

Abstract
Recurrent rearrangements of Chromosome 8p23.1 are associated with congenital heart defects and developmental delay. The complexity of this region has led to inconsistencies in the current reference assembly, confounding studies of genetic variation. Using comparative sequence-based approaches, we generated a high-quality 6.3-Mbp alternate reference assembly of an inverted Chromosome 8p23.1 haplotype. Comparison with nonhuman primates reveals a 746-kbp duplicative transposition and two separate inversion events that arose in the last million years of human evolution. The breakpoints associated with these rearrangements map to an ape-specific interchromosomal core duplicon that clusters at sites of evolutionary inversion (P = 7.8 × 10−5). Refinement of microdeletion breakpoints identifies a subgroup of patients that map to the same interchromosomal core involved in the evolutionary formation of the duplication blocks. Our results define a higher-order genomic instability element that has shaped the structure of specific chromosomes during primate evolution contributing to rearrangements associated with inversion and disease.
The Chromosome 8p23.1 region is one of the most structurally dynamic regions of the human genome associated with both normal and disease-causing variation. It contains a common, 4.2-Mbp polymorphic inversion that is highly stratified among human populations (Antonacci et al. 2009; Salm et al. 2012). The allele frequency of the inversion ranges from 60% in Africans to as low as 20% in Asian populations. Recurrent microdeletions between large blocks of segmental duplications (SDs) are associated with congenital heart defects, microcephaly, and developmental delay (Devriendt et al. 1999). Reciprocal duplications and smaller atypical deletions have also been described in individuals that present with a wide range of phenotypic features, including developmental delay, mild dysmorphism, and congenital anomalies (Devriendt et al. 1999; Barber et al. 2007). The breakpoints associated with these rearrangements, including the inversion polymorphism, map to large, complex blocks of high-identity SDs located at either end of the critical region. It has been proposed that heterozygous carriers of the inverted haplotype are particularly predisposed to unequal crossover resulting in recurrent rearrangements at Chromosome 8p23.1 and susceptibility to disease (Giglio et al. 2001; Giorda et al. 2007).
Structural variants, including copy number polymorphic loci, map to SDs that contain a cluster of six beta-defensin genes and the defensin-related gene, sperm associated antigen 11B (Giglio et al. 2001; Hollox et al. 2003). The beta defensins vary substantially in copy number, and this common variation has been associated with multiple immune-related phenotypes, including psoriasis and Crohn's disease (Cantsilieris and White 2013). Despite the importance of the Chromosome 8p23.1 locus in human health and disease, the existence of large alternative structural haplotypes, combined with a complex organization of SDs, has led to inconsistencies in the current human reference assembly that have not yet been resolved (Bakar et al. 2009). Moreover, such genomic complexity represents a significant challenge that has impeded studies of genetic association, including susceptibility to disease (Hollox 2012).
In this study, we sought to understand the contemporary and evolutionary structural instability associated with this locus, first by constructing a high-quality alternate reference sequence of the inverted 8p23.1 haplotype (6.3 Mbp) through SMRT (single-molecule, real-time) sequencing and de novo assembly of large-insert clones from the CHM1 hydatidiform bacterial artificial clone (BAC) library. The new alternate reference facilitated detailed phylogenetic, population genetic, and copy number variation analyses, providing mechanistic insights into the evolutionary instability of this locus as well as a framework for understanding its disease susceptibility in the human species.
Results
Sequence and assembly of the Chromosome 8p23.1 inverted haplotype
The current representation of the Chromosome (Chr) 8p23.1 locus (GRCh37 and GRCh38) has been defined as the direct orientation. It is incompletely assembled, with gaps present at both the distal and proximal SD clusters (referred to as REPD and REPP). We generated a high-quality, alternate reference assembly using large-insert clones (Supplemental Table 1) from the CHORI-17 (CH17) BAC library created from a hydatidiform (haploid) mole-derived human cell line, CHM1hTERT (Kajii and Ohama 1977). The absence of allelic variation allowed us to unambiguously sequence and assemble 68 CH17 BAC clones using SMRT sequencing (after the inclusion of three additional clones from the NCBI clone repository resource [https://www.ncbi.nlm.nih.gov/clone/]) to generate a contiguous 6.3-Mbp alternate assembly representative of the 8p23.1 inverted haplotype (Supplemental Table 1). The alternate CHM1 assembly represents an inverted haplotype, which we refer to as H2 to distinguish it from the structural haplotype represented by the human reference genome (H1).
We assessed the quality of the Chromosome 8p23.1 H2 haplotype by combining paired-end sequence data, read-depth analysis, and high-quality SMRT sequencing (Supplemental Section 1). Our initial clone-based assemblies identified two positions of increased read depth indicative of a tandem duplication that is incompletely assembled (referred to as a “collapsed duplication”). Sequence analysis revealed that this corresponded to a tandem repeat array of 6.12 kbp located at REPD and REPP. The extent of the collapse predicted by read depth at REPD was 30 kbp and was expanded to >80 kbp at REPP (Supplemental Section 1.2). These positions of sequence collapse remain unresolved in our final CHM1 assembly. Remapping of paired-end sequence data from large-insert (BAC and fosmid) clones shows that the underlying assembly is supported by a uniform distribution of concordant end sequence pairs, tight insert-size distributions, and an alignment identity >99.99% for paired-end mappings (Supplemental Table 2; Supplemental Sections 1.3, 1.4). SMRT sequencing of fosmid clones (24 fosmids), predicted by discordant end sequence pileups, detected several regions of structural variation, including a ∼19-kbp deletion at the alpha-defensin cluster found in two individuals (NA19240 and NA12878) (Supplemental Section 1.5). As a final validation, we constructed a BioNano Genomics fingerprint (Lam et al. 2012) map from the original CHM1 source material. A comparison of the long-molecule restriction map confirmed the order and orientation of the H2 assembly with the exception of the two collapses indicated above (Supplemental Section 1.6).
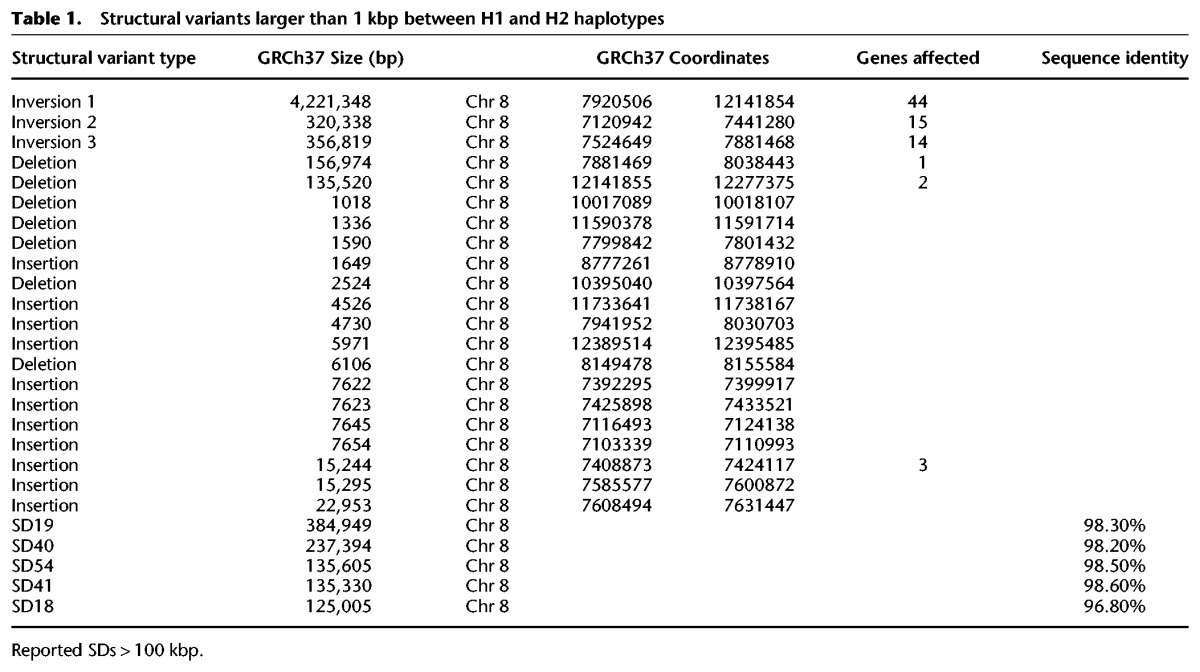
We compared the two human genome assemblies of Chromosome 8p23.1 (Fig. 1). Unlike the GRCh37 H1 haplotype, which contains two large gaps, each estimated at 50 kbp, there are no gaps present in the alternate H2 reference. Sequence comparison between the H1 and H2 haplotypes revealed that our assembly differed from the GRCh37 reference by 21 structural differences >1 kbp in size (Table 1; Fig. 1; Supplemental Fig. 2; Supplemental Tables 11, 13). These differences affect the copy number and/or orientation of 79 genes in the region (Supplemental Section 1.7; Supplemental Figs. 3, 4; Supplemental Table 12). Notably, eight of the differences were >10 kbp in length (Table 1; Supplemental Table 3), including three large inversions ranging from ∼300 kbp to 4.2 Mbp in size. Inversion 1 corresponds to the previously cytogenetically visible common inversion polymorphism of 4.2 Mbp. Inversion 2 (320 kbp), while previously uncharacterized, is supported by a BioNano Genomics fingerprint map and by clone-end sequence mapping, which identifies concordant placements across the interval. The third inversion (inversion 3, estimated at 357 kbp) maps to the proximal side of the distal gap in the H1 haplotype (Fig. 1). Paired-end sequence analysis does not confirm the organization of the corresponding region in H1 but does support the organization of H2 (Supplemental Section 1). Due to the proximity of the distal gap and previous experimental data indicating that the distal beta-defensin cluster 2 should in fact be located at the REPP cluster (Bakar et al. 2009), we conclude that inversion 3 represents an assembly artifact. To this end, we reconstructed the tiling path of RP11 clones underlying the H1 reference assembly and found that clone overlaps within the inversion 3 region in H1 were shorter on average (<20-kbp overlaps) and corresponded to high-identity regions where there is evidence that a hybrid assembly between H1/H2 occurred within the REPP gap region (Supplemental Section 2; Supplemental Figs. 5, 6). This likely contributed to the large-scale error of 357 kbp in the human reference genome represented by inversion 3.
Figure 1.

Structural differences between the reference and the inverted 8p23.1 human haplotypes. The human reference genome sequence (GRCh37 representing the direct haplotype H1) is compared to the inverted haplotype (H2 alternate reference assembly) using Miropeats (Parsons 1995). Joining lines indicate homologous sequence between the two haplotypes with the extent and orientation of segmental duplications (SDs) shown in the context of REPD (GRCh37 Chr 8: 6839961–8100062) and REPP (GRCh37 Chr 8: 11860821–12573597). Colored arrows represent the size and orientation of pairwise SDs >100 kbp. The H2 haplotype contains the largest duplication in direct orientation (385 kbp) (SD19 colored blue) creating susceptibility to unequal crossover. Colored bars represent duplication blocks defined using whole-genome assembly comparison (WGAC). These duplication blocks mediate recurrent microdeletion associated with disease. Three large-scale inversions are depicted: two polymorphisms including the large 4.2-Mbp inversion 1 (purple) and a smaller (∼320 kbp) inversion 2 (green) as well as an artifact of the GRCh37 assembly (inversion 3 indicated in blue). A core duplicon (orange bar) defines the inversion breakpoints and are indicated here as inversion-associated repeats (IARs). The locations of alpha- and beta-defensin copies as well as the tiling path of clones sequenced and assembled are depicted. Sequenced clones that could not be fully resolved due to collapse of large internal duplications are highlighted with an asterisk. Clones confirmed to contain small inserts based on fingerprinting are annotated by a dagger. Redundant clones in the tiling path that were sequenced but incompletely assembled are designated with an x. Although a complete 6.3-Mbp assembly was generated, the schematic focuses on SD blocks contained within the flanking sequence of the H2 assembly (breaks in the unique sequence are shown). The higher-level sequence organization of the new assembly was confirmed by a BioNano Genomics map and BAC-end sequence mapping.
Table 1.
Structural variants larger than 1 kbp between H1 and H2 haplotypes
Inversion breakpoint analysis
We estimate the length of the inversion 1 polymorphism as 4.2 Mbp, refining previous estimates (Fig. 1; Giglio et al. 2001; Antonacci et al. 2009). The inversion is flanked by large, highly identical (>98%) SD blocks of ∼960 kbp at REPD and ∼770 kbp at REPP, with individual duplications mapping in both direct and inverted orientation. To identify the breakpoints associated with inversion 1, we created a ∼400-kbp multiple sequence alignment (MSA) using the distal (REPD) and proximal (REPP) sequences from both the H2 and H1 haplotypes (Fig. 2A; Supplemental Section 2.1). Using a hidden Markov model (HMM), we mapped the most likely breakpoint transition region to a 79.7-kbp stretch of sequence (GRCh37 distal Chr 8: 7920506–7998357 and proximal Chr 8: 12091855–12141854) (Fig. 2A). The absence of contiguous sequence located at the proximal gap in the H1 haplotype and the presence of high-identity sequence with few distinguishing breakpoints made it impossible to refine the breakpoint any further.
Figure 2.

Sequence refinement of 8p23.1 inversion breakpoints within homologous sequences. (A) A 400-kbp multiple sequence alignment (MSA) of homologous sequence between H1 and H2 haplotypes (GRCh37 Chr 8: 7886497–8286302 and Chr 8: 11993961–12171854) was used to refine the breakpoint of inversion 1. Sequence differences mapping inside (black) and outside (yellow) the inversion are depicted in the MSA (top panel). Regions of perfect sequence identity >100 bp are highlighted in dark gray. Breakpoint intervals (dashed red box) are refined (transition region from yellow to black) to a 79.7-kbp region contained within IAR2 and IAR3. The 4.2-Mbp inversion is depicted using Miropeats (lower panel) (GRCh37 Chr 8: 7906497–12171854) with the relative positions of HERVK, IAR, and sequence gaps indicated. The presence of a sequence gap in the H1 assembly (red) prevents further refinement of the inversion 1 breakpoint. (B) Similarly, a schematic of a 100-kbp MSA shows a transition region from yellow to black lines for inversion 2 (GRCh37 Chr 8: 7090000–7175000 and Chr 8: 7370000–7474000). It localizes to a 449-bp region within 33 kbp of IARs 1 and 2. The availability of complete sequence (GRCh37 Chr 8: 6990000–7500000) from H1 and H2 haplotypes allows fine-scale resolution of the inversion 2 breakpoint.
Inversion 2 is estimated to be 320.3 kbp and is completely contained within a high-identity duplication mapping to REPD (SD19 384.94 kbp and >98% identity). The inversion encompasses 15 genes, including the beta-defensin gene family. We narrowed inversion 2 breakpoints to a 449-bp interval in SD19 (GRCh37 Chr 8: 7120942–7121391 distal, Chr 8: 7440831–7441280 proximal) (Fig. 2B; Supplemental Section 2.2). Sequence characterization of the breakpoint interval demonstrated that it was positioned within a 6.12-kbp higher-order tandem repeat unit with multiple copies mapping at both the distal and proximal breakpoints, consistent with previous findings that highly repetitive elements often localize at nonallelic homologous recombination (NAHR)-mediated breakpoints (Kidd et al. 2010). Inversion 2 creates a duplication architecture potentially more susceptible to recurrent rearrangements because the inversion flips a ∼320-kbp duplication (Ottolini et al. 2014) of >98% sequence identity, creating significantly larger blocks (∼385 kbp) of homology that can drive NAHR between REPD and REPP. While the presence of inversion 2 is confirmed by our BioNano Genomics restriction map of CHM1 (Supplemental Fig. 1A), we note that an African sample (NA19240) represents the direct (H1) orientation of this region (Supplemental Fig. 1B), suggesting that this inversion is also polymorphic in the human population. However, high-quality sequence and assembly will be required to confirm whether inversion 2 is in fact polymorphic on both H1 and H2 haplotypes.
An inversion and evolutionary rearrangement instability element
A comparison of the inversion 1 and inversion 2 breakpoint intervals revealed an extended region of homology that was shared among the inversion breakpoints (Supplemental Section 3). We identified a ∼65-kbp duplication particularly enriched for interspersed repetitive elements (74% common repeats; LINEs, SINEs, and LTRs) mapping at the breakpoint of inversion 1 and in close proximity (33 kbp) to inversion 2 (Fig. 2). Due to the proximity of this duplication to Chromosome 8p23.1 inversion events, we referred to this duplication as an inversion-associated repeat (IAR).Within the human reference genome (GRCh37), we discovered 15 copies of these repeats mapping to seven human chromosomes (3, 4, 7, 8, 11, 12, and 16) (Fig. 3A; Supplemental Table 4). The overall sequence identity among the copies was high (94.6%), and in many cases IARs were associated with larger, more complex duplications. These repeat elements correspond to one of the few interchromosomal core duplicons (Newman and Trask 2003; Jiang et al. 2007) not specifically associated with pericentromeric or subtelomeric regions of the genome. Many of the IAR map locations correspond to breakpoint regions associated with evolutionary inversion breakpoints (Darai-Ramqvist et al. 2008). We used BAC end sequence mapping data from nonhuman primate species (chimpanzee, gorilla, and orangutan) (Supplemental Section 3.6) to identify updated map locations of chromosomal evolutionary inversions (Ventura et al. 2011) and overlaid these sites with genomic IAR coordinates. There are 15 IAR map locations within the human reference genome, and we find that 6/27 of the inversions associate with these elements, representing an estimated 7.4-fold enrichment. We note, for example, that IAR cores are localized to the sites of three evolutionary inversion events specifically contributing to the orientation of 3p12.3 and 3q22.1 within the human lineage (Supplemental Section 3.6). Additionally, we identified IAR cores localized to the breakpoints of a fourth evolutionary inversion at Chromosome 11q13.4 and 11p15.4, within the orangutan lineage. We tested by simulation whether IAR-containing duplication blocks significantly clustered with breakpoints of evolutionary inversions (Supplemental Section 3.6). Using a random distribution of duplication blocks within a chromosome, we computed the median distance between the midpoint of the duplication block and the midpoint of the closest inversion breakpoint. As expected, the observed association between IAR duplication blocks and inversion breakpoints is significant (P = 1 × 10−6). Since SDs have previously been shown to be associated with evolutionary rearrangements (91% of inversion breaks), we repeated the simulation, fixing the inversion breakpoints and shuffling the IARs within SD blocks. Even after this restriction, we still find a significant association between IARs and evolutionary inversions in the context of the specific chromosomes (P = 7.8 × 10−5).
Figure 3.

Phylogenetic and comparative analysis of IAR core duplicons. (A) Phylogeny of the core duplicon based on an unrooted neighbor-joining tree (MEGA5) from a 9.2-kbp MSA constructed from 21 IAR core sequences obtained from human and nonhuman primate genome assemblies. Allelic H2 IARs overlapping 8p23.1 inversion breakpoint regions are highlighted in red. The phylogeny shows that the core originated from 16p13.3 and subsequently expanded to various chromosomes early in the ape lineage after divergence from the Old World monkey (see Supplemental Table 10 for timing estimates for nodes A–O, and bootstrap support is indicated). Repeat analysis of a 79-kbp segment associated with the inversion (highlighted) shows that 80% of the structure consists of various classes of common repeats, including LINEs (green), LTRs (orange), and SINEs (purple). (B) FISH analysis using a probe containing core IAR sequences from the gibbon BAC library (CH271-9G12) for human, gibbon (Nomascus leucogenys), and macaque (Macaca mulatta) chromosomal metaphase spreads. Gibbon signals are observed on Chromosomes 4 and 8, which are partially homologous to human Chromosomes 8 (8p21.3–23.3, displayed in light green) and 16 (16p13.1–13.3, displayed in light blue). Macaque signals on Chromosome 20 are homologous to human Chromosome 16p13.3.
We designed a series of single-color metaphase fluorescence in situ hybridization (FISH) experiments and tested for the presence of IARs in lymphoblastoid cell lines obtained from the macaque, gibbon, and human. The macaque showed a single FISH signal syntenic to human Chromosome 16p13.3 consistent with the phylogenetic analysis that this location represents the most likely ancestral locus of the core (Fig. 3; Supplemental Fig. 12). FISH analysis on gibbon metaphase spreads revealed limited duplication, with copies at two locations syntenic to human Chromosome 16p13.3 and Chromosome 8p21.3-23.3 (Fig. 3B; Supplemental Section 3.8). To estimate the timing and order of when this core duplicon expanded during primate evolution, we constructed a phylogenetic tree using a 9-kbp MSA consisting of sequences specific to all interchromosomal cores using a single-copy sequence from macaque and marmoset as an outgroup. Using a locally calibrated molecular clock and an estimated macaque/human divergence time of 25 million years ago (mya) (Gibbs et al. 2007), we estimated that the ancestral core on Chromosome 16p13.3 duplicated to Chromosome 4p16.1 ∼19 mya (Fig. 3A), confirming our initial FISH experiments (Fig. 3B). This initial duplication was followed by rapid expansion of this repeat to Chromosomes 11, 12, and 3 prior to the divergence of the great apes 11–15 mya. We estimate that more recent expansions of the IAR appear to have occurred on Chromosome 8p23.1 (0.55–1 mya), distributing copies both at REPD and REPP (Fig. 3A; Supplemental Table 10).
In order to focus on the evolutionary history of the Chromosome 8p23.1 region, we sequenced, using the Illumina short-read platform, a total of 211 nonhuman primate BAC clones mapping to the orthologous regions. From these data, we selected 71 clones (16 chimpanzee, 34 gorilla, and 21 orangutan) for high-quality sequence and assembly using SMRT sequencing (Supplemental Table 1). The generation of high-quality sequence assemblies from these large-insert clones was critical, given the poor construction (misassembly and gapped sequence) of the Chromosome 8p23.1 locus within nonhuman primate reference genome assemblies. Using these data and the human H1 and H2 references, we constructed a series of phylogenetic trees to estimate the timing of the two large inversion events (Supplemental Sections 3.2, 3.3; Supplemental Figs. 7–9). Sampling two locations within inversion 1 totaling ∼286 kbp, we estimate that the two haplotypes diverged between 0.37 and 0.52 ± 0.03 mya, consistent with previous estimates (0.39 ± 0.07 and 0.59 ± 0.09 mya) (Salm et al. 2012). Similarly, we estimate that inversion 2 arose ∼0.55–0.69 ± 0.02 mya based on analysis of 85 kbp of aligned sequence. We conclude that these inversions arose in concert over a narrow evolutionary period (400–600 thousand years ago [kya]).
To understand the ancestral organization of Chromosome 8p23.1, we created a new reference haplotype of the REPD and REPP regions using large-insert clones from the orangutan BAC library (CH276) (Supplemental Section 4.1). At the distal end, REPD, we sequenced and assembled 10 orangutan BAC clones to generate four contigs totaling 1.56 Mbp anchored to the unique flanking regions (Fig. 4A). Like the human reference (H1), we determined that inversion 2 maps in direct orientation in the orangutan (Supplemental Fig. 14). The REPD locus in orangutan is >200 kbp larger than human, in part due to the presence of lineage-specific expansions of defensin genes. Compared to the CHM1 reference, we observe an ∼80-kbp expansion of the alpha-defensin cluster, with the orangutan containing six full-length copies of the 19-kbp tandem repeat array (each repeat array sharing 98% sequence identity). In contrast to humans, where the theta copies are pseudogenized, our analysis shows that five out of the six orangutan theta-defensin 1 copies maintain an open reading frame and are likely functional (Supplemental Section 4.1; Supplemental Fig. 13). In addition, two different segments harboring beta-defensin genes have expanded in the orangutan lineage. This includes a ∼77-kbp tandem duplication involving DEFB130 and ZNF705D that shares 96% sequence identity and a more recent ∼154-kbp duplication of DEFB134, DEFB135, and DEFB136 that shares 99.1% sequence identity. Copy number variation analysis shows that the DEFB135-136 segment is also duplicated in chimpanzee and bonobo but absent in human and gorilla, likely as a result of a lineage-specific expansion (Supplemental Section 4.1; Supplemental Fig. 15).
Figure 4.

Orangutan sequence assembly of REPD and REPP orthologous regions. (A) A 1.56-Mbp region in the orangutan orthologous to REPD in humans was sequenced and assembled from 10 finished BAC clones and compared using Miropeats (see Fig. 1, H2 for annotation). Lines connecting the two sequences indicate regions of homology and line colors highlight differences between the orangutan and human contig. With the exception of inversion 2, which is in direct orientation (dark blue), the orangutan locus is orientated similarly to human H2. It is >200 kbp larger than human REPD, in part due to a 80-kbp expansion of the alpha-defensin cluster (aqua) and two lineage-specific expansions of beta-defensin (DEFB130 to DEFB136) genes (light blue). (B) A 615-kbp region orthologous to REPP was sequenced and assembled from four finished BAC clones. Comparison with the human H2 assembly using Miropeats shows that the orangutan is missing almost all the duplications (∼746 kbp) present on the human proximal H2 haplotype, including the beta-defensin gene cluster. Black lines connect regions of unique sequence that are shared between orthologous regions in the orangutan and human assemblies. An additional ∼28.8 kbp of ancestral repeat-rich sequence (>65% common repeats) is identified in the orangutan (orange box). (C) A schematic of the proximal insertion site is shown comparing human and orangutan. Sequence analysis of the breakpoint region shows an AluS element mapping on the telomeric side of the 746-kbp duplication block, which is flanked by the 28.8 kbp of ancestral sequence that was lost from the primate ancestor. The centromeric breakpoint maps to a ∼18-kbp core duplicon (Xiao element) that contains OR7E, a gene flanked by SATR2, SATR1, and ERV1 repeat sequences. (D) A schematic description of the DA core duplicon. DupMasker annotation maps the ancestral duplicons to Chr 16p13.3 and Chr 21q22.11; these duplicons form part of a larger complex duplication block referred to as DA. The Xiao core duplicon demarcates the integration site of the SD and is part of the larger DA composite repeat which defines the inversion-associated breakpoint sequences.
At REPP, we constructed a 616.5-kbp sequence contig in orangutan anchored in at least ∼150 kbp of unique sequence flanking the REPP cluster at Chromosome 8p23.1. Sequence analysis demonstrated that the orangutan locus was in the H2 inverted orientation (inversion 1). Notably, the orthologous locus completely lacked 746.1 kbp of SD content found on the CHM1 haplotype, including the entirety of the proximal beta-defensin cluster (Fig. 4B). Instead, the orangutan REPP contained an additional 28.8 kbp of sequence not present in the CHM1 or GRCh37 human genome reference assemblies. Copy number variation analysis identified this ∼28-kbp segment as unique in orangutan and mapping to the orthologous region of Chromosome 8p23.1 in gibbon and macaque. These results indicate that the locus was deleted either prior to or during the duplicative transposition of the 746 kbp from REPD to REPP. Using the molecular clock described above along with orangutan outgroup sequence, we estimate that this large duplicative transposition occurred within the human lineage ∼0.84 mya ± 0.99 (Supplemental Section 4.2). However, we cannot rule out the effect of gene conversion acting on beta-defensin paralogs.
Accounting for this 28.8 kbp of additional sequence, we were able to identify the breakpoints of the duplicative transposition at the base-pair level. The integration breakpoints map at one end to an Alu repeat flanked by an 18.54-kbp SD (termed a “Xiao” element) that is present at high copy in the human reference genome (43 locations across 10 chromosomes) (Supplemental Section 4.2). Intersection of these locations with 15 duplication blocks containing the IAR found that, with the exception of the ancestral 16p13.3 locus, the Xiao element is present at all SDs carrying the IAR (Supplemental Section 4.2). The Xiao element is in fact part of a larger composite ∼200-kbp repeat (termed “DA”) (Ji and Zhao 2008; Li et al. 2009) that includes the IAR SD and corresponds to a larger interchromosomal core duplicon network (M1) identified previously (Supplemental Section 4.2; Supplemental Figs. 16–20; Jiang et al. 2007). Thus, all major evolutionary rearrangements involving inversion and duplicative transposition events of Chromosome 8p23.1 associate with this complex interchromosomal core duplicon (Jiang et al. 2007). In the case of the duplicative integration, we map the distal breakpoints to satellite-associated repeats (SATR1 and SATR2), which define the canonical repeat structure of the Xiao element (Fig. 4C; Ji and Zhao 2008). Similar to the inversion breakpoint regions, sequence analysis of the 28.8-kbp pre-integrated DNA in the orangutan assembly reveals that the common repeat content exceeded 65% (Fig. 4C). This suggests that the proximity of repeat-rich DNA adjacent to a Xiao SD core made it a preferential target for duplicative transposition bringing a larger DA SD that promoted subsequent large-scale inversions (Fig. 4D).
Human sequence diversity analyses
Regions of extended haplotype homozygosity
To explore the patterns of sequence diversity within human, we initially generated a ∼3.6-Mbp pairwise alignment between the H1 and H2 haplotypes that included the Chromosome 8p23.1 critical region (Supplemental Section 5.2). Our analysis identified several unusual patterns of sequence diversity, including six regions of near-perfect sequence identity between the two haplotypes and an additional five regions showing elevated signals of nucleotide diversity (Supplemental Fig. 21). To investigate these regions in more detail, we subsampled these sequences using fosmid libraries from seven diverse humans and sequenced clones to high quality using SMRT sequencing (Supplemental Table 1). The inversion 1 genotype status of these seven individuals had been previously determined by cytogenetic analysis (Antonacci et al. 2009). For each of the seven regions (including of two control regions), we constructed a phylogenetic tree using orthologous regions from the chimpanzee, gorilla, and orangutan reference assemblies (Supplemental Fig. 22). Interestingly, we identified a single ∼25-kbp region completely contained within XKR6 (GRCh37 Chr 8: 10815626–10839946) that demonstrated a tree topology that perfectly separated the H1 and H2 structural haplotypes (Supplemental Section 5.2). Sequence analysis of individuals confirmed for inversion 1 status (H1 direct: NA18956, GRCh37, and inverted H2: CHM1, NA19240, NA12878) identified 36 single-nucleotide variants (SNVs) that perfectly segregated with inversion status (Supplemental Fig. 22).
We also assessed whether Chromosome 8p23.1 shows any regions of extended haplotype homozygosity (eHH)—a potential signal of a recent selective sweep (Sabeti et al. 2002). Using phased SNV and indel calls generated as part of the Human Genome Diversity Project (HGDP) and 1000 Genomes Project, we calculated eHH in >2500 diverse humans and compared this with individuals stratified for inversion 1 status (Supplemental Section 5.2; Supplemental Figs. 23–27). The analysis identified a striking polar pattern of eHH between the direct and inverted haplotypes (Fig. 5). Haplotype bifurcation diagrams show evidence of long-range linkage disequilibrium (Fig. 5B) on the inverted haplotype, extending proximally and distally from the core single-nucleotide polymorphism (SNP) (rs4841222) with a correspondingly high eHH value (0.75) (Fig. 5). The ∼115-kbp block of eHH (GRCh37 Chr 8: 9484432–9599827) is completely housed within TNKS, a gene previously implicated in behavior anomalies as phenotypic features of Chromosome 8p23.1 rearrangements. On the direct H1 haplotype, we identified a more significant ∼75-kbp block of eHH (0.93 eHH) (GRCh37 Chr 8: 10878357–10953092) ∼1.2 Mbp downstream from the inverted eHH block (Fig. 5). Analysis of eHH independent of inversion status showed that the eHHD block is enriched in Asian populations, consistent with the high frequency of the direct haplotype in that population (∼80%). The eHHD block maps to XKR6, a gene previously shown to be under strong signals of selection (Deng et al. 2008).
Figure 5.

Genetic diversity within the 8p23 inversion. (A) Patterns of extended haplotype homozygosity (eHH) (each point = 20 SNV window) are shown for homozygous direct (H1/H1) and inverted (H2/H2) haplotypes from HGDP samples (Sudmant et al. 2015a). Two regions of eHH are highlighted, eHHI (red box) and eHHD (blue box), with the maximal eHH SNP windows (red and blue dots) and a SNP (rs4841222, blue dashed line) commonly associated with the inverted haplotype. Black bars (a–g) represent haplotypes that were fully resolved by sequencing fosmids from seven individuals (Supplemental Table 14). (B) A bifurcation diagram depicting haplotype sharing from rs4841222 (a marker for the inverted H2 haplotype) as a function of genomic distance from Phase III 1000 Genomes Project. The green bifurcation diagram depicts extensive haplotype sharing consistent with linkage to the H2 haplotype distribution. The frequency of rs4841222 (T) is 0.763 for the PFIDO inverted haplotype and 0.374 for the PFIDO direct haplotype. (C) A principal component analysis for the 1000 Genomes Project for the eHHI block shows almost complete discrimination of the haplotypes in that region irrespective of the human population (shapes). The color denotes the inversion status.
Copy number variation
Given the role of SDs in promoting recurrent rearrangement associated with disease in Chromosome 8p23.1, we also assessed the H2 sequence for the presence of high-identity duplications in REPP and REPD that might promote recurrent microdeletion and rearrangement. We find that the inverted H2 haplotype contains multiple, highly identical SDs in direct orientation that flank the disease-associated critical region (Supplemental Table 5; Supplemental Section 5.1); the largest of which, SD19 (∼385 kbp of >98% sequence identity), corresponds to the beta-defensin gene family cluster. We assessed the extent of normal copy number variation of this duplicated segment by measuring the aggregate beta-defensin copy number in 236 high-coverage human and 56 great ape genomes sequenced as part of the HGDP (Sudmant et al. 2015a) and the Great Ape Genome Project (Prado-Martinez et al. 2013). All great apes were diploid for this segment with the exception of human, chimpanzee, and bonobo. Read-depth analysis predicts discrete diplotypes ranging from 2 to 8 in human, consistent with previous experimental analyses (Fig. 6A; Hollox et al. 2003; Hardwick et al. 2011). Extrapolating from the extremes, we estimate that some humans may differ by as much as ∼1.2 Mbp based on SD content differences between REPD and REPP. Africans had a significantly higher mean beta-defensin copy number than non-Africans (F(1,99) = 6.602; P = 0.012) (Fig. 6B). Analyzing a larger number of human genomes (n = 2504) sequenced as part of the 1000 Genomes Project (Sudmant et al. 2015b) confirmed this observation. In particular, individuals of African ancestry harbor significantly more beta-defensin copies (>7 diploid copy number) relative to individuals of non-African ancestry (Fisher's exact test, P = 6.3 × 10−6) (Supplemental Section 5.1).
Figure 6.

Copy number diversity of the beta-defensin cluster. (A) Copy number estimates for the beta-defensin segmental duplication (SD19) based on sequence read depth from 236 humans and 56 great ape genomes. Copy number represents the diploid aggregate (combined proximal and distal beta-defensin segments at REPP and REPD). (B) Box plots show that Africans have a significantly higher mean beta-defensin copy number compared to non-Africans (P = 0.012). No copy number difference is observed between homozygous direct (H1/H1) versus inverted (H2/H2) haplotypes (P = 0.854). Nonhuman great apes show a copy number consistent with no duplication (diploid copy number 2), with the exception of chimpanzee and bonobo where independent expansions of the beta defensins are predicted to have occurred. The less discrete diplotypes in great apes (i.e., noninteger) are likely a reflection of fewer reads mapping between ape and human reference sequences.
Given that inversion 1 status was predicted to play a critical role in susceptibility to recurrent Chromosome 8p23.1 microdeletion (Giglio et al. 2001), we investigated whether Chromosome 8p23.1 inversion status correlated with a higher aggregate beta-defensin copy number, potentially creating a better substrate for NAHR. We used the subset of samples with known inversion status (55 homozygous H1, 15 homozygous H2, 34 heterozygous H1/H2) genotyped by FISH and/or inferred using the PFIDO algorithm (Salm et al. 2012) to examine copy number differences in the beta-defensin gene cluster among inversion statuses and continental group. Contrary to our initial expectations, we found that inversion status has no significant effect on total beta-defensin copy number (F(2,99) = 0.159; P = 0.854), and the interaction between ancestral group and inversion status was not significant (F(1,99) = 0.439; P = 0.509) (Fig. 6B). We expanded this analysis to include paralog-specific copy number measurement using variants that uniquely distinguished the proximal and distal beta-defensin paralogs (Sudmant et al. 2010). Consistent with our aggregate copy number estimates, we found that only population group had a significant effect on paralog-specific copy number (Wilks lambda = 0.882; F(2,98) = 6.5506; P = 0.0021); however, there was no significant effect on copy number and inversion status (MANOVA: Wilks λ = 0.9859; F(4,196) = 0.3484; P = 0.8449). These data suggest that copy number variation of the beta-defensin gene cluster has evolved largely independently on both the H1 and H2 haplotypes and that both REPD and REPP show similar ranges of copy number change.
Chromosome 8p23.1 microdeletion patient breakpoint analysis
Using the new H2 reference as a guide, we attempted to identify the breakpoints associated with disease in patients with recurrent rearrangements. We obtained DNA samples from 13 individuals with a suspected Chromosome 8p23.1 microdeletion who presented with congenital abnormalities, structural heart defects, and/or developmental delay. We designed a customized microarray and performed array comparative genomic hybridization (CGH) for each patient's DNA (Fig. 7). We confirmed 13 cases of Chromosome 8p23.1 microdeletion (Supplemental Table 6), including seven typical cases that contained the canonical ∼3.6-Mbp deletion mediated by the flanking SD clusters at REPD and REPP (Fig. 7). To further refine the breakpoints of these seven individuals with greater precision, we generated a residual best-fit model of array profiles representing the signal depletion expected for each unique and duplicated array probe across the H2 haplotype under the assumption of NAHR between each of the SD pairs (Supplemental Tables 7, 8). We compared the per-probe copy number difference to the median number for a population control group of 23 HapMap individuals of European ancestry (Supplemental Section 6.1; Supplemental Figs. 28, 29).
Figure 7.

Patient microdeletion breakpoint analysis. Array CGH data for 13 of the 8p23.1 microdeletion cases (seven typical and six atypical) associated with congenital heart defects mapped against the H2 sequence assembly. The microdeletion breakpoints for seven typical microdeletion cases (SD-mediated) map to clusters of SDs located at REPD and REPP. Breakpoints were refined to specific SDs by using a model for expected signal depletion under recombination between specific directly orientated SD pairs (SD19 and SD20/21/25 and SD41) (see Supplemental Section 6). Two types of breakpoints were identified, with three patients mapping to SD19 (highlighted with dashed red box) and four patients mapping within SD20/21/25 (highlighted by the dashed blue box). The latter breakpoints map to the DA core duplicon associated with the inversion polymorphism.
Focusing only on SDs in direct orientation, we found that we had robust statistical power to differentiate two distinct groups of patients (Supplemental Tables 7, 8): Group 1 patients (n = 3) showed a pattern consistent with their breakpoints mapping to SD19, while group 2 patients (n = 4) carried a larger deletion mapping to SD20/21/25, estimated to be ∼5.2 Mbp (Fig. 7; Supplemental Section 6.1; Supplemental Table 9). Notably, SD19 represents the largest (∼385 kbp) and most highly identical (>98.3%) SD mapping to Chromosome 8p23.1 (Fig. 8). This segment also contains the beta-defensin cluster which itself is highly copy number variable among humans (Fig. 6A). These findings are consistent with the observation that orientation, length, and sequence identity are important parameters mediating NAHR. In contrast, SD20/21/25 is a ∼177-kbp SD with substantially lower sequence identity (∼95.5%), which is present multiple times in direct orientation at REPP and REPD. This breakpoint region corresponds to the same interchromosomal core harboring the DA and Xiao SDs (Figs. 7, 8). Thus, the same genomic instability element that defines the breakpoints associated with the evolution of this locus is promoting rearrangements in approximately half of the patients with microdeletion and developmental delay.
Figure 8.

Chromosome 8p23.1 evolutionary and instability model. The model shows the evolution and organization of SDs at Chromosome 8p23.1. Colored arrows represent the largest and most highly identical SDs spanning the critical region. At the top, a schematic of the likely primate ancestral organization of the region based on the sequence assembly of the orangutan REPD and REPP units excluding lineage-specific expansions. A ∼746-kbp duplicative transposition or gene conversion to the proximal side of REPP occurred ∼0.84 mya ± 0.99 (see Supplemental Section 4.2 for timing estimate) and inserted into the Xiao core duplicon at REPP. Two large-scale inversions led to the formation of H2 and H1 in the human lineage (0.5–0.6 mya) with breakpoints associated with the DA/Xiao core duplicons. The H1/H2 configuration promotes interchromosomal nonallelic homologous recombination (NAHR) between directly orientated SDs (SD19, blue arrow) that flank the disease-critical region. The size (385 kbp) and large number of tracts of perfect sequence identity suggest that these rearrangements are driven by NAHR in a subset of patients (n = 3). The second group of patients (n = 4) shows breakpoints mapping to a second pair of duplications (SD20-25, red arrow) corresponding to the DA/Xiao core duplicon. The pair is smaller with only three tracks of perfect sequence identity (>500 bp). We propose that group 2 patients arise as a result of recombination between DA/Xiao core instability elements. Colored arrows represent the length and orientation of SD pairs on each haplotype >100 kbp. The positions of the DA, Xiao, and IAR elements are indicated beneath the SDs (green, purple, blue, and red triangles).
Discussion
We present a high-quality assembly (6.3 Mbp) of the human Chromosome 8p23.1 inverted haplotype (H2). We resolved ∼1.8 Mbp of duplicated sequence that was incompletely assembled within the current human reference genome, including two large-scale inversion polymorphisms (∼320 kbp and 4.2 Mbp) (Fig. 1). Our data show the structure and orientation of a ∼385-kbp copy number polymorphism containing a cluster of beta-defensin genes. This organization has remained largely unresolved in successive reference genome builds since the initial sequencing of the human genome (Hollox et al. 2003; Ottolini et al. 2014). Extrapolating from the extremes, we estimate that some humans may differ by as much as ∼1.2 Mbp based on SD content differences at REPD and REPP, largely driven by copy number changes in the beta-defensin cluster (Fig. 6A). We surmise that the existence of large-scale, alternate structural configurations and copy number polymorphic loci resulted in misassignment of paralogous beta-defensin copies in the H1 organization (Bakar et al. 2009), confounding complete assembly of the Chromosome 8p23.1 locus.
The construction of a high-quality reference assembly as well as targeted sequencing of the locus in nonhuman primate genomes allowed us to reconstruct the evolutionary history of the Chromosome 8p23.1 region (Fig. 4). Remarkably, we identified the same interchromosomal core duplicon (DA) (Newman and Trask 2003; Ji and Zhao 2008; Li et al. 2009) at the breakpoints of every large evolutionary change that has reshaped the region during human evolution (Supplemental Fig. 10). The large duplication that transposed ∼746 kbp from REPD to REPP (estimated ∼1 mya), for example, maps within this interchromosomal core— the breakpoint occurring within a satellite-associated repeat (SATR2) that composes its higher-order structure (Supplemental Section 4.2; Ji and Zhao 2008; Li et al. 2009). Similarly, both large inversion polymorphisms (estimated to have occurred 400–600 kya) map adjacent or within the boundaries of the interchromosomal core (Fig. 2; Supplemental Section 3.4). Although our breakpoint precision ranged from 449 bp to ∼80 kbp and could not be further refined due to the absence of sequence in the H1 haplotype, our sequence analysis strongly implicates these elements as opposed to the HERVK repeats as previously suggested (Salm et al. 2012).
Our phylogenetic and comparative FISH analysis confirms that this interchromosomal core has expanded over the last ∼25 million years of ape evolution. Within the human genome, there are at least 15 copies of this interchromosomal core duplicon dispersed to seven human chromosomes (Fig. 3; Supplemental Section 3.5). Interestingly, the core element is enriched for interspersed repetitive elements (Fig. 3; Supplemental Section 3.5). Based on updated maps of evolutionary chromosomal rearrangements between humans and apes, we find the same interchromosomal cores localized at the sites of 6/27 (22%) evolutionary inversions on Chromosomes 3, 7, and 11, including two inversions that are specific to the human lineage on Chromosome 3p12.3 and 3q22.1 (Supplemental Section 3.6). Our permutation tests strongly indicate this association is significant (Supplemental Fig. 11). In total, the data are compelling that this particular interchromosomal core represents a preferential site of genomic instability, a property we have previously observed for core duplicons identified on 16p.12.1, 17q21.31, and 15q13.3 (LCR16a, LRRC37, GOLGA) (Zody et al. 2008; Antonacci et al. 2010, 2014).
Over the years, this particular interchromosomal core has been recognized (albeit with various monikers) in association with different forms of genomic instability. A 1.3-kbp portion of the core duplicon was originally identified by Newman and Trask (2003) because it carried a subfamily of the olfactory receptor gene family (called the 7E-containing SDs). Although the human genome was still work in progress, the authors provided evidence of rampant gene conversion and noted its distribution as unusual because it was one of the few interchromosomal blocks not biased toward pericentromeric or subtelomeric regions of chromosomes. Using a graph-theory and comparative approach, we identified the core sequence as defining a single network (M1) of highly inter-related SDs (Jiang et al. 2007). It was hypothesized to represent one of 24 core duplicons that drove the “punctuated” expansion of SDs in the human–great ape lineage (Jiang et al. 2007). Darai-Ramqvist later identified these elements as tumor break-prone segmental duplications (TBSDs) because of their observation that they were more than three times more likely to be involved in carcinoma-associated rearrangements (Darai-Ramqvist et al. 2008), especially somatic unbalanced translocations. They also found it preferentially mapping to regions of evolutionary breakpoint reuse during primate chromosomal evolution. Zhao and colleagues successfully delineated the structure of the core, distinguishing the smaller (∼30 kbp) LTR-enriched SD, dubbed Xiao (meaning “small” in Chinese), as part of a larger composite (∼300 kbp) higher-order repeat, called DA (meaning “large”). They concluded that these elements evolved in a series and had been extremely active since the divergence of the Old World monkey and ape lineages but had subsequently become quiescent before the divergence of the chimpanzee and human lineage (Li et al. 2009). Our results, in contrast, clearly indicate that these elements continue to restructure human chromosomes.
Although the mechanism by which these elements are driving genomic instability remains a matter of future investigation, sequence analysis reveals some important clues. The duplicative transposition of 746 kbp involved the coordinated loss of extremely repeat-rich sequence at the point of integration (a property that has been observed for other core duplicons) (Johnson et al. 2006). The cores themselves are also repeat-rich, consisting of various classes of common repeats in addition to a diagnostic set of satellite-associated repeats (SATR1-SATR2) that demarcate the point of SD integration at REPP (Supplemental Section 4.2). The interchromosomal DA core duplicons, in general, are preferentially associated with inversions both within and between species, although this may be a consequence of selection operating more efficiently against other forms of genetic variation. Zuffardi and colleagues suggested, for example, that sequences within these duplicated clusters might underlie the second most common constitutional translocation in humans: t(4;8)(p16;p23) (Giglio et al. 2002). Although there are at least 15 DA copies distributed across the genome, it is interesting that eight of these occur as intra-chromosomal “twin signatures,” where the DA repeats are separated by 4.5–5.5 Mbp (Supplemental Section 3.6). Three of these four chromosomal locations have also been associated with inversion polymorphisms representing some of the largest inversion polymorphisms in the human genome (Giglio et al. 2001, 2002; Hurle et al. 2011; Ma and Amos 2012). Analysis of primate genomes suggests that such events have occurred recurrently (i.e., the presence of both direct and inverted haplotypes in the bonobo) (Antonacci et al. 2009). Several of the more active events occur in regions of sharp GC transition in the genome (Darai-Ramqvist et al. 2008) and a comparison with RepliSeq (Hansen et al. 2010) data indicates that ∼30% of the DA elements map to regions of late replication. One possibility may be that these cores represent areas of preferential stalled replication forks (Lee et al. 2007) that frequently reinitiate at paralogous sites, leading to large-scale deletion, duplication, inversion, and unbalanced translocation events.
Our breakpoint analysis of the patients carrying recurrent microdeletions associated with developmental delay and congenital heart defects suggests two distinct classes of breakpoints. Approximately half of patient microdeletion breakpoints map to the polymorphic ∼385-kbp SD (SD19). At this location, the H2 assembly contains the largest, most highly identical SDs in direct orientation (98.3%) with 125 tracts of >500 bp of perfect sequence identity between REPP and REPD. This is consistent with the notion that NAHR is driving recurrent rearrangements in these patients. Our assembled H2 haplotype is particularly predisposed to NAHR because the ∼320-kbp inversion at REPD creates a larger block of directly orientated SDs of ∼385 kbp (98% sequence identity) flanking the disease-associated critical region. Although the frequency of inversion 2 or its phase with respect to the larger inversion cannot be determined because it is completely embedded within SDs, it is interesting that heterozygous carriers would be particularly prone to NAHR under this model (Fig. 8). These structural differences between H1 and H2 may explain why parents of patients are enriched for the heterozygous genotype with respect to the larger Chromosome 8p23.1 inversion (i.e., H1/H2 maternal carriers) (Giglio et al. 2001). It is interesting that our eHH shows patterns consistent with possible positive selection for both H2 and H1 haplotypes at Chromosome 8p23.1 (Fig. 5). The maintenance of such a large inversion polymorphism in all human populations despite its susceptibility to disease, thus, may be a direct consequence of a selective advantage conferred by both haplotypes (Deng et al. 2008; Salm et al. 2012).
The second class of patients (4/7) carries a slightly larger deletion, with breakpoints mapping to a smaller (177 kbp) set of SDs (SD20/SD21/SD25). Group 2 patients would delete ∼283 kbp of additional sequence, removing two annotated genes (FAM86B2 and LOC100506990). Although the SDs mediating this event are in direct orientation, the sequence identity of this block is relatively low (95.5%) and typically below the level of homology that drives the most common recurrent microdeletions in the human species (Cooper et al. 2011; Coe et al. 2014). In contrast to SD19, for example, there are only three perfect sequence identity tracts >500 bp in length. Remarkably, this breakpoint cluster sequence corresponds to the same 250-kbp repeat element (DA/Xiao interchromosomal core) that drove the evolutionary formation of the locus. The fact that approximately half of the patients are mediated by this element suggests that such events are relatively common and factors other than sequence homology are contributing to the genetic instability associated with human disease. Our findings suggest that the DA and Xiao core duplicons not only played a fundamental role in shaping the architecture of the H1 and H2 haplotypes but continue to predispose it to disease-causing rearrangements.
Methods
Sequence and assembly of BAC clones
DNA from CH17, CH251, CH276, and CH277 BAC clone libraries were isolated, prepped into bar-coded genomic libraries, and sequenced (PE101) on a MiSeq using a Nextera protocol (Steinberg et al. 2012). Sequencing data (∼300-fold coverage) were mapped with mrsFAST (Hach et al. 2010) to the reference genome, and singly unique nucleotide (SUN) identifiers were used to discriminate between highly identical SDs (Sudmant et al. 2010). PacBio (Pacific Biosciences, Inc.) SMRTbell libraries were prepared and sequenced using RSII C2P4 chemistry or RSII C2P6 chemistry. Inserts were assembled using QUIVER and HGAP (Chin et al. 2013). Contig assembly was performed using Sequencher (Gene Codes Corporation) and compared to the human reference genome (GRCh37) using Miropeats (Parsons 1995) and BLAST (Altschul et al. 1990). (See Supplemental Section 1 for detailed methods.)
FISH analysis
Single-color metaphase FISH was performed using lymphoblast cell lines obtained from one human individual (GM12878), one macaque (MMU, Macaca mulatta, 3238) from The Biomedical Primate Research Centre of Rijswijk, and one gibbon (Nomascus leucogenys). FISH experiments were performed using a gibbon BAC clone (CH271-9G12) directly labeled by nick-translation with Cy3-dUTP (PerkinElmer) as described previously (Lichter et al. 1990) with minor modifications (Supplemental Section 3.8).
Phylogenetic analyses
We estimated the evolutionary timing of SDs and inversion events by generating MSAs representative of human, chimpanzee, gorilla, orangutan, and macaque orthologous and paralogous sequences using MAFFT (Katoh et al. 2002). We constructed an unrooted phylogenetic tree using the neighbor-joining method (MEGA5) (Tamura et al. 2011). Genetic distances were computed using the Kimura two-parameter method with standard error estimates and interior branch test of phylogeny (n = 500 bootstrap replicates). Tajima's relative rate test (MEGA5) was used to assess branch length neutrality. We estimated the coalescence of time using the equation R = K/2T, assuming a chimpanzee–human divergence time (T) of 6–7 mya for chimpanzee, 15 mya for the orangutan, and 25 mya for the macaque (Supplemental Sections 3.2, 3.3).
Copy number variation analysis
Copy number genotyping was performed using the sequence read-depth method (Sudmant et al. 2010) with whole-genome sequence data from humans (n = 236) (Sudmant et al. 2015b) and nonhuman primates (n = 56) (Supplemental Section 5.1; Prado-Martinez et al. 2013). The duplication content was determined using whole-genome shotgun sequence detection (WSSD) and whole-genome assembly comparison (WGAC) as previously described (Bailey et al. 2002). Patient DNA was obtained from Chromosome 8p23.1 microdeletions (Coriell) and assessed for 8p23.1 rearrangements using array CGH (Supplemental Section 6.1). Array CGH experiments were performed on 14 of the 8p23.1 microdeletion cases using a custom, high-density oligonucleotide 4 × 180K Agilent chip targeted to “genomic hotspots” with a density of 500 bp for SD regions and 1 kbp for unique regions.
Population genetic analysis
We used SNV/indel calls from the Human Genome Diversity Project or HGDP (Sudmant et al. 2015a) and the Phase III 1000 Genomes Project (Sudmant et al. 2015b) mapped to GRCh37. HGDP genomes were phased using BEAGLE 4.0 (r1399). Observed heterozygosity (oHET), extended haplotype homozygosity, integrated haplotype score (iHS), and FST were calculated using the genotype–phenotype association toolkit (GPAT), within VCFLIB (https://github.com/vcflib/vcflib). All smoothed data were generated with GPAT's smoothing program. For the FST and oHET analyses, we used the 1000 Genomes Project data without filtering. Haplotype analyses were performed with biallelic variants with global frequencies between 0.1 and 0.9. The PCA was performed using phased haplotypes between 9.0 Mbp and 10.25 Mbp for Chromosome 8p23.1. The “plotHaps” tool in GPAT was used to convert the phased VCF to a haplotype matrix; data were imported into R and the “prcomp” was used to calculate the principal components (Supplemental Section 5.2).
Data access
All sequenced clones from this study have been submitted to the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under the accession numbers listed in Supplemental Table 1. The sequences are also available, along with the H2 haplotype assembly (6.3 Mbp), at NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA306877.
Competing interest statement
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc. and is a consultant for the Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program.
Supplementary Material
Acknowledgments
We thank T. Brown for assistance with manuscript preparation. This work was supported, in part, by grants from the US National Institutes of Health (NIH grant 2R01HG002385 to E.E.E. and U41HG007635 to R.K.W. and E.E.E.). This work was supported by an American Asthma Foundation Senior Investigator award (to E.E.E., #10-0159). S.C. was supported by a National Health and Medical Research Council (NHMRC) CJ Martin Biomedical Fellowship (#1073726). E.E.E. is an Investigator of the Howard Hughes Medical Institute.
Author contributions: K.M., S.C., and E.E.E. designed the study. K.M. performed the library construction for Illumina and SMRT sequencing, array CGH experiments and sequence analysis. S.C. performed the library construction for Illumina sequencing and sequence analysis. J.H. performed sequence analysis and simulations. B.J.N. performed copy number variation analysis. B.P.C. performed array CGH experiments, sequence analysis, and analysis of patient breakpoints. C.D.C. performed analysis for Illumina sequencing and analysis for CH17 BAC clones. C.B. and L.H. performed BAC hybridization experiments. K.M.M. performed library construction for SMRT sequencing. Z.N.K. performed population genetic analysis. M.K. performed BioNano Genomics experiments. A.R. performed sequence analysis and BES/FES mapping. C.R.C. performed the FISH experiments. T.A.G. and R.K.W. performed the capillary sequencing and analysis of nonhuman primate BAC clones. M.V. performed the evolutionary analysis. K.M., S.C., and E.E.E. contributed to the data interpretation. K.M., S.C., and E.E.E. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.211284.116.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Ventura M, Siswara P, Jiang Z, Eichler EE. 2009. Characterization of six human disease-associated inversion polymorphisms. Hum Mol Genet 18: 2555–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, Girirajan S, Alkan C, Campbell CD, Vives L, Malig M, et al. 2010. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet 42: 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M, et al. 2014. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet 46: 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bakar SA, Hollox EJ, Armour JAL. 2009. Allelic recombination between distinct genomic locations generates copy number diversity in human β-defensins. Proc Natl Acad Sci 106: 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber JCK, Maloney VK, Huang S, Bunyan DJ, Cresswell L, Kinning E, Benson A, Cheetham T, Wyllie J, Lynch SA, et al. 2007. 8p23.1 duplication syndrome; a novel genomic condition with unexpected complexity revealed by array CGH. Eur J Hum Genet 16: 18–27. [DOI] [PubMed] [Google Scholar]
- Cantsilieris S, White SJ. 2013. Correlating multiallelic copy number polymorphisms with disease susceptibility. Hum Mutat 34: 1–13. [DOI] [PubMed] [Google Scholar]
- Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LELM, et al. 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. 2011. A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darai-Ramqvist E, Sandlund A, Müller S, Klein G, Imreh S, Kost-Alimova M. 2008. Segmental duplications and evolutionary plasticity at tumor chromosome break-prone regions. Genome Res 18: 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Zhang Y, Kang J, Liu T, Zhao H, Gao Y, Li C, Pan H, Tang X, Wang D, et al. 2008. An unusual haplotype structure on human chromosome 8p23 derived from the inversion polymorphism. Hum Mutat 29: 1209–1216. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Matthijs G, Van Dael R, Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J, Bröndum-Nielsen K, Marynen P, et al. 1999. Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am J Hum Genet 64: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: 222–234. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, et al. 2001. Olfactory receptor–gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 68: 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, et al. 2002. Heterozygous submicroscopic inversions involving olfactory receptor–gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet 71: 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorda R, Ciccone R, Gimelli G, Pramparo T, Beri S, Bonaglia MC, Giglio S, Genuardi M, Argente J, Rocchi M, et al. 2007. Two classes of low-copy repeats comediate a new recurrent rearrangement consisting of duplication at 8p23.1 and triplication at 8p23.2. Hum Mutat 28: 459–468. [DOI] [PubMed] [Google Scholar]
- Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, Sahinalp SC. 2010. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat Methods 7: 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. 2010. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci 107: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick RJ, Machado LR, Zuccherato LW, Antolinos S, Xue Y, Shawa N, Gilman RH, Cabrera L, Berg DE, Tyler-Smith C, et al. 2011. A worldwide analysis of beta-defensin copy number variation suggests recent selection of a high-expressing DEFB103 gene copy in East Asia. Hum Mutat 32: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ. 2012. The challenges of studying complex and dynamic regions of the human genome. Methods Mol Biol 838: 187–207. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Armour JAL, Barber JCK. 2003. Extensive normal copy number variation of a β-defensin antimicrobial-gene cluster. Am J Hum Genet 73: 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurle B, Marques-Bonet T, Antonacci F, Hughes I, Ryan JF, Eichler EE, Ornitz DM, Green ED. 2011. Lineage-specific evolution of the vertebrate Otopetrin gene family revealed by comparative genomic analyses. BMC Evol Biol 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Zhao S. 2008. DA and Xiao—two giant and composite LTR–retrotransposon-like elements identified in the human genome. Genomics 91: 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. 2007. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet 39: 1361–1368. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Cheng Z, Morrison VA, Scherer S, Ventura M, Gibbs RA, Green ED, Eichler EE. 2006. Recurrent duplication-driven transposition of DNA during hominoid evolution. Proc Natl Acad Sci 103: 17626–17631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajii T, Ohama K. 1977. Androgenetic origin of hydatidiform mole. Nature 268: 633–634. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. 2010. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. 2012. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotech 30: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CMB, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. [DOI] [PubMed] [Google Scholar]
- Li X, Slife J, Patel N, Zhao S. 2009. Stepwise evolution of two giant composite LTR-retrotransposon-like elements DA and Xiao. BMC Evol Biol 9: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter P, Tang C, Call K, Hermanson G, Evans G, Housman D, Ward D. 1990. High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247: 64–69. [DOI] [PubMed] [Google Scholar]
- Ma J, Amos CI. 2012. Investigation of inversion polymorphisms in the human genome using principal components analysis. PLoS One 7: e40224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman T, Trask BJ. 2003. Complex evolution of 7E olfactory receptor genes in segmental duplications. Genome Res 13: 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolini B, Hornsby MJ, Abujaber R, MacArthur JAL, Badge RM, Schwarzacher T, Albertson DG, Bevins CL, Solnick JV, Hollox EJ. 2014. Evidence of convergent evolution in humans and macaques supports an adaptive role for copy number variation of the β-defensin-2 gene. Genome Biol Evol 6: 3025–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JD. 1995. Miropeats: graphical DNA sequence comparisons. Comput Appl Biosci 11: 615–619. [DOI] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O'Connor TD, Santpere G, et al. 2013. Great ape genetic diversity and population history. Nature 499: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Salm MPA, Horswell SD, Hutchison CE, Speedy HE, Yang X, Liang L, Schadt EE, Cookson WO, Wierzbicki AS, Naoumova RP, et al. 2012. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res 22: 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, et al. 2012. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet 44: 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. 2010. Diversity of human copy number variation and multicopy genes. Science 330: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. 2015a. Global diversity, population stratification, and selection of human copy-number variation. Science 349: aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015b. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Catacchio CR, Alkan C, Marques-Bonet T, Sajjadian S, Graves TA, Hormozdiari F, Navarro A, Malig M, Baker C, et al. 2011. Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Res 21: 1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung H-C, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. 2008. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 40: 1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.