

Abstract
Lentiviral vectors (LVs) pseudotyped with vesicular stomatitis virus envelope glycoprotein (VSV-G) have demonstrated great promise in gene therapy trials employing hematopoietic stem cell and T-cells. The VSV-G envelope confers broad tropism and stability to the vector but is toxic when constitutively expressed, which has impeded efforts to generate stable producer cell lines. We previously showed that cocal pseudotyped LVs offer an excellent alternative to VSV-G vectors because of their broad tropism and resistance to human serum inactivation. In this study, we demonstrate that cocal LVs transduce CD34+ and CD4+ T-cells more efficiently than VSV-G LVs and share the same receptor(s) for cell entry. 293T-cells stably expressing the cocal envelope produced significantly higher LV titers than VSV-G expressing cells. We developed cocal pseudotyped, third-generation, self-inactivating LV producer cell lines for a GFP reporter and for a WT1 tumor-specific T-cell receptor, which achieved concentrated titers above 108 IU/ml and were successfully adapted for growth in suspension, serum-free culture. The resulting LVs were at least as effective as standard LVs in transducing CD34+ and CD4+ T-cells. Our stable cocal LV producer cell lines should facilitate the production of large-scale, high titer clinical grade vectors.
Introduction
Lentiviral vectors (LVs) are currently considered the gold standard for hematopoietic stem cell (HSC) gene therapy and for immunotherapies with genetically modified T-cells (as reviewed in ref. 1).
These vectors were first developed in the early 1990s and are typically made by transient transfection of helper and vector plasmids into cells that support the assembly of LV virions. LVs have commonly been pseudotyped with the heterologous vesicular stomatitis virus envelope glycoprotein (VSV-G), which confers broad tropism and stability to the vector. However, VSV-G is inactivated by human serum complement, making it unsuitable for in vivo delivery when vector amount is limiting. Most notably, VSV-G is cytotoxic when stably expressed in human cells, which has impeded efforts to develop LV producer cell lines.2
Lentivirus producer cell lines that stably express all the different components required for the assembly of LV have several advantages over traditional production methods that use transient transfection of plasmids: (i) reproducibility and consistency in vector titer and quality; (ii) safety: the absence of DNA in the preparation avoids the risk of recombination between transfected plasmids and the production of replication-competent lentiviruses; (iii) cost: clinical grade plasmid DNA is expensive and considerably adds to the cost of the vector; and (iv) scale-up: producer cells can be adapted to grow in suspension cultures suitable for bioreactors.
Our laboratory has previously developed and used a cocal vesiculovirus envelope glycoprotein to pseudotype LV.3 The cocal envelope glycoprotein shares 71.5% identity at the amino acid level with the VSV-G Indiana envelope, and cocal pseudotyped LVs (cocal LVs) were found to have broad tropism and to be more resistant to inactivation by human serum than VSV-G pseudotyped LVs (VSV-G LVs). In addition, cocal LVs could be produced at high titers and efficiently transduced human, nonhuman primate (NHP), and canine hematopoietic stem cells.3 In this study, we describe the development of high titer third-generation self-inactivating (SIN) LV producer cell lines based on the cocal envelope.
Results
Stable expression of the cocal envelope in HEK 293T-cells results in the production of over 10-fold more LV as compared to VSV-G expression
Stable LV producer cells have several advantages over transient vector production. However, constitutive expression of large amount of viral proteins required for assembly of LV particles has proven difficult. Expression of the human immunodeficiency virus (HIV) Gag-Pol and of the VSV-G envelope has previously been associated with high levels of cytotoxicity,2,4 and has largely contributed to the challenges associated with the development of high titer, stable LV producer cell lines. To determine if the cocal envelope is a better choice than VSV-G for constructing a producer cell line, we stably expressed either the cocal or the VSV-G envelope in human embryonic kidney (HEK) 293T-cells and measured the resulting LV titers from each cell line. Plasmid pMD2.G was modified so that both the hygromycin resistance gene and the envelope encoding gene (cocal or VSV-G) were expressed from the same promoter using a 2A self-cleaving polypeptide (Figure 1a, pMD2.hyg.2A.cocal and pMD2.hyg.2A.VSV-G) to ensure that hygromycin resistant cells will concomitantly express the envelope proteins. Following transfection of envelope plasmids and hygromycin selection to allow for stable expression, LV production was induced in these cells by transfection of all other required third-generation helper plasmids and of the SIN configured LV plasmid pRRLSIN.cPPT.PGK-GFP.WPRE (Figure 1a). This plasmid encodes the same SIN LV backbone used in multiple clinical trials of gene transfer (National clinical trials (NCT) identifiers NCT02343666 and NCT01331018) with a human phosphoglycerate kinase (PGK) promoter driving expression of an enhanced green fluorescent protein (eGFP) for ease of monitoring transduction. For simplicity, this plasmid will be referred to as PGK-eGFP. LV titer was determined by transducing the HT1080 fibrosarcoma cell line and by measuring expression of the transgene in transduced cells using flow cytometry (see Materials and Methods). Although we found no significant difference in titer when cocal LV and VSV-G LV were produced in 293T-cells by the standard protocol using transient transfection (Figure 1b, left), stable expression of the cocal envelope in 293T-cells produced ~10-fold more infectious particles as compared to VSV-G expression (Figure 1b, right). No LV production was detected in 293T-cells that did not express envelope proteins (data not shown). Notably, the cocal and VSV-G cell lines were developed in parallel using a similar protocol, and LV production was measured from bulk cultures, not from selected producer cell clones, explaining why the titers obtained with the cell lines are lower than titers obtained with transient transfections. Using SYBR Green-based quantitative PCR (qPCR), we found that cocal cells expressed twofold to fourfold more envelope mRNA transcripts as compared to VSV-G cells (Figure 1c), which may explain the difference in titers. The cocal gene was previously codon-optimized,3 whereas the VSV-G gene is made of the native sequence, so it is possible that the higher performance of the cocal envelope is due to “codon-optimization”. To rule out this possibility, we have substituted the original VSV-G gene in pMD2.G plasmid with a codon-optimized VSV-G (VSV-G CO) for expression in human cells (see Materials and Methods). Stable expression of plasmids pMD2.Cocal-G, pMD2.G, and pMD2.G CO was achieved in 293T-cells by cotransfection with a hygromycin-resistant plasmid and selection. After inducing LV production in these cells, we found that VSV-G CO expression did not improve vector titer as compared to VSV-G; on the contrary, production was reduced by 38% in VSVG CO cells as compared to VSV-G cells (Supplementary Figure S1). Overall, stable expression of the cocal envelope results in higher LV titer as compared to VSV-G, and may be the preferred envelope for the generation of LV producer cell lines.
Figure 1.

Stable expression of cocal and VSV-G envelopes in 293T-cells. (a) Schematic representation of cocal and vesicular stomatitis virus envelope glycoprotein (VSV-G) envelope plasmids and lentiviral transfer plasmids used for the generation of Lentiviral vector (LV) producer cells. (b) Unconcentrated LV titer from standard vector production using transient transfection (left), or from cells stably expressing each envelope (right). Mean titer is given as infectious units per ml (IU/ml) from cells transduced in triplicates. (c) SYBR Green-based quantitative PCR measurements of envelope transcripts in two independent cocal or VSV-G cell lines. Envelope mRNA expression was calculated relative to expression of the endogenous gene glyceraldehyde 3-phosphate dehydrogenase from reactions run in triplicates. Cell line A was used to generate the titers given in b. Error bar shows standard error (SE) of the mean. CMV, human cytomegalovirus promoter; hBGint, human beta globin intronic sequence; 2A,self-cleaving peptide sequence; hBpA, human beta globin polyA sequence; ΔLTR, long terminal repeat containing short deletion; RSV, Rous sarcoma virus promoter; PGK, phosphoglycerate kinase 1 promoter (human); eGFP, enhanced green fluorescent protein; Wpre, woodchuck hepatitis virus regulatory element; MSCV, murine stem cell virus promoter; TCRα, T-cell receptor alpha chain; TCRβ, T-cell receptor beta chain.
Cocal-pseudotyped LVs outperform VSV-G vectors in the transduction of human and NHP CD34+ and CD4+ T-cells
Our laboratory previously demonstrated that cocal LVs have a broad tropism and efficiently transduce HSCs from different species at low multiplicities of infection (MOI).3 Here, we show that cocal LVs transduce both NHP and human CD34+ cells at higher efficiencies than VSV-G LVs when matched by multiplicity of infection (Figure 2a,b). Cocal LV-transduced human CD34+ exhibited similar differentiation potential compared to VSV-G LV-transduced CD34+ cells as determined by the colony-forming assay (Figure 2c). Extending these studies, cocal LVs transduced both NHP and human CD4+ T-cells more efficiently than VSV-G LV at low MOI (Figure 2d,e). Overall, these results validate the use of cocal LVs for efficient gene transfer in HSCs and T-cells and suggest higher potency than VSV-G LVs.
Figure 2.

Transduction of primary cells by VSV-G and cocal LV.(a) Transduction efficiency of nonhuman primate (NHP) CD34+ cells (n = 3) exposed to lentiviral vectors (LVs) (PGK.eGFP, generated with standard protocol) using two doses of vector of 6 hours each at multiplicities of infection (MOI) of five. Mean percentage of eGFP+ cells was determined at 10 days post-transduction. PGK, phosphoglycerate kinase (1 promoter human); eGFP, enhanced green fluorescent protein. (b) Mean transduction efficiency of human CD34+ cells (n = 2) exposed to LVs at an MOI of 5. The percentage of eGFP+ cells was determined 6 days post transduction. (c) Human CD34+ cells from one of the donor described in b were plated for colony-forming cells (CFCs). The fraction of progenitor cells for different lineages was enumerated and compared between mock, VSV-G or cocal LV transduction. E, erythroid; M, monocyte; GM, granulocyte/macrophage; GEMM, granulocyte/erythrocyte/macrophage/megakaryocyte. (d) Transduction efficiency in NHP peripheral blood from two donors enriched for CD4+, activated for 3 days and exposed to LVs at MOI of 5. The percentage of eGFP+ cells was determined at 3 days post transduction from reactions run in duplicates. (e) Transduction efficiency in human CD4+ cells exposed to two different LV preparations (LV1 and LV2) at MOI of 1. The percentage of eGFP+ cells was determined at 3 days post transduction. In all experiments, error bars show SE of the mean.
Soluble LDL receptor inhibits both VSV-G and cocal LV transduction and suggests overlap in cellular receptors usage
Although the cocal envelope glycoprotein shares 71.5% identity at the amino acid level with the VSV-G envelope, it is not known whether the two envelopes use the same receptor(s) for cell entry. The low-density lipoprotein receptor (LDLR) and its family members were previously identified as cellular receptor for VSV-G.5 We used purified recombinant human soluble LDLR (sLDLR), which correspond to the ligand-binding domain of LDLR, to determine its effect on transduction efficiency by VSV-G LV, cocal LV or by foamy virus vector (FV). We found that sLDLR inhibited both VSV-G and cocal LV transduction of HT1080-cells in a dose-dependent manner, with a less pronounced effect seen for cocal LV (Figure 3a). In contrast, FV transduction was not affected by sLDLR, consistent with the notion of distinct cellular receptor used by the foamy envelope.6,7 Inhibition by sLDLR was also observed in NHP CD34+ cells, where transduction by both VSV-G and cocal LV was inhibited at a similar level, whereas no effect was detected for FV transduction (Figure 3b). These results demonstrate that sLDLR blocks transduction by both cocal and VSV-G LVs, suggesting that both envelopes use the same receptor(s) for cell entry.
Figure 3.

sLDLR inhibits VSV-G and cocal LV transduction. (a) Dose-dependent inhibition of vesicular stomatitis virus envelope glycoprotein (VSV-G) and cocal pseudotyped LV (cocal LV) transduction of HT1080 cells by human soluble LDLR (sLDLR). 100x concentrated foamy virus vector (FV), VSV-G LV and cocal-LV (all PGK.eGFP, generated with standard transfection protocol) were preincubated with different concentrations of sLDLR (in μg/ml) and used to transduce HT1080 cells. The percentage of eGFP+ cells was determined at 3 days post transduction from reactions run in triplicates, and results show the percentage of infectivity in comparison to untreated cells. (b) Transduction of nonhuman primate (NHP) CD34+ cells by FV, VSV-G LV and cocal LV in the presence (10 μg/ml) or absence of sLDLR. Results show mean % infectivity taken from two time points (2 and 6 days post transduction) relative to untreated cells. In all experiments, error bars show SE of the mean.
Development of cocal and VSV-G third-generation LV packaging cell lines
We derived the cocal or VSV-G-expressing cell lines (Figure 1b) into third-generation LV packaging cell lines by sequential transfection of plasmids encoding the gagpol and rev helper genes along with plasmids encoding antibiotic resistance genes as described in Figure 4a. We found that a molar ratio of 1 : 5 of selection plasmid to helper plasmid, respectively, was optimal for LV production (Supplementary Figure S2). Stable expression of GagPol and Rev proteins was achieved by selection with puromycin and blasticidin, respectively, for ~3 weeks. We subsequently induced LV production in the resulting cell lines, now expressing all helper genes (referred to as packaging cells), by transfection of the lentiviral plasmid PGK-eGFP. As shown in Figure 4b (red bar), bulk cocal packaging cells produced on average eightfold more LV as compared to bulk VSV-G packaging cells (1.2 × 105 IU/ml for cocal versus 1.5 × 104 IU/ml for VSV-G). To further increase vector titer, we isolated single cocal or VSV-G packaging cell clones using a limiting dilution approach. Single cells were seeded in a 96-well plate and expanded to six-well plates under selective conditions, and the resulting LV titers produced by each clone was quantified. Screening of over a dozen individual clones for each cell line identified the best cocal packaging clone that generated unconcentrated titers of 1.9 × 106 IU/ml and the best VSV-G packaging clone that generated 2.9 × 104 IU/ml (Figure 4b, open circles). Similar titers could be achieved from these best clones when scaling up culture conditions to 15-cm plates (Figure 4c). Upon concentration by centrifugation (100×), the best cocal packaging clone produced a LV titer that averaged 1.2 × 108 IU/ml, whereas the best VSV-G packaging clone produced 1.1 × 106 IU/ml. Treatment with sodium butyrate (NaBu) during the production phase was necessary as titers were significantly lower in the absence of treatment (Supplementary Figure S3a). The titer obtained with the highest performing cocal packaging cell line is equivalent to the titer we routinely obtain using our standard LV production protocol by transient transfection (Figure 1b, left). The greatest utility of this cell line would be the achievement of stable vector production of equivalent titer over an extended period of time in culture. To verify the stability of these packaging cells, we measured LV production after culture under selection for 2–3 months, and we observed consistent titers over time (Figure 4d). In summary, we have successfully developed a stable LV packaging cell line using the cocal envelope, which produces significantly higher titers than VSV-G packaging cells generated with an identical approach.
Figure 4.

Generation of VSV-G and cocal LV packaging cell lines. (a) Diagram highlighting the steps and plasmids used toward the generation of lentiviral vector (LV) packaging/producer cell lines. Stable expression was achieved by plasmid DNA transfection followed by selection with the respective drugs. (b) LV titer determined by flow cytometry for single cocal and VSV-G packaging clones (open circles) or from bulk cells (red bar). Unconcentrated titer is given as infectious units/ml (IU/ml). (c) Unconcentrated (1×) and concentrated (100×) LV titers obtained from the best producer cocal or VSV-G clones identified in b grown in 15-cm plates. Mean titers are from one representative experiment using four different dilutions of the vector for transduction. Error bars show SE of the mean. (d) Stability of cocal and VSV-G packaging cell lines as determined by unconcentrated titers produced following long-term culture. To induce LV production, the PGK.eGFP transfer plasmid was transiently transfected into cocal or VSV-G packaging cells.
Generation of a high-titer cocal producer cell line for PGK-eGFP LV
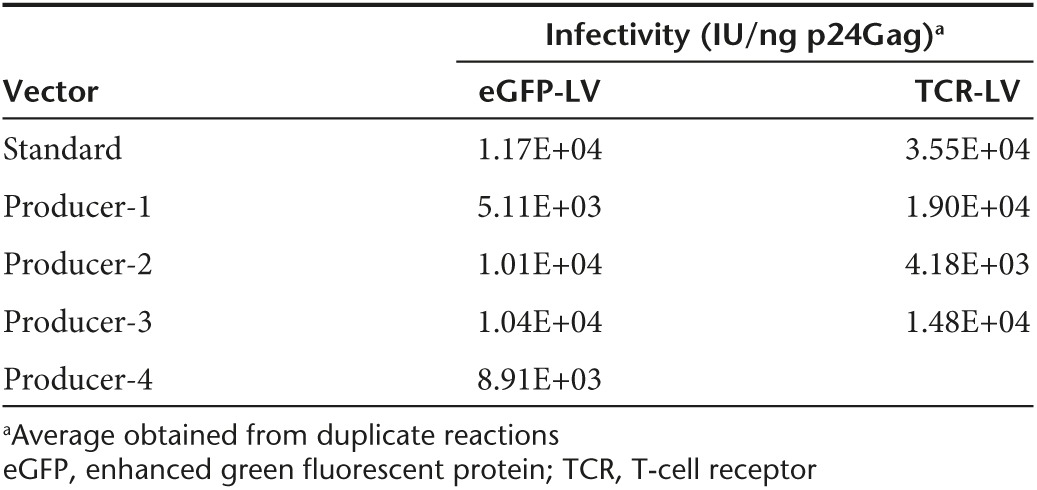
We next sought to stably express lentiviral plasmids in packaging cells to obtain LV producer cells. As cocal packaging cells produced a LV titer 100-fold higher than VSV-G packaging cells (Figure 4c), we focused on cocal cells to generate a LV producer cell line. As a proof of principle experiment, we used the PGK-eGFP SIN lentiviral construct (Figure 1a). Cocal packaging cells were transfected with both the lentiviral construct and a plasmid encoding zeocin resistance, and selection was carried out in zeocin-containing media for ~3 weeks to allow for stable expression of the plasmid. Flow cytometry analysis of the producer cells revealed that only ~50% of the cells were eGFP positive, with variable levels of expression (Figure 5a). In an effort to increase vector production, we first sorted cells based on the mean fluorescence intensity of eGFP expression as assessed by flow cytometry, and analyzed LV production in low-, intermediate-, and high-eGFP+ subpopulations (Figure 5b, upper). We found a direct correlation between the levels of eGFP expressed by the cells and the resulting LV titer. In the high eGFP-expressing cells, an unconcentrated titer of 1 × 105 IU/ml was achieved while low-expressing cells produced a titer of < 1 × 103 IU/ml (Figure 5b, lower). As an alternative approach to increase LV titer from producer cells, we isolated and screened single producer clones similarly to what we have described earlier for packaging cells. Analysis of two dozens clones identified six clones that produced LV titers greater than bulk cells (compare open circles with bar, Figure 5c, left). LV titer in the best cocal producer clone, which was named eGFP 2–12, was confirmed from culture in 15-cm plates and resulted in unconcentrated titer (1×) above 1.0 × 106 IU/ml (± 1.7 × 104 IU/ml) and concentrated titer (100×) reaching 5.3 × 107 IU/ml (± 6.4 × 106 IU/ml) (Figure 5d). LV titer generated from eGFP 2–12 cells was stable after serial passages under selective conditions for about 4 months (Figure 5e). We were able to further increase LV titer in eGFP 2–12 cocal cells following a second round of single clone screening (Figure 5c, right), which now generated concentrated titer above 108 IU/ml. To determine which helper component was most limiting in eGFP 2–12 cells for LV production, we transfected cells with each helper plasmid separately and measured the corresponding titer. Transfection with the cocal envelope plasmid gave the highest increase in titer (30%) followed by gagpol (18%) and rev (15%) (Supplementary Figure S4). We also quantified levels of HIV-1 p24Gag in LV produced by eGFP 2–12 cells or by the standard transfection protocol and correlated these values to the functional titer obtained from transduction of HT1080-cells. For most (three out of four) vectors, we found a good correlation between LV made by the standard transfection protocol with LV made by the producer cells (Figure 5f). Accordingly, the infectivity, as defined by IU/ng p24Gag, was comparable in most eGFP LV preparations (Table 1, left). We next assessed the performance of the cocal eGFP vector made with the producer cell line as compared to identical cocal eGFP vector made using the standard transfection protocol for the transduction of NHP CD34+ cells. Cells were transduced with three different MOI, and transduction efficiency was measured after 6 days based on the fraction of GFP+ cells. As shown in Figure 5g, we found that the vector generated with producer cells resulted in considerably higher transduction efficiencies at MOI of 10 as compared to vector generated by transient transfection but transduced CD34+ cells equally at MOI of two. Therefore, eGFP LV generated with our cocal producer cell line shows superior transduction efficiencies of NHP HSCs as compared to LV made with the standard protocol.
Figure 5.

Generation of a GFP cocal LV producer cell line. (a) Flow cytometry analysis of bulk cocal producer cells expressing varying levels of enhanced green fluorescent protein (eGFP) fluorescence, represented by flow chart (left) or histogram (right). (b) Sorting of cocal producer cells from a based on eGFP fluorescence intensity (histogram) and corresponding mean lentiviral vector (LV) titer measured in each subpopulation (bar chart). Mean titer is obtained from two different dilutions of the vector and error bars show SE of the mean. (c) LV titer measured in single eGFP producer clones (open circles) or bulk cells (bar) after the first or second round of screening. Unconcentrated titer is given as infectious units/ml (IU/ml). (d) Unconcentrated (1×) and concentrated (100×) LV titers from the best eGFP producer clone identified in c from the first screen. Bar graph shows mean titer from one representative experiment using four different dilutions of the vector for transduction and error bars show SE of the mean. (e) Stability of eGFP LV producer cell line based on unconcentrated titer after long-term culture. (f) Functional titers plotted against HIV-1 p24 concentration (ng/ml) for different vector preparation made with the standard protocol (black square) or with producer cells (X). (g) Transduction of nonhuman primate (NHP) CD34+ cells by LV generated with the standard protocol (black) or with producer cells (gray). Different multiplicities of infection (MOI) of vector were used and cells were analyzed by flow cytometry at 6 days post transduction. Mean transduction efficiencies are from three independent experiments and error bars show SE of the mean.
Table 1. Infectivity of lentiviral vector (LV).
Generation of a cocal SIN LV producer cell line encoding a WT1-specific T-cell receptor
Our findings that cocal LVs transduce CD4+ T-cells at higher efficiencies than VSV-G LVs (Figure 2d,e) prompted us to develop a cocal producer cell line that could be used in immunotherapy applications. We thus employed the LV construct, pRRLSIN.C4b17_P2A_aWPRE-1, which encodes the α and β chains of an HLA-A2-restricted T-cell receptor (TCR) specific for the cancer antigen WT1 (Figure 1a).8,9 We used a similar procedure to the one described earlier for the eGFP producer cell line, starting with cocal packaging cells, with the difference that LV titer was now measured either by qPCR or by antibody staining for surface receptor expression in T-cells. After selecting cells that stably expressed the TCR lentiviral plasmid, we measured titer in a dozen single producer cell clones. Two clones produced LV titers that were higher than the bulk cell population as measured by qPCR (compare open circles with bar, Figure 6a), and the best producer clone generated unconcentrated titer reaching 3.8 × 106 IU/ml, and was named C4 1–9. Repeated rounds of LV production carried out with the C4 1–9 producer clone showed an average unconcentrated titer of 3.0 × 106 IU/ml (± 6.1 × 105 IU/ml) and a concentrated titer of 1.0 × 108 IU/ml (± 4.5 × 107 IU/ml) (Figure 6b). In addition, LV titer generated by C4 1–9 cells was stable for over 100 days when cultured under selection (Figure 6c). We measured HIV-1 p24 levels in different LV preparations made by C4 1–9 and found that the infectivity of these vectors was slightly lower than the infectivity of vectors made with the standard protocol (Table 1, right). Next, we sought to directly compare the performance of these vectors by transduction of both the H9 lymphoma cell line and of primary human CD4+ T-cells with different amounts of each vector. Transduction efficiency was quantified by TCR surface staining with a TCRVβ17 antibody conjugated to phycoerythrin. For this experiment, we used LV preparations with comparable concentrated titers of 2 × 108 IU/ml prepared either by producer cells or by transient transfection. Following transduction of H9 cells or CD4+T-cells, we found that TCR expression was nearly identical between the cells transduced with cocal LVs generated by producer cells versus the cells transduced with LVs made with the standard protocol (Figure 6d,e). In CD4+ T-cells, we found that TCR expression levels induced by LV transduction were indistinguishable from endogenous TCR levels, as indicated by mean fluorescence intensity (Figure 6f). These results demonstrate that cocal LV generated with our producer cell line is equivalent to LV generated with the standard protocol, even in the context of primary human T-cells. Similarly to what we described earlier for the eGFP producer cells, we were able to further increase LV titers from C4 1–9 cells by carrying out a second round of single clone screening, now by measuring LV titers from transduction and TCR expression of H9 cells. Several clones generated significantly higher LV titers than the original cell line, with an increase in titer by as much as threefold (Supplementary Figure S5). In conclusion, we have successfully developed a third-generation cocal LV producer cell line for the WT1-specific TCR that is capable of achieving concentrated titers above 1 × 108IU/ml, thus, making it a suitable system for the generation of clinical-grade vector.
Figure 6.

Generation of a cocal producer cell line for WT1-specific TCR LV. (a) Lentiviral vector (LV) titer measured by quantitative PCR (qPCR) in single cocal T-cell receptor (TCR) producer clones (open circles) or bulk cells (bar). Unconcentrated titer is given as infectious units/ml (IU/ml). (b) Unconcentrated (1×) and concentrated (100×) LV titer measured by qPCR from multiple experiments (n = 5) using the best producer clone identified in a. Bar graph shows mean titer and error bars show SE of the mean. (c) Stability of cocal TCR LV producer cell line based on unconcentrated titer after long-term culture. (d) TCR expression measured in H9 cell line transduced with different amounts of LV made with the standard protocol or with the producer cell line. TCR expression was detected by surface antibody staining at 3 days post transduction and the fraction of TCR positive cells was quantified by flow cytometry. (e) TCR expression measured in human CD4+ cells transduced with different amounts of vector as described in d. (f) Histogram showing levels of TCR expression measured in live human CD4+ cells from e transduced with 5μl LV made with producer cells (gray line) or with standard conditions (black line), as compared to untransduced/stained cells (red line) and to unstained cells (gray shade).
Adaptation of cocal producer cells to suspension/serum-free cultures for large-scale LV preparations
Current LV gene therapy trials often involve the treatment of a large number of patients, which, in turn, require sizeable volumes of clinical grade vector. To initiate efforts toward the scaling-up of vector production, we adapted cocal producer cells eGFP 2–12 and C4 1–9 for growth in suspension/serum-free cultures. Adherent producer clones were grown in 10-ml shaker flasks in Freestyle 293 Expression serum-free medium, with media changes performed every 3 days. After 2 weeks of culture under these conditions, both producer cell lines grew robustly in suspension cultures in the absence of selection, with a doubling time average of 41 hours as compared to 29 hours for 293T-cells (Figure 7a). We measured both LV production and viable cell counts in these suspension producer cell lines over time following induction with NaBu. We found a steady increase in vector produced by 2–12 eGFP producer cells monitored over 5 days, reaching an unconcentrated titer of about 5 × 105 IU/ml (Figure 7b). LV titer in the C4 1–9 producer cells was slightly higher, reaching an unconcentrated titer of about 5.5 × 105 IU/ml by day 4 (Figure 7c). Viable cell count increased steadily during the first 2 to 3 days of culture but dropped thereafter due to cytotoxicity associated with NaBu in the media. We increased LV production to 100 ml cultures using spinner bottles, later to 1 l using a bioreactor. On average, suspension cells achieved 100× concentrated titers of 2.0 × 107 IU/ml (± 2.7 × 106 IU/ml) for 2–12 eGFP LV and 4.0 × 107 IU/ml (± 5.9 × 106 IU/ml) for C4 1–9 LV. These titers are threefold to fivefold lower than titers obtained from adherent cells, and production will require further optimization. However, these data demonstrate the possibility of adapting our producer cell lines to a suspension culture system, which should have significant advantages for future large-scale LV production.
Figure 7.

Adaptation of cocal producer cell lines to growth in suspension/serum free media. (a) Growth rate of 293T-cells, eGFP 2–12 and C4 1–9 producer cells adapted to growth in suspension/serum free media. Cells were passaged every 3–4 days, and viable count was determined by trypan blue staining. (b) Unconcentrated LV titer (flow cytometry, bar graph) and viable cell count (line graph) measured in 2–12 eGFP LV producer cells grown in suspension following induction with sodium butyrate (NaBu) (T0). (c) Unconcentrated LV titer quantitative PCR (qPCR, bar graph) and viable cell count (line graph) measured in C4 1–9 LV producer cells grown in suspension following induction with NaBu (T0).
Discussion
In this study, we report the development of two high-titer, third-generation, LV producer cell lines employing the cocal glycoprotein envelope for a standard eGFP transgene and for a TCR specific for the tumor antigen WT1. The use of the cocal envelope was a key component for successfully achieving high-titer LV because stable expression of the commonly used VSV-G envelope led to significantly lower titers. These lower titers were correlated with reduced (twofold to fourfold) VSV-G mRNA transcripts as compared to cocal transcripts, suggesting that low VSV-G expressing cells are selected in our procedure, possibly because of the reported toxicity associated with stable expression of this envelope. This result is certainly consistent with previous studies that relied on inducible promoters to drive VSV-G expression for the construction of LV packaging/producer cell lines.10,11,12,13,14 In contrast, we were able to achieve stable, high LV titers in our producer cell lines by constitutive expression of the cocal envelope. Additional advantages of cocal LVs include resistance to human serum inactivation,3 and higher transduction efficiency of human and NHP HSCs and CD4+ T-cells at low vector doses. We also provide evidence for shared receptor usage by cocal and VSV-G envelopes as transduction by LV containing either envelope was inhibited by soluble LDL receptors.5 This result is consistent with the high amino acid sequence conservation shared by the two envelope proteins and implies that cocal and VSV-G LVs use similar uptake and internalization pathways for cell entry.
Although we cannot exclude that cocal LVs utilize other cell receptors, our observations of more efficient transduction of HSCs and CD4+ T-lymphocytes for cocal LVs as compared to VSV-G LVs may be explained by higher binding affinity with the LDL receptor or more efficient cell membrane fusion for cocal envelope as compared to VSV-G. Alternatively, cocal LV may display a higher density of envelope molecules per virion than VSV-G LV, resulting in greater avidity for the entry receptor. This hypothesis is corroborated by our findings showing that higher cocal expression is achieved in producer cells as compared to VSV-G expression. The lower levels of sLDLR-mediated inhibition in transduced HT1080 cells by cocal LV as compared to VSV-G LV also agree with this hypothesis. Additional experiments will be required to differentiate between these possibilities.
Our generation of a high-titer cocal packaging cell line provides a great foundation for the construction of additional LV producer cell lines, simply by stable expression of a lentiviral transfer plasmid and selection of best producer single clones, which can be completed within 2 months. We first used a standard eGFP lentiviral plasmid to streamline and optimize our protocol, which we applied later to the development of the WT1-specific TCR LV cell line. The performance of LV was assessed by determining the infectivity (IU/ng p24 Gag) of several preparations. For eGFP LV, we found comparable infectivity levels between LV made with the standard protocol versus LV made with producer cells. For TCR LV, infectivity was slightly lower for LV made with producer cells. Nevertheless, TCR LV generated by producer cells performed as well than LV made with our standard protocol for the transduction of human CD4+ T-cells, and eGFP LV made by producer cells transduced CD34+ more efficiently at higher MOI. Future work will compare the engraftment potential of HSCs transduced with LV made with producer cells with that of cells transduced with standard LV.
We have used the histone deacetylase inhibitor NaBu to achieve high LV titers and thus, efficient transduction of HSCs and T-cells. Previous studies showed that inhibition of histone deacetylases by sodium butyrate increases LV titer using a transient transfection protocol.15,16 This induction in LV titer can probably be accounted for, at least in part, by an increase in steady-state levels of viral vector RNA within the producer cells. Our observations that show increased production of HIV-1 p24 in the presence of sodium butyrate (Supplementary Figure S3b) confirm this hypothesis. In the absence of NaBu treatment, viral proteins expression and LV production are kept at minimal levels, thus limiting cytotoxicity and allowing producer cells to grow robustly. This strategy is analogous to other inducible systems that were used in the construction of LV producer cell lines such as the tetracycline-regulated promoter10 and the cumate inducible system.11
In current gene therapy HSC trials, roughly 1–5 × 109 IU of vector per patient is required. Based on our LV titers, this would necessitate the production of 0.5 l–2.5 l of unconcentrated vector per patient. Although production of such LV preparation is certainly feasible using adherent producer cells, the use of suspension producer cells would greatly facilitate this process, especially with cultures grown in bioreactors. As of now, we have shown that we could adapt our producer cells to grow in suspension, serum-free media and we were able to produce LV using a 1 l bioreactor. Although LV titers obtained from these cells are currently threefold to fivefold lower than those obtained with adherent cells, culture parameters will further be optimized to increase LV production.
The majority of gene therapy clinical trials employ LV produced by transient transfections of plasmids, with known limitations in safety, cost, and reproducibility. Although several LV packaging/producer cell lines have been generated,4,11,12,17,18 to our knowledge, only one LV producer cell line derived from GPRG cells is currently used in clinical trials aimed at curing SCID-X1.10,13 Our new cocal third-generation SIN LV packaging cell line was successfully derived into two independent LV producer cell lines that generate clinically usable titers. The broad applicability of our cocal packaging cell line offers a promising tool toward efforts aimed at generating large-scale, clinical grade LV.
Materials and Methods
Construction of plasmids. pMD2.G, pMDLg/pRRE, and pRSV-Rev were gifts from Didier Trono (Addgene plasmid # 12259, 12251, and 12253, Cambridge, MA).14 The Hygromycin.2A.cocal and Hygromycin.2A.VSV-G cassettes were derived from plasmids pTK-Hyg (Clontech Laboratories, Mountain View, CA), plasmids pMD2.G, and pMD2.Cocal-G,3, assembled by overhang PCR using High Fidelity Platinum Taq polymerase (Invitrogen, Carlsbad, CA) and subcloned into the XhoI/HindIII restriction sites of plasmid Bluescript SK+ (Stratagene California, La Jolla, CA). Primers sequences are given in Supplementary Table S1. Integrity of the sequences was confirmed by capillary sequencing (FHCRC, Genomics shared resources, Seattle, WA). The inserts were then extracted with restriction enzymes NotI/XhoI (New England Biolabs (NEB), Ipswich, MA), XhoI was blunted with DNA pol I (NEB), and the resulting product was ligated into the NotI/PmlI sites of pMD2.Cocal-G to create pMD2.hyg.2A.cocal and pMD2.hyg.2A.VSV-G. For VSVG CO, VSV-G was codon-optimized for Homo sapiens using the IDT codon optimization tool (Integrated DNA Technologies, Coralville, IA), ordered as gBlocks Gene Fragments, and cloned into the PmlI/MscI restriction sites of pMD2.G to generate pMD2.G CO. Puromycin, Blasticidin, and Zeocin selections were carried out using plasmid pPUR (Clontech), pSELECT-blasti (InvivoGen, San Diego, CA) and pSELECT-zeo (InvivoGen), respectively. The SIN LV transfer vector pRRLSIN.cPPT.PGK-GFP.WPRE was a gift from Didier Trono (Addgene plasmid # 12252) and pRRLSIN.C4b17_P2A_aWPRE-1 was generated by cloning the α and β chains of the WT1-specific TCR C4, separated by a P2A element, into pRRLSIN.cPPT.MSCV/GFP.WPRE, replacing GFP. The pRRLSIN.cPPT.MSCV/GFP.WPRE vector was a kind gift from Richard Morgan.19
Generation of stable cell lines. Stable expression of the different helper genes was achieved sequentially by plasmid(s) transfection, followed by selection with the appropriate drugs. Cells were seeded in a six-well plate and transfected with lipofectamine LTX (Thermo Fischer Scientific, Waltham, MA) following the manufacturer's instructions for the transfection of 293T-cells, and using a 1 : 5 ratio of selection plasmid to helper plasmid. Cells were expanded to a 10-cm culture dish and selection drugs were added to the media 3–5 days post-transfection. Selection media was changed every 3 days and cells were passaged each time they reached confluence for a total of 3 weeks before LV production was induced in these cells. After achieving stable expression, individual clones were isolated using a limiting dilution approach by seeding single cells in a 96-well plate and expanding them to 12-well plate, later to 6-well plates under selective conditions.
qPCR measurement of envelope expression. Envelope expression was analyzed from cocal and VSV-G expressing cells using the SYBR Green Expression assay (Thermo Fischer Scientific). Briefly, total RNA was isolated cells stably expressing the envelopes using the RNeasy Mini Kit (Qiagen N.V., Hilden, Germany) and converted to cDNA using the Superscript III first strand synthesis system (Thermo Fischer Scientific). The SYBR Green reaction was run following the manufacturer protocol using 25 ng template using the Applied Biosystems (Foster City, CA) 7500 Real-Time PCR system (Thermo Fischer Scientific). Relative mRNA expression was calculated by the comparative Ct method using glyceraldehyde 3-phosphate dehydrogenase as reference gene. Primer sequences are given in Supplementary Table S1.
Standard lentivirus and foamy virus vector production. In the standard protocol, third-generation LVs were produced by four plasmid-polyethylenimine transfection in HEK 293T-cells. HEK 293T-cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% Hyclone Cosmic Calf serum (Thermo Fischer Scientific), 1% sodium pyruvate, nonessential aminoacids, L-Glutamine, and 1% penicillin/streptomycin. HEK 293T-cells were adapted to growth in suspension using Freestyle 293 expression media (Thermo Fischer Scientific). During passaging, LV producer cells were grown in the presence of hygromycin (150 μg/ml, Thermo Fischer Scientific), puromycin (1 μg/ml, Life Technologies, Carlsbad, CA), blasticidin (7.5 μg/ml, Life Technologies) and zeocin (100 μg/ml, Life Technologies). For LV production in 15-cm plates, cells were plated on 0.1% gelatin at a density of 1.8 × 107 cells/plate and transfected with 27 μg transfer vector construct, 6 μg pMDLg-pRRE, 12 μg pRSC-Rev, and 6 μg pMD2.G for VSV-G pseudotyped LV or 3 μg pMD2.Cocal-G for cocal-pseudotyped LV. When 10-cm plates or six-well plates were used to produce LV, the number of cells and amount of DNA used was scaled down proportionally to the surface area of the plate. The next day, cells were washed with 1× Dulbecco's phosphate buffered (Thermo Fischer Scientific) and treated with 15 ml media containing 10 mmol/l sodium butyrate (Sigma-Aldrich, St. Louis, MO) for about 8 hours. Cell supernatant was harvested and combined with two additional harvests carried out over a time span of 48 hours. The supernatant was filtered through a 0.8 μm-pore-size filter, concentrated 100-fold by centrifugation for 15−20 hours at 4 °C at 5,000×g, and stored at −80 °C. FV was produced by polyethylenimine transfection of HEK 293T-cells as previously described20 using the transfer vector pFV.PGK.GFP.
Lentivirus vector generated with producer cell lines. For LV generated in 15-cm plates using adherent producer cells, cells were plated on 0.1% gelatin at a density of 3.0 × 107 cells/plate and media was replaced with 25 ml fresh media containing 10 mmol/l NaBu the next day. A single harvest was carried out at 48 hours post media change, and the resulting LV was purified and concentrated as described in the standard protocol. Producer cells were adapted for growth in suspension by culture in 10-ml shaker flasks in Freestyle 293 Expression serum-free medium (Thermo Fischer Scientific), with media changes performed every 3 days. LV production was induced from these cells after culture in suspension for 2 weeks by adding 10mmol/l NaBu to the media. Both LV titer and viable cell count were measured over time using Trypan blue staining and a countess cell counter (Thermo Fischer Scientific).
Determination of LV titer. The titer of the vector preparations was determined by adding different amounts of LV to the human fibrosarcoma cell line HT1080. HT1080 cells were grown in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and were plated at 1 × 105 cells/ml in a 12-well plate one day before transduction. For 100× concentrated LV, volumes used in the transduction were 1, 0.3, 0.1, 0.03 μl following serial dilutions of the vector. For unconcentrated LV, 20 and 100 μl were used for transduction. Protamine sulfate was added to the cells at a final concentration of 8 μg/ml. For vectors containing GFP, cells were analyzed by flow cytometry 3 days post transduction and the percentage of GFP-expressing cells was used to calculate the number of infectious units (IU) per ml of vector. For LV not containing a fluorophore, titer was determined by a TaqMan assay. Briefly, HT1080 cells were transduced as described above and were passaged every 3–4 days for a total of 10 days, after which genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen N.V.). LV DNA was measured by the TaqMan assay (Applied Biosystems) using probes specific for the vector and for the endogenous gene beta globin, and values were compared to a standard curve to determine the number of IU per ml.
Primary cell culture and transduction. All LV transductions were carried in presence of 8 μg/ml protamine sulfate. Human CD34+ cells were collected from volunteers under an institutional review board-approved protocol. Human and NHP CD34+ were isolated, cultured, and transduced as described earlier.3 Briefly, CD34+ cells were cultured in Iscove's Modified Dulbecco's Medium with 10% FBS containing 100 ng/ml each of Flt-3 ligand, stem cell factor, and granulocyte colony-stimulating factor. For transduction, cells were plated on CH-296 fibronectin (Takara, New York, NY) at 2 μg/ml, and exposed to the vector at various MOI as determined by HT1080 titer. In the colony-forming experiments, CD34+ cells were plated in Methocult H4230 (Stem Cell Technologies, Vancouver, Canada) supplemented with the following cytokines: 100 ng/ml each of stem cell factor, TPO, GM-CSF, granulocyte colony-stimulating factor, IL-3, IL-6, and 4 U/ml erythropoietin. After 12 days, plates were scored for hematopoietic colony phenotypes (CFU-E, CFU-M CFU-GM, and CFU-GEMM) and for GFP fluorescence under a microscope. The H9 cell line is a clonal derivative of the Hut 78 cell line (see ATCC TIB-161, Manassas, VA) and was grown in Dulbecco's Modified Eagle Medium supplemented with 10% FBS and 1% penicillin/streptomycin. NHP and human CD4+ cells were isolated and cultured using a similar protocol to the one described previously for human CD4+ cells.21 Briefly, blood was drawn from consented adult healthy donors at Seattle Puget Sound Blood Center and primary CD4 cells were isolated from whole blood using Rosette Sep enrichment or using a magnetic selection kit from Miltenyi Biotec from peripheral blood mononuclear cells, which were isolated from whole blood using density-gradient centrifugation, as per manufacturer's instructions. Cells were grown in RPMI (Invitrogen) supplemented with 20% FBS, 50 ng/ml human IL-2, 5 ng/ml IL-7, and 5 ng/ml IL-15 (Peprotech, Rocky Hill, NJ) and cultured at 1–3 × 106 cells/ml unless otherwise noted. Cells were activated using CD3/28 beads (Dynabeads, Thermo Fischer Scientific) for 48 hours at a 1:1 cell to bead ratio. Following transduction with the TCR LV, expression was detected by flow cytometry following extracellular staining with the TCRVβ17 antibody conjugated to phycoerythrin (Beckman Coulter, Brea, CA) at a 1:50 dilution. A recombinant soluble form of human LDLR was purchased from R&D Systems, (Minneapolis, MN) and preincubated at 4 °C for 4 hours with lenti or foamy vectors at final concentrations of 0, 2.5, 10, and 25 μg/ml prior to transduction.
HIV-1 p24 ELISA. Physical LV production was obtained by measuring HIV-1 p24 concentration from serially diluted supernatant collected from producer cells using the Retrotek HIV-1 p24 Antigen enzyme-linked immunosorbent assay (ELISA) 2.0 (ZeptoMetrix Corporation, Buffalo, NY) following the manufacturer's instructions.
SUPPLEMENTARY MATERIAL Figure S1. LV production from cells stably expressing different envelopes. Figure S2. Effect of helper to selection plasmid ratios on LV titer. Figure S3. Induction of LV titer and p24 expression with sodium butyrate. Figure S4. Determination of limiting factors for LV production by individual transfection of each helper plasmid into the eGFP best producer cells Figure S5. Second round of screening for best TCR LV producer clones. Table S1. Primer sequences.
Acknowledgments
This work was supported in part by grants AI097100, DK056465, and HL116217, from the National Institutes of Health, Bethesda, MD. H.-P.K. is a Markey Molecular Medicine Investigator and recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research. We thank Daniel Humphrys and Biswajit Paul for technical assistance. We thank Didier Trono for supplying reagents. We thank Jennifer Adair and Stefan Radke for critical reading of the manuscript. We also acknowledge the assistance of Helen Crawford in preparing the manuscript. H.-P.K. and O.H. have filed a provisional patent application regarding technology described here; otherwise, the authors have no conflicts of interests.
Supplementary Material
References
- Naldini, L (2015). Gene therapy returns to centre stage. Nature 526: 351–360. [DOI] [PubMed] [Google Scholar]
- Ory, DS, Neugeboren, BA and Mulligan, RC (1996). A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA 93: 11400–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge, GD, Wu, RA, Hansen, M, Ironside, C, Watts, KL, Olsen, P et al. (2010). Cocal-pseudotyped lentiviral vectors resist inactivation by human serum and efficiently transduce primate hematopoietic repopulating cells. Mol Ther 18: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanber, KS, Knight, SB, Stephen, SL, Bailey, R, Escors, D, Minshull, J et al. (2015). Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep 5: 9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein, D, Werman, A, Novick, D, Barak, S and Rubinstein, M (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA 110: 7306–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plochmann, K, Horn, A, Gschmack, E, Armbruster, N, Krieg, J, Wiktorowicz, T et al. (2012). Heparan sulfate is an attachment factor for foamy virus entry. J Virol 86: 10028–10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasimuzzaman, M and Persons, DA (2012). Cell Membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol Ther 20: 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis, AG, Ragnarsson, GB, Nguyen, HN, Chaney, CN, Pufnock, JS, Schmitt, TM et al. (2013). Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 5: 174ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, TM, Aggen, DH, Stromnes, IM, Dossett, ML, Richman, SA, Kranz, DM et al. (2013). Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 122: 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throm, RE, Ouma, AA, Zhou, S, Chandrasekaran, A, Lockey, T, Greene, M et al. (2009). Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 113: 5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussau, S, Jabbour, N, Lachapelle, G, Durocher, Y, Tom, R, Transfiguracion, J et al. (2008). Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol Ther 16: 500–507. [DOI] [PubMed] [Google Scholar]
- Wielgosz, MM, Kim, YS, Carney, GG, Zhan, J, Reddivari, M, Coop, T et al. (2015). Generation of a lentiviral vector producer cell clone for human Wiskott-Aldrich syndrome gene therapy. Mol Ther Methods Clin Dev 2: 14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, MR, Lockey, T, Mehta, PK, Kim, YS, Eldridge, PW, Gray, JT et al. (2012). Transduction of human CD34+ repopulating cells with a self-inactivating lentiviral vector for SCID-X1 produced at clinical scale by a stable cell line. Hum Gene Ther Methods 23: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull, T, Zufferey, R, Kelly, M, Mandel, RJ, Nguyen, M, Trono, D et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, BL, Potts, PR and Porteus, MH (2011). Creating higher titer lentivirus with caffeine. Hum Gene Ther 22: 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, JC and Sechelski, J (1995). Use of sodium butyrate to enhance production of retroviral vectors expressing CFTR cDNA. Hum Gene Ther 6: 1195–1202. [DOI] [PubMed] [Google Scholar]
- Ikeda, Y, Takeuchi, Y, Martin, F, Cosset, FL, Mitrophanous, K and Collins, M (2003). Continuous high-titer HIV-1 vector production. Nat Biotechnol 21: 569–572. [DOI] [PubMed] [Google Scholar]
- Stornaiuolo, A, Piovani, BM, Bossi, S, Zucchelli, E, Corna, S, Salvatori, F et al. (2013). RD2-MolPack-Chim3, a packaging cell line for stable production of lentiviral vectors for anti-HIV gene therapy. Hum Gene Ther Methods 24: 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S, Peng, PD, Yang, S, Hsu, C, Cohen, CJ, Zhao, Y et al. (2009). Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther 20: 630–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiem, HP, Wu, RA, Sun, G, von Laer, D, Rossi, JJ and Trobridge, GD (2010). Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther 17: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather, BD, Romano Ibarra, GS, Sommer, K, Curinga, G, Hale, M, Khan, IF et al. (2015). Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med 7: 307ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.