

Abstract
Background.
Hypoplastic left heart syndrome (HLHS) is an etiologically multifactorial congenital heart disease affecting one in 5,000 newborns. Thirty years ago there were no treatment options for this pathology and the natural course of the disease led to death, usually within the first weeks of life. Recently surgical palliative techniques have been developed allowing for a five-year survival in more than half the cases.
Materials and methods.
We reviewed literature available on HLHS, specifically its anatomy, embryology and pathophysiology, and treatment. The Pubmed and ClinicalKey databases were searched using the key words hypoplastic left heart syndrome, foetal aortic valvuloplasty, foetal septoplasty, Norwood procedure, bidirectional Glenn procedure, Fontan procedure, hybrid procedure. The relevant literature was reviewed and included in the article. We reported a case from Children’s Clinical University Hospital, Riga, to illustrate treatment tactics in Latvia.
Results.
There are three possible directions for therapy in newborns with HLHS: orthotopic heart transplantation, staged surgical palliation and palliative non-surgical treatment or comfort care. Another treatment mode – foetal therapy – has arisen. Staged palliation and full Fontan circulation is a temporary solution, however, the only means for survival until heart transplantation. Fifty to 70% of patients who have gone through all three stages of palliation live to the age of five years.
Conclusions.
The superior mode of treatment is not yet clear and the management must be based on each individual case, the experience of each clinic, as well as the financial aspects and will of the patient’s parents.
Keywords: hypoplastic left heart syndrome, Norwood procedure, bidirectional Glenn procedure, Fontan procedure, hybrid procedure, balloon valvuloplasty
Abstract
KAIRIOSIOS ŠIRDIES HIPOPLAZIJOS SINDROMAS: APŽVALGA
Santrauka
Prielaida. Kairiosios širdies hipoplazijos sindromas (KŠHS) etiologiniu požiūriu yra daugiaveiksnė įgimta širdies liga, kuria suserga vienas iš 5000 naujagimių. Prieš trisdešimt metų gydyti šią patologiją galimybių nebuvo ir paprastai pirmosiomis gyvenimo savaitėmis ligos eiga natūraliai baigdavosi mirtimi. Neseniai atsiradę paliatyviosios chirurgijos intervencijos metodai ligos atveju leidžia išgyventi penkerius metus daugiau nei pusei pacientų.
Medžiagos ir metodai. Peržvelgėme literatūrą apie KŠHS, jos anatomiją, embriologiją, patofiziologiją ir gydymą. Atliktos paieškos PubMed ir ClinicalKey duomenų bazėse naudojant raktinius žodžius kairiosios širdies hipoplazijos sindromas, vaisiaus aortos valvuloplastika, vaisiaus septoplastika, Norwood procedūra, dvikryptė Gleno procedūra, Fontan procedūra, hibridinė procedūra. Analizuota atitinkama literatūra, aptikta informacija panaudota straipsnyje. Kaip taikomo gydymo pavyzdį Latvijoje aprašėme ligos atvejį Rygos vaikų klinikinėje universitetinėje ligoninėje.
Rezultatai. Galimos trys sergančių KŠHS naujagimių gydymo terapijos kryptys: ortotopinė širdies transplantacija, sudėliotas chirurginis lengvinantis gydymas ir paliatyvioji ne chirurginė terapija arba komforto priežiūra. Pradėtas taikyti naujas gydymo būdas – vaisiaus terapija. Lengvinantis gydymas ir pilna Fontan cirkuliacija yra laikinas sprendimas, tačiau tai vienintelė išgyvenimo priemonė iki širdies transplantacijos. Nuo 50 iki 70 % pacientų, kurie ištvėrė visus tris lengvinančio gydymo etapus, gyveno iki penkerių metų.
Išvados. Aukščiausio režimo gydymas dar nėra aiškus ir ligos valdymas turi būti pagrįstas kiekvienu konkrečiu atveju, klinikos patirtimi, taip pat finansiniais aspektais ir paciento tėvų valia.
Raktažodžiai: kairiosios širdies hipoplazijos sindromas, Norwood procedūra, dvikryptė Gleno procedūra, Fontan procedūra, hibridinė procedūra, balioninė valvuloplastika
Introduction
Hypoplastic left heart syndrome (HLHS) is an etiologically multifactorial congenital heart disease affecting one in 5,000 newborns. Thirty years ago there were no treatment options for this pathology and the natural course of the disease led to death, usually within the first weeks of life. Recently surgical palliative techniques have been developed allowing for a five-year survival in more than half the cases.
This article reviews the pathophysiology, treatment options and outcomes of the disease, as well as the current situation of the pathology in Latvia. We report a case to illustrate the treatment tactics in Latvia.
Embryology, anatomy, physiology
Hypoplastic left heart syndrome is a set of congenital and pathogenically closely related malformations, where the central element is an atresia or critical stenosis of either mitral or aortic valve with sequential left ventricle (LV), ascending aorta and aortic arch hypoplasia (1).
Fig. 1.
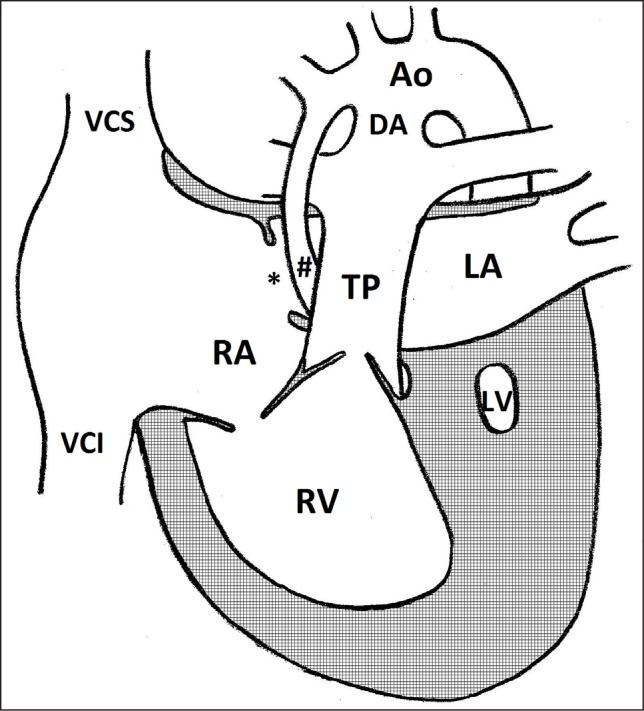
Anatomy of hypoplastic left heart syndrome. Ao – transverse aorta; DA – ductus arteriosus; LA – left atrium; LV – left ventricle; RA – right atrium; RV – right ventricle; TP – truncus pulmonalis; VCI – vena cava inferior; VCS – vena cava superior; # – hypoplastic ascending aorta; * – foramen ovale
The aetiology and pathophysiology of HLHS is variable, but leads to the same outcome – underdevelopment of the structures of the left heart. The leading factors that determine this peculiar anatomy are divided in groups of obstruction of outflow from the LV (aortic valve stenosis (AVS) or atresia (AVA), left ventricular outflow tract obstruction) or obstructed flow into the LV (restrictive foramen ovale (FO), deviation of septum primum, mitral valve stenosis (MVS) or atresia (MVA)) (2). Defective aortic valve is the most common cause for decreased flow in the LV and increased pressure in it. The most credible scenario of evolution of hypoplasia in this case would be that the increased afterload contributes to LV hypertrophy with ensuing dilation and decreased contractility (3). At this point the LV would appear to be an appropriate size for gestational age on an echocardiogram (4, 5). The reduced flow is an inhibitor of further growth of the LV, and, as the foetus continues to increase in size, the left heart structures become hypoplastic (3–6). The increasing pressure in the left atrium causes the flow through FO to become bidirectional or even reverse contributing even more to the flow decrease in the LV. Another factor that inhibits growth of the left heart is the enlarged structures of the right side (6). In the case of MVS or MVA the hypoplasia is caused by the decreased preload and pressure in the LV (3, 6). Dilated cardiomyopathy and endocardial fibroelastosis might be independent factors causing HLHS (7–9).
HLHS is a progressive pathology with features that vary in time. It cannot always be excluded based on non-existent anatomic changes on the foetal echocardiography; deviations from the normal physiology must be taken into account, such as reverse flow in the transverse aorta, monophasic flow through the mitral valve, left-to-right flow through the FO, LV dysfunction (10).
Pulmonary veins usually drain to the left atrium, which is in communication with the right atrium through atrial septal defect, most commonly persistent FO. Seldom anomalous pulmonary vein drainage directly to the right atrium or either vena cava is observed (11–14). The ductus arteriosus in HLHS is typically variable in size, but of normal anatomical location (12). Morphologically the ductal tissue extends proximally and distally into the aortic wall, which is thought to be the cause of the frequently associated coarctation of the aorta (12, 15, 16).
HLHS is a ductus arteriosus dependent cyanotic heart disease, where parallel pulmonary and systemic circulation is maintained by the single right ventricle. The LV in a child with HLHS is a non-functional anatomical structure (14). The systolic volume is ejected into the truncus pulmonalis, where the blood flow is divided. The pulmonary circulation, alike in normal anatomy, is supplied through left and right pulmonary arteries. The systemic circulation, however, is perfused through a common arterial structure composed of truncus pulmonalis, ductus arteriosus and descending aorta (15). This is the reason for ductus dependency of the systemic blood flow in HLHS. The hypoplastic ascending aorta serves as a common coronary artery, thus a pre-ductal aortic coarctation may compromise an adequate perfusion of the hypertrophied right heart muscle (15). Since the blood flow to and from the left ventricle is minute or absent, the atrial septal defect provides the only significant exit from the left atrium for the pulmonary venous blood flow (14). Because of systemic and pulmonary venous blood flow merging in the right atrium, the approximate arterial blood oxygen saturation is about 80%.
The aberrant anatomy of parallel, rather than in series, arranged pulmonary and systemic circulation is the cause of precarious distribution of cardiac output to pulmonary, systemic and coronary blood flow (12). In the foetus the pulmonary vascular resistance is far greater than systemic vascular resistance. Normal systemic and coronary perfusion pressure is maintained as a result of most of the cardiac output entering the descending aorta and cerebral and coronary circulation through the ductus arteriosus (1). The said aberrant anatomy in the foetus does not lead to a significant impairment of the systemic blood supply. The transition of circulation after birth that results in normal postnatal cardiorespiratory physiology in a healthy child – the decline in pulmonary vascular resistance and the spontaneous closure of ductus arteriosus – causes serious haemodynamic consequences in a child with HLHS. The spontaneous closure of ductus arteriosus compromises the blood flow in the systemic circulation including the cerebral and coronary pool. The decreased pulmonary vascular resistance reallocates the cardiac output in favour of pulmonary circulation, and the blood flow in the lungs gradually rises. In an attempt to maintain an adequate perfusion in the systemic circulation the cardiac output increases, however, with the consequent increase in cardiac effort, the compensatory mechanisms are short-lived. With rapid development of hypoxia and tissue ischemia metabolic acidosis ensues (1, 14, 15). A significant pulmonary blood flow limiting factor is the left atrial pressure, which is for the most part determined by the size of the atrial septal defect. As a result, a moderately restrictive flow from the left to right atrium with retained adequate pulmonary venous drainage might be advantageous (14).
Treatment options
There are three possible directions for therapy in newborns with HLHS: orthotopic heart transplan transplantation (OHT), surgical staged palliation and palliative non-surgical treatment or comfort care. Another treatment mode – foetal therapy – has arisen and its options and results are being actively studied.
Staged surgical palliation was first described by Norwood in the 1980s. Initially the results were not promising, although it was the only mode of treatment with the possibility of an increased lifespan. Shortly thereafter the first OHTs were performed and the mortality rates were lower compared to the Norwood’s method. In the thirty years since, results after both methods have increased dramatically and a new surgical technique has been developed – the hybrid procedure – encompassing the elements of palliative surgery and a transcatheter approach. It is not clear as to which method is the superior one but staged palliation is employed most often. Besides palliative surgery the options remaining are OHT, the use of which is restricted by the limited number of available donors, comfort care or termination of pregnancy.
Foetal therapy
The purpose of foetal aortic valvuloplasty (FAV) is to halt the development of HLHS through altering the pathologic anatomy and physiology before severe arrest in growth of the left heart structures has occurred. FAV is a percutaneous procedure performed on about the 24th gestational week (17). The procedure is performed while the mother is under regional or local anaesthesia; the foetus requires anaesthesia as well and is paralysed. The foetal intrauterine position is adjusted. The mother’s abdominal wall, uterus, and then the foetal thoracic wall is punctured. The foetal LV is approached from the apex. The guide-wire and the balloon are introduced through the stenotic aortic valve. The balloon is inflated repeatedly, gradually dilating the valve (3, 18, 19).
FAV is performed on patients with aortic stenosis, physiologic aberrations leading to HLHS and a LV with a potential for functioning. As to the characteristics of patients with a potentially salvageable LV, they remain to be specified. For now FAV is indicated in patients with AVS (and not AVA), bidirectional or reverse flow in the transverse aorta, retrograde flow through the atrial septum or intact septum, monophasic flow through the mitral valve and decreased LV function. The establishment of a biventricular (BV) circulation is considered successful with enlargement of the left heart structures and increased pressure in the LV (17, 20, 21).
At the moment the relative success of FAV is reflected in two large one-centre studies as well as a registry consisting of data from several smaller one-centre studies. The results are similar. The procedure was a technical success in 80% of cases. The success rate was lower at first, but the tendency was to improve due to a learning curve. A BV circulation was established in 43–66.7% of live-born neonates (17, 19, 21). Different patient groups, increased gestational age at the time of the procedure and a larger balloon used were the considered factors for a better outcome in the centre with better results (21). Despite the promising results foetal mortality – 11–18.6% – must be considered. The mortality rate was higher in the multicentre study where the learning curve might be slower due to a smaller number of cases (19).
There are no long-term results as of yet, but one study describes mid-term results. FAV was performed on 100 foetuses that were stratified in biventricular and univentricular outcome groups. There was a significant difference in neonatal mortality – 12.3% in the HLHS group and 0% in the BV group. Although early mortality was higher in the HLHS group, in seven years the mortality rates in both groups were practically the same. The cardiac mortality rate in patients born with BV circulation (and not corrected to BV after birth) was 0%. The overall 10-year survival for the whole group was 72%; this is comparable to the survival after staged palliation (17).
The early post-procedural complications included foetal bradycardia, haemopericadium, LV thrombus, rupture of the balloon or failure to retract it and intrauterine foetal death. No maternal complications were reported (17, 19–21). Late complications were further valvular stenosis, valvular insufficiency, endocardial fibroelastosis, and myocardial fibrosis. LV remodelation with the decreased systolic and diastolic function has been observed in patients with BV circulation (17, 19–22).
Even after achieving BV circulation cardiac intervention was often necessary. In 74% of patients there had been at least one cardiac catheterization, at least one surgical intervention was required in 84%. Overall, 97% of patients with BV circulation after birth needed further invasive therapy (17). Balloon valvuloplasty and valvular replacement were the procedures most often performed amongst these patients (17, 19–21).
Another modality of foetal therapy is atrial septoplasty performed on patients with established HLHS and a restrictive or an intact atrial septum. This procedure might prevent or alleviate pulmonary venous hypertension, allow for effective pulmonary blood flow and prevent postnatal hypoxemia (23). The atrial septum is approached from the right atrium. If needed, the septum is perforated, afterwards a balloon is introduced and inflated. A stent is implanted in the case of a thick atrial septum (3).
The number of cases with an intact septum is small (6–19% of all the HLHS cases), and it is not yet clear as to which is the most appropriate tactic and when should it be applied. In one study the procedure was performed in 21 foetuses on a mean gestational age of 29 weeks. Procedural related death occurred in two cases. Twelve patients required urgent left atrial decompression after birth; the condition was stable in seven patients with medical therapy (24). To prevent closure of the interatrial connection, a stent may be implanted (25, 26).
Norwood procedure
Stage I palliation in HLHS is the Norwood procedure. Its aims are to 1) allow for an unobstructed blood flow from the right ventricle to the systemic circulation, 2) establish a connection between the systemic and the restricted pulmonary circulation, 3) provide an unobstructed flow from the pulmonary veins through the atrial septum to the right atrium (19). The procedure is performed shortly after birth. It has some variations based on individual anatomy. The ductus arteriosus is ligated, the truncus pulmonalis is separated and the distal end is sutured. The hypoplastic native aorta is longitudinally incised beyond the opening of ductus arteriosus. A neo-aorta is constructed from the native aorta, the proximal end of truncus pulmonalis and a pulmonary homograft or an auto-pericardium; this is the connection between the RV and the systemic circulation. Septum primum is excised establishing an unobstructed junction between the atria. The pulmonary circulation is ensured through a modified Blalock-Taussig (BT) shunt, which connects a pulmonary artery with a brachiocephalic or a subclavian artery, or a shunt from the right ventricle to a pulmonary artery (13, 27, 28).
Fig. 2.
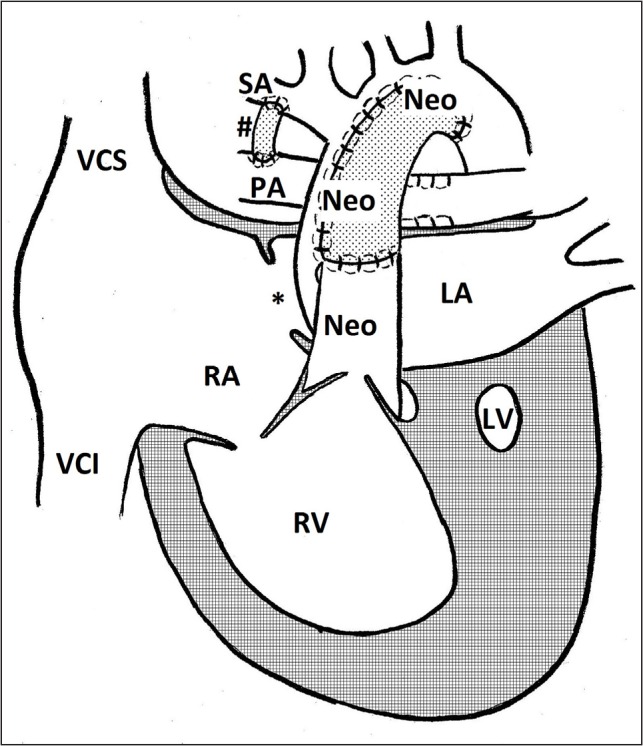
Norwood procedure. Neo – neoarta; LA – left atrium; LV – left ventricle; PA – pulmonary artery; RA – right atrium; RV – right ventricle; SA – subclavian artery; VCI – vena cava inferior; VCS – vena cava superior; # – modified Blalock-Taussig shunt; * – foramen ovale
Hybrid procedure
The hybrid procedure is an alternative choice to the first stage of the classic staged repair technique and is performed during the first week of a child’s life. It consists of a bilateral pulmonary artery banding and a stent placement in ductus arteriosus to secure the right to left shunt (1, 29). This grants a restrictive blood flow to the lungs, while maintaining a systemic perfusion. The hybrid procedure is frequently performed simultaneously with the trans-catheter enlargement of the atrial septal defect or interatrial stent placement. During the hybrid procedure, unlike in the Norwood operation, cardiopulmonary bypass and cardiac arrest, which are both significant insult to the child’s not yet fully matured myocardium, is not required. For this reason, the hybrid procedure is the surgical method of choice for high risk (premature, low birthweight, CNS developmental defect, multiple organ failure, restrictive atrial septal defect, severe ventricular dysfunction, severe atrioventricular regurgitation) HLHS patients (1, 2, 12, 16, 29). The persistent risk of a haemodynamically significant preductal aortic coarctation with compromised coronary artery perfusion is a major flaw of the hybrid procedure. The stage two procedure succeeding the hybrid procedure is a combination between the Norwood and the bidirectional Glenn operation (1, 16).
Fig. 3.
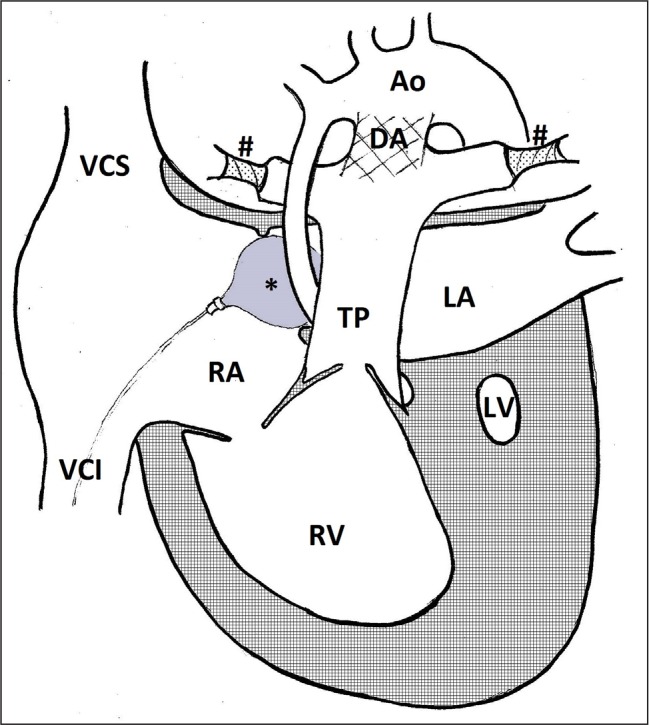
Hybrid procedure. Ao – transverse aorta; DA – stent in ductus arteriosus; LA – left atrium; LV – left ventricle; RA – right atrium; RV – right ventricle; TP – truncus pulmonalis; VCI – vena cava inferior; VCS – vena cava superior; # – pulmonary artery banding; * – balloon in foramen ovale
Bidirectional Glenn procedure
Initially the Fontan operation was performed during the first year of life as the second stage after a successful Norwood operation, however, this approach was associated with high morbidity and mortality rates. Accordingly, an inter-stage was introduced – a bidirectional superior cavo-pulmonary anastomosis (Glenn or hemi-Fontan operation) (13). The second stage is a transition from a high pressure arterial supply to the pulmonary blood flow to a venous one, through creating an anastomosis between the superior vena cava and the pulmonary artery. The original parallel conduit of pulmonary and systemic circulations is rearranged into series. During the Glenn operation the superior vena cava is separated from the right atrium and connected to the pulmonary artery, the superior vena cava ostium and the BT shunt is closed. Thus the pulmonary circulation is perfused with the fully desaturated, rather than mixed, blood from the systemic venous flow, while the pulmonary venous flow is conveyed to the systemic circulation alone. The pulmonary perfusion is achieved passively owing to the pressure difference between the superior vena cava and pulmonary veins. In the hemi-Fontan technique, the superior vena cava is not fully detached from the right atrium, but its inlet is closed by a patch (1, 2, 13). The second stage operation is performed at 3 (4) to 6 months of age (1, 2, 12, 13).
Fig. 4.
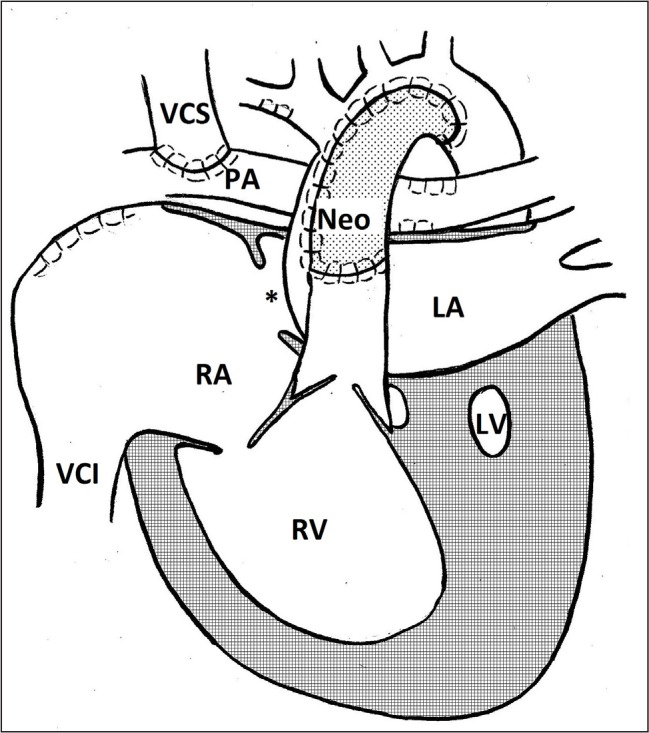
Bidirectional Glenn procedure. Neo – neoarta; LA – left atrium; LV – left ventricle; PA – pulmonary artery; RA – right atrium; RV – right ventricle; VCI – vena cava inferior; VCS – vena cava superior; * – foramen ovale
Fontan procedure
In the third stage a total cavo-pulmonary anastomosis or full Fontan circulation is formed. The Fontan operation, in which the systemic arterial and venous blood flow is completely separated, is the final stage, and the full Fontan circulation is the end goal of the palliative surgical therapy. During the Fontan operation the inferior vena cava is connected to the pulmonary artery bypassing the heart, thereby the entire systemic venous flow enters the pulmonary circulation rather than the right atrium. Thus the systemic circulation is perfused exclusively with fully saturated blood, enhancing oxygen delivery. The operation is performed using either one of the following techniques: 1) by creating a lateral tunnel using a patch, that connects the inlets of the inferior and superior vena cava or 2) a modification of the original Fontan technique using an extra-cardiac conduit – vascular prosthesis – to connect the inferior vena cava to the pulmonary artery (2, 13, 30). At the moment there is no unanimous attitude towards the most appropriate age for completion of the full Fontan circulation. Generally, the Fontan operation is performed at 18 to 36 months, however, in some programs the final stage is delayed until the manifestation of failing Glenn circulation – desaturation and cyanosis during physical activity – thus completing the staged repair at 3 to 5 years of age (1, 2, 13).
Fig. 5.
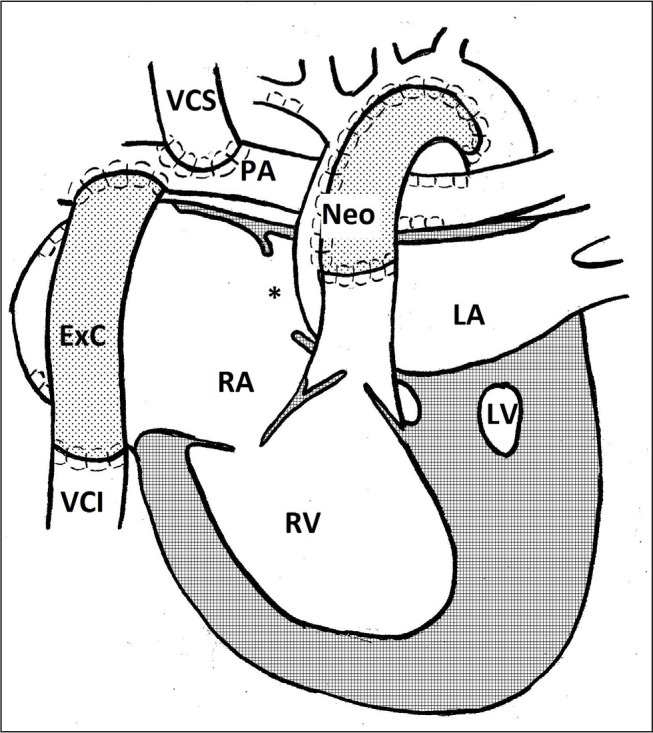
Fontan procedure. ExC – extra-cardiac conduit; Neo – neoarta; LA – left atrium; LV – left ventricle; PA – pulmonary artery; RA – right atrium; RV – right ventricle; VCI – vena cava inferior; VCS – vena cava superior; * – foramen ovale
Outcome of staged palliation
Staged palliation is the only means for survival until OHT. The six-year survival after the Norwood procedure is 62%. Both early and 6-year survival was higher if the right ventricle to the pulmonary artery shunt was used compared to the modified BT shunt (31). In the leading cardiac surgery centres the 30-day survival is 85–90% (28, 32). The mortality rate from the 30th day of life until stage II palliation is 12–22% (28). The mental and psychomotor development has been proven to be lower in patients after the Norwood procedure (33). Fifty to 70% of patients who have gone through all three stages of palliation live to the age of 5 years (2).
Over the past decade, an increasing attention has been paid to the long-term outcome of staged palliation of HLHS. It is a surgical therapy, with an intent to create the full Fontan circulation, that is based on a single functioning right ventricle and a common circulatory loop with pulmonary and systemic circulations arranged in series (14, 12, 34). The described haemodynamics is associated with a gradual change in the pulmonary vessel structure and function, and therefore increasing pulmonary vascular resistance, with a leading role in the late Fontan circulatory failure, thus making Fontan physiology a temporary solution, on the average lasting 4.9 years, that inevitably requires an OHT (35). Fontan circulation is associated with a considerably reduced exercise capacity and such potentially lethal complications as protein-losing enteropathy, plastic bronchitis, arrhythmias and progressive heart failure. Furthermore, the said physiology is thought to have the most rapid decline in efficiency during the first 6 months (36). In surveys adults with Fontan circulation report inferior exercise capacity, mental health, ability to integrate into society and health overall (37).
The exact cause of the protein-losing enteropathy in patients with Fontan circulation is not clear, however, it is thought to be associated with insufficient intestinal mucosal blood supply, chronic inflammation and impaired lymphatic drainage (2, 36, 38). The reported prevalence of the protein-losing enteropathy is 3 to 24% in the Fontan circulation patients (30, 36, 38, 39). The characteristic clinical presentation consists of a hypoproteinaemia syndrome – peripheral oedema, ascites and transudate in the pericardial and pleural cavities (2, 36). The protein-losing enteropathy following the Fontan operation is associated with increased morbidity, and the 5-year mortality approaches 50% (30).
Plastic bronchitis is a rare complication observed in children with Fontan circulation. Characteristic features include excessive sputum and viscous secretion collection in the bronchi with consequent bronchial obstruction and ventilation impairment. The pathogenesis is not clear, however, it is believed to involve increased systemic and pulmonary venous pressure and hindered lymphatic drainage, and therefore an increased amount of protein entering small airways (2, 36).
One of the leading non-perioperative causes of death in Fontan circulation patients is heart rhythm disturbances with an incidence of 7 to 50%, furthermore, 20 years following completion of the Fontan circulation more than half of the patients suffer supraventricular tachyarrhythmias (39). Various arrhythmias have been reported, including supraventricular tachycardia, atrial fibrillation and sinus node dysfunction. Lateral tunnel is associated with an increased risk of cardiac arrhythmias (30 to 50%) compared to extra-cardiac conduit (8 to 10%) (2).
The mental development of patients has been evaluated to be normal with a mean intelligence quotient (IQ) of 94 points (40). In a survey of parents of children with HLHS they report their children’s academic achievements to be the average (42%), and an equal amount reported their child’s achievements to be above the average. The most common complaint was difficulty in concentrating (37). Limitation of the mental function post transplantation is equal to that of post staged palliation with IQ < 70 in 18% of cases (2). A correlation has been found between the duration of stay in the transplant waiting list and a lower IQ – a decrease of 4.5 points per month. The social skills of children after staged palliation are not impaired (2). In a survey of adults with HLHS limitations in physical capacity and the overall health were reported, but the level of everyday energy was noted to be similar to the general population. There was no indication of a higher chronic pain level. Sixty-five percent of the responders were employed (37).
There are reports of pregnancies in patients with Fontan circulation. In a study of 14 pregnant women two had a cardiovascular complication (supraventricular tachycardia and heart failure) although in both cases the outcome of labour and birth was successful (2).
Orthotopic heart transplantation
OHT is a corrective, rather than palliative, surgical technique, based on restoring normal cardiac anatomy and near normal cardiovascular physiology (34). In the long term, transplantation is the better choice to assure the best quality of life, comparable to general population, and the highest survival. The average 5-year survival in children treated with OHT as a first line therapy is 65%, which is similar to that for staged repair, 50 to 70% (2, 34). However, it should be taken in account that OHT performed in newborns is associated with much better results when compared to other age groups – 16-year survival in these patients is 74% – which is probably due to development of selective immune tolerance to the transplant. Ten years following the OHT there is no exercise limitation in 93% (41). Nevertheless, a significant obstacle to OHT and the reason for necessity of staged repair is the lack of heart donors. The transplant must fit the size of the recipient’s thoracic cavity, thus only a newborn heart can be used, which substantially limits the number of donors. This makes OHT during the first 30 days of life rarely attainable, despite the excellent results. Transplantation performed after the first 30 days of life has a considerably worse outcome – 10-year survival not reaching 50% (34). Infants and children on the heart transplant waiting list have the highest mortality rates – 17 to 25% – compared with other transplant waiting lists owing to the long waiting time (2, 42). Furthermore, the said mortality has increased over the past two decades (43). One opinion towards OHT is that it should be reserved for patients with no other options of therapy than transplantation, for example, congenital cardiomyopathies (44).
Comfort care
Considering the feeble prognosis in HLHS and the costs of surgical therapy, a non-surgical palliative therapy is often opted for. It is a passive approach – the child is being anesthetized and sedated, but the prostaglandin therapy is withheld – with a lethal outcome usually during the first week of life. A satisfactory outcome of staged repair is for the most part reliant on the compliance of parents, and high perioperative and inter-stage medical care quality, but when lacking in this regard, the expected survival and quality of life may not be in accordance with the encouraging results from the leading cardio surgery centres reported in literature. In addition, 15 percent of HLHS cases are associated with a genetic syndrome or a serious extra cardiac malformation (16).
Medical care of HLHS in Latvia
In the time period of 2010–2014, twenty-six cases of HLHS were diagnosed antenatally. Of these, 17 pregnancies were terminated. In the time period of 2006–2014, twenty-four patients with HLHS were treated in Children’s Clinical University Hospital, Riga. At the moment 5 children are being observed: one after the hybrid procedure and four post stage II palliation. Two hybrid procedures were performed in Latvia with stage II performed in the United Kingdom. Two patients were transferred to Germany after birth where the Norwood and Glenn procedures were performed. Eight children did not reach the staged palliative repair, eleven died during the hybrid or Norwood procedure or in the early postoperative period.
CASE REPORT
A female at 40-week gestation was born to a primigravida 18-year-old mother in spontaneous labour. Despite having no pregnancy risk factors for a cardiac anomaly, a congenital heart disease was suspected on a routine week 22 sonogram and the patient was referred to a paediatric cardiologist where an echocardiogram was performed. No flow through the mitral and the aortic valve was visualised. The left ventricle and the ascending aorta was hypoplastic. Retrograde flow in the ascending aorta via ductus arteriosus and through foramen ovale was visualised. The diagnosis of HLHS was confirmed.
The baby was born in a delivery centre specialising in abnormal pregnancies. The birthweight was 3,060 g, the Apgar score was 8 and 9 in the first and fifth minute, respectively. After initiating prostaglandin E1 infusion 0.05 mcg/kg/min, the newborn was transferred to the NICU in Children’s Clinical University Hospital in Riga. An echocardiogram was performed in which a hypoplastic left ventricle with no flow through the mitral and the aortic valve was visualised. The annulus fibrosus was 2–3 mm, the pulmonary artery diameter was 10–12 mm, there was a patent ductus arteriosus 5–6 mm wide, the foramen ovale was 10 mm in diameter with a non-restrictive left to right shunt. Clinically the newborn corresponded to NYHA class II–III cardiovascular failure. After physical and an extensive instrumental evaluation no other congenital malformations were found.
Fig. 6.

Echocardiography image. Apical four-chamber view. The left atrium (LA) is much smaller than the right atrium (RA). Rudimentary left ventricle (LV), aortic and mitral valve atresia. The right ventricle (RV) occupies the cardiac apex
A hybrid procedure was scheduled as an initiation for staged repair. Preoperatively the patient was managed with no sedation, neither inotropic nor respiratory support, receiving continuous prostaglandin E1 infusion, as well as furosemide, spironolactone and carvedilol for heart failure treatment, and omeprazole for stress ulcer prophylaxis. A hybrid procedure was performed in accordance with the technique described previously.
After the procedure the child received inotropic support with dopamine, thrombosis prophylaxis with heparin, followed by aspirin and clopidogrel, and sedation with fentanyl. On the echocardiography imaging there was a non-restrictive left to right shunt through the foramen ovale. The flow in the PDA was intensive and retrograde to the aorta with arterial blood saturation on pulse oxymetry (SpO2) 76 to 85%. The right ventricle ejection fraction was 58%. The overall condition was stable and the patient was extubated on the fifth postoperative day. On the seventh postoperative day the patient’s condition deteriorated rapidly with respiratory distress and cardiovascular compromise (recapilarization time 4 sec, mean blood pressure 37 mmHg, metabolic acidosis with anion gap 40.8 mmol/l). An echocardiography imaging was performed; the ejection fraction was 45% while the SpO2 had increased to 93% (FiO2 = 0.21). A preductal aortic coarctation was suspected which was confirmed on the CT angiography. Concurrently stenting of the coarctation was performed.
Fig. 7.

Echocardiography image. Suprasternal long-axis view. Preductal coarctation of the aorta (Co Ao). Retrograde flow from stented ductus to transverse arch (Ao arch)
There was an immediate improvement (ejection fraction 63%). The patient was extubated on the second postoperative day and required no supplemental oxygen therapy from the fourth postoperative day. As the condition remained relatively stable during the first postoperative week, the patient was transferred to the Neonatal Care Unit. Despite continuous adjustment of therapy for heart failure, the overall condition slowly deteriorated with decreasing the ejection fraction and progressive systemic and pulmonary congestion. On the 54th day of life the overall condition worsened rapidly and the patient was transferred back to the NICU where inotropic and ventilatory support was initiated. The therapy was ineffective and shortly thereafter cardiac arrest occurred. Cardiopulmonary resuscitation was unsuccessful and the patient died.
CONCLUSIONS
The advancements in surgical management and improvements in intensive care of patients with HLHS allow for a better outcome for this pathology compared to the recent past. Still, the superior mode of treatment is not yet clear and the management must be based on each individual case, the experience of each clinic, as well as the financial aspects and will of the patient’s parents.
Roberts Gobergs, Elza Salputra, Ingūna Lubaua
References
- Park MK. Cyanotic congenital heart defects. In: Park MK. ed. Park’s Pediatric Cardiology for Practitioners. 6th ed Philadelphia, PA: Elsevier, Saunders; 2014. p. 206–89. [Google Scholar]
- Feinstein JA, Benson W, Dubin AM, Cohen MS, Maxey DM, Mahle WT, et al. Hypoplastic left heart syndrome: current considerations and expectations. J Am Coll Cardiol. 2012; 59 Suppl 1: S1–42. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AC. Cardiac catheterization and fetal intervention. In: Sellke FW del Nido PJ Swanson SJ editors. Sabiston and Spencer Surgery of the Chest. 9th ed. Philadelphia, PA: Elsevier, Saunders; 2016. p.1915–36. [Google Scholar]
- Allan LD, Sharland G, Tynan MJ. The natural history of the hypoplastic left heart syndrome. Int J Cardiol. 1989; 25(25): 341–3. [DOI] [PubMed] [Google Scholar]
- Danford DA, Cronican P. Hypoplastic left heart syndrome: progression of left ventricular dilation and dysfunction to left ventricular hypoplasia in utero. Am Heart J. 1992; 123(123): 1712–3. [DOI] [PubMed] [Google Scholar]
- Frommelt MA. Challenges and controversies in fetal diagnosis and treatment: hypoplastic left heart syndrome. Clin Perinatol. 2014; 41(41): 787–98. . [DOI] [PubMed] [Google Scholar]
- Webb GD, Smallhorn JF, Therrien J. Redington AN. In: Mann DL, Zipes DP, Libby P, Bonow RO, Braunwald E. editors. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 10th ed Philadelphia, PA: Elsevier, Saunders; 2015. p. 1391–445. [Google Scholar]
- Mc Elhinney DB, Vogel M, Benson CB, Marshall AC, Wilkins-Haug LE, Silva V, Tworetzky W. Assessment of left ventricular endocardial fibroelastosis in fetuses with aortic stenosis and evolving hypoplastic left heart syndrome. Am J Cardiol. 2010; 106(106): 1792–7. . [DOI] [PubMed] [Google Scholar]
- Friedman KG Schidlow D Freud L Escobar-Diaz M Tworetzky W.. Left ventricular diastolic function and characteristics in fetal aortic stenosis. Am J Cardiol. 2014; 114(114): 122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkikallio K, Mc Elhinney DB, Levine JC, Marx GR, Colan SD, Marshall AC, et al. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: patient selection for fetal intervention. Circulation. 2006; 113(113): 1401–5. [DOI] [PubMed] [Google Scholar]
- Fraser CD, Carberry KE. Congenital Heart Disease. In: Townsend CM, Beauchamp RD, Evers BM, Mattox KL. editors. Sabiston Textbook of Surgery. 19th ed Philadelphia, PA: Elsevier, Saunders; 2012. p. 1611–49. [Google Scholar]
- Graham EM, Shirali GS.Hypoplastic Left Heart Syndrome. In: Crawford MH DiMarco JP Paulus WJ editors. Cardiology. 3rd ed. Philadelphia, PA: Elsevier Ltd.; 2010. p. 1519–28. [Google Scholar]
- Mettler BA, Pigula FA. Hypoplastic Left Heart Syndrome. In: Sellke FW del Nido PJ Swanson SJ editors. Sabiston and Spencer Surgery of the Chest. 9th ed Philadelphia, PA: Elsevier, Saunders; 2016. p. 2295–312. [Google Scholar]
- Steven JM, Marino BS, Jobes DR. Hypoplastic Left Heart Syndrome. In: Nichols DG, Cameron DE. editors. Critical Heart Disease in Infants and Children. 2nd ed Philadelphia, PA: Elsevier, Mosby; 2006. p. 823–44. [Google Scholar]
- Perloff JK, Marelli JA. Hypoplastic Left Heart. In: Perloff JK, Marelli JA. editors. Clinical Recognition of Congenital Heart Disease. 6th ed Philadelphia, PA: Elsevier, Saunders; 201. 2. p. 522–9. [Google Scholar]
- Wernovsky G, Dominguez TE, Gruber PJ, Anderson RH. Hypoplasia of the Left Heart. In: Price G. editor. Paediatric Cardiology. 3rd ed Philadelphia, PA: Elsevier Ltd., Churchill Livingstone; 2010. p. 625–45. [Google Scholar]
- Freud LR McElhinney DB Marshall AC Marx GR Friedman KG del Nido PJ et al. . Fetal aortic valvuloplasty for evolving hypoplastic left heart syndrome: postnatal outcomes of the first 100 patients. Circulation. 2014; 130(130): 638–45. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tworetzky W Wilkins-Haug L Jennings RW van der Velde ME Marshall AC Marx GR et al. . Balloon dilation of severe aortic stenosis in the fetus: potential for prevention of hypoplastic left heart syndrome: candidate selection, technique, and results of successful intervention. Circulation. 2004; 110(110): 2125–31. [DOI] [PubMed] [Google Scholar]
- Moon-Grady AJ, Morris SA, Belfort M, Chmait R, Dangel J, Devlieger R, et al. International Fetal Cardiac Intervention Registry: A Worldwide Collaborative Description and Preliminary Outcomes. J Am Coll Cardiol. 2015; 66(66): 388–99. [DOI] [PubMed] [Google Scholar]
- Mc Elhinney DB, Marshall AC, Wilkins-Haug LE, Brown DW, Benson CB, Silva V, et al. Predictors of technical success and postnatal biventricular outcome after in utero aortic valvuloplasty for aortic stenosis with evolving hypoplastic left heart syndrome. Circulation. 2009; 120(120): 1482–90. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzt W, Wertaschnigg D, Veit I, Klement F, Gitter R, Tulzer G. Intrauterine aortic valvuloplasty in fetuses with critical aortic stenosis: experience and results of 24 procedures. Ultrasound Obstet Gynecol. 2011; 37(37): 689–95. . [DOI] [PubMed] [Google Scholar]
- Friedman KG Freud L Escobar-Diaz M Banka P Emani S Tworetzky W.. Left ventricular remodeling and function in children with biventricular circulation after fetal aortic valvuloplasty. Pediatr Cardiol. 2015; 36(36): 1502–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SM. Fetal cardiac interventions: an update of therapeutic options. Rev Bras Cir Cardiovasc. 2014; 29(29): 388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AC, Levine J, Morash D, Silva V, Lock JE, Benson CB, et al. Results of in utero atrial septoplasty in fetuses with hypoplastic left heart syndrome. Prenat Diagn. 2008; 28(28): 1023–8. . [DOI] [PubMed] [Google Scholar]
- Chaturvedi RR Ryan G Seed M van Arsdell G Jaeggi ET. Fetal stenting of the atrial septum: technique and initial results in cardiac lesions with left atrial hypertension. Int J Cardiol. 2013; 168(168): 2029–36. . [DOI] [PubMed] [Google Scholar]
- Kalish BT Tworetzky W Benson CB Wilkins-Haug L Mizrahi-Arnaud A McElhinney DB et al. . Technical challenges of atrial septal stent placement in fetuses with hypoplastic left heart syndrome and intact atrial septum. Catheter Cardiovasc Interv. 2014; 84(84): 77–85. . [DOI] [PubMed] [Google Scholar]
- Kouchoukos NT, Blackstone EH, Hanley FL, Kirklin JK. Aortic atresia and other forms of hypoplastic left heart physiology. In: Kouchoukos NT, Blackstone EH, Hanley FL, Kirklin JK. editors. Kirklin/Barratt-Boyes Cardiac Surgery. 4th ed Philadelphia, PA: Elsevier, Saunders; 2013. p. 1780–812. [Google Scholar]
- Ohye RG, Sleeper LA, Mahony L, Newburger JW, Pearson GD, Lu M, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010; 362(362): 1980–92. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bardino DJ, Gomez-Arostegui J, Kemp A, Raviendran R, Hegde S, Devaney EJ, et al. Intermediate results of hybrid versus primary Norwood operation. Ann Thorac Surg. 2015; 99(99): 2141–9. . [DOI] [PubMed] [Google Scholar]
- Kanter KR. Management of single ventricle and cavopulmonary connections. In: Sellke FW, del Nido PJ, Swanson SJ. editors. Sabiston and Spencer Surgery of the Chest. 9th ed Philadelphia, PA: Elsevier, Saunders; 2016. p. 2313–29. [Google Scholar]
- Wilder TJ McCrindle BW Phillips AB Blackstone EH Rajeswaran J Williams WG et al. . Survival and right ventricular performance for matched children after stage-1 Norwood: Modified Blalock-Taussig shunt versus right-ventricle-topulmonary- artery conduit. J Thorac Cardiovasc Surg. 2015; 150(150): 1440–1452.e8. . [DOI] [PubMed] [Google Scholar]
- Barron DJ, Kilby MD, Davies B, Wright JG, Jones TJ, Brawn WJ. Hypoplastic left heart syndrome. Lancet. 2009; 374(374): 551–64. . [DOI] [PubMed] [Google Scholar]
- Newburger JW, Sleeper LA, Bellinger DC, Goldberg CS, Tabbutt S, Lu M, et al. Early developmental outcome in children with hypoplastic left heart syndrome and related anomalies: the single ventricle reconstruction trial. Circulation. 2012; 125(125): 2081–91. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey LL. Transplantation is the best treatment for hypoplastic left heart syndrome. Cardiol Young. 2004; 14 Suppl 1: 109–14. [DOI] [PubMed] [Google Scholar]
- Bernstein D, Naftel D, Chin C, Addonizio LJ, Gamberg P, Blume ED, et al. Outcome of listing for cardiac transplantation for failed Fontan: a multi-institutional study. Circulation. 2006; 114(114): 273–80. [DOI] [PubMed] [Google Scholar]
- Goldberg DJ, Dodds K, Rychik J. Rare problems associated with the Fontan circulation. Cardiol Young. 2010; 20 Suppl 3: 113–9. [DOI] [PubMed] [Google Scholar]
- van den Bosch AE, Roos-Hesselink JW, Van Domburg R, Bogers AJ, Simoons ML, Meijboom FJ. Long-term outcome and quality of life in adult patients after the Fontan operation. Am J Cardiol. 2004; 93(93): 1141–5. [DOI] [PubMed] [Google Scholar]
- Silvilairat S, Cabalka AK, Cetta F, Grogan M, Hagler DJ, O’Leary PW. Protein-losing enteropathy after the Fontan operation: associations and predictors of clinical outcome. Congenit Heart Dis. 2008; 3(3): 262–8. [DOI] [PubMed] [Google Scholar]
- Khairy P, Mercier LA. Late complications following the Fontan operation. In: Gatzoulis MA, Webb GD, Daubeney PEF. editors. Diagnosis and Management of Adult Congenital Heart Disease. 2nd ed Philadelphia, PA: Elsevier, Saunders; 2011. p. 104–9. [Google Scholar]
- Pike NA Evangelista LS Doering LV Koniak-Griffin D Lewis AB Child JS. Health-related quality of life: a closer look at related research in patients who have undergone the Fontan operation over the last decade. Heart Lung. 2007; 36(36): 3–15. [DOI] [PubMed] [Google Scholar]
- Aurora P, Edwards LB, Kucheryavaya AY, Christie JD, Dobbels F, Kirk R, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirteenth official pediatric lung and heart-lung transplantation report – 2010. J Heart Lung Transplant. 2010; 29(29): 1129–41. . [DOI] [PubMed] [Google Scholar]
- Morales DL, Dreyer WJ, Denfield SW. Over two decades of pediatric heart transplantation: how has survival changed? J Thorac Cardiovasc Surg. 2007; 133(133): 632–9. [DOI] [PubMed] [Google Scholar]
- Chrisant MR, Naftel DC, Drummond-Webb J. Fate of infants with hypoplastic left heart syndrome listed for cardiac transplantation: a multicenter study. J Heart Lung Transplant. 2005; 24(24): 576–82. [DOI] [PubMed] [Google Scholar]
- Kon AA. Ethics of cardiac transplantation in hypoplastic left heart syndrome. Pediatr Cardiol. 2009; 30(30): 725–8. . [DOI] [PMC free article] [PubMed] [Google Scholar]