996. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 86: 379–389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.