

Abstract
Hepatitis B virus (HBV) causes chronic infections that cannot yet be cured. The virus persists in infected hepatocytes, because covalently closed circular DNA (cccDNA), the template for the transcription of viral RNAs, is stable in nondividing cells. Antiviral therapies with nucleoside analogues inhibit HBV DNA synthesis in capsids in the cytoplasm of infected hepatocytes, but do not destroy nuclear cccDNA. Because over 200 million people are still infected, a cure for chronic hepatitis B (CHB) has become one of the major challenges in antiviral therapy. As a first step toward the development of curative therapies, we previously demonstrated that the CRISPR/Cas9 system can be used to functionally inactivate cccDNA derived from infectious HBV. Moreover, some evidence suggests that certain cytokines might induce an APOBEC-mediated cascade leading to the destruction of cccDNA. In this report we investigated whether a combination of the two mechanisms could act synergistically to inactivate cccDNA. Using next generation sequencing (NGS), we determined the complete spectrum of mutations in cccDNA following Cas9 cleavage and repair by nonhomologous end joining (NHEJ). We found that over 90% of HBV DNA was cleaved by Cas9. In addition our results showed that editing of HBV DNA after Cas9 cleavage is at least 15,000 times more efficient that APOBEC-mediated cytosine deamination following treatment of infected cells with interferon alpha (IFNα). We also found that a previously used method to detect cytosine deaminated DNA, termed 3D-PCR, overestimates the amount and frequency of edited HBV DNA. Taken together, our results demonstrated that the CRISPR/Cas9 system is so far the best method to functionally inactivate HBV cccDNA and provide a cure for CHB.
Introduction
Hepatitis B virus (HBV) is a DNA virus belonging to the Hepadnaviridae family of viruses that replicate by reverse transcription of an RNA intermediate.1,2 HBV has a small 3.2 kb-long relaxed circular (rc) genome with overlapping open reading frames that also contain binding sites for transcription factors that are required for expression of the viral proteins. Infection of hepatocytes results in the conversion of the rcDNA genome into a covalently closed circular (ccc) DNA that resides in the cell nucleus. cccDNA is the template for the transcription of the viral genes. It is the means for persistence of HBV in the infected liver and hence, represents the molecular basis for chronic hepatitis B (CHB).
A cure from CHB has become one of the major challenges in the field of antiviral research. It entails elimination or functional inactivation of cccDNA from infected hepatocytes. CRSIPR/Cas9 has been invoked as a possible solution because it can cleave and functionally inactivate cccDNA produced from infectious HBV.3 Cytokines have been proposed as mediators of an innate immune response that could culminate in the destruction of cccDNA. This idea was postulated more than 15 years ago based on data providing estimates for the dynamics of viral DNA intermediates during the course of acute, self-limiting HBV infections in chimpanzees.4 A hallmark of such infections is that previously infected hepatocytes become virus free, indicating that mechanisms exist to cure cells from infection. However, since resolution from natural infections is always accompanied by destruction and proliferation of infected hepatocytes, it is possible that cccDNA is lost as a consequence of dilution during multiple rounds of cell division, rather than by an active elimination process.5,6 So far, direct evidence for non-cytolytic destruction of cccDNA still has not been forthcoming.
A recent report claimed that deamination of cytosine residues on the minus strand of HBV cccDNA by cytokine-induced APOBEC3A (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) and APOBEC3B could induce an enzymatic cascade leading to the destruction of cccDNA.7 This model was based on four assumptions. First, enzymes belonging to the family of APOBEC proteins gain access to the transcribed minus strand of cccDNA. Second, essentially every cytosine residue within a cytosine-rich segment in cccDNA can be deaminated to yield uracil. Third, uracil-DNA glycosylase removes the uracil base. Fourth, removal of the base causes destruction of cccDNA rather than repair by the base excision repair (BER) pathway as with chromosomal DNA.8 Hence, this model provided hope that a better understanding of the mechanism of the pathway leading to cccDNA degradation could be exploited for therapies leading to the selective destruction of cccDNA and, perhaps in combination with other strategies, such as the CRISPR/Cas9 approach, to a cure of CHB.7,9
Our previous work demonstrated that cccDNA is a target for CRISPR-Cas9.3 Cloning of PCR fragments derived from Hirt extracts10 of HBV infected HepG2 cells revealed that Cas9-inudced cleavage can lead to repair of cccDNA favoring single nucleotide insertions and deletions over longer deletions. However, a limitation of our study was the relatively small number of cloned PCR fragments analyzed with conventional Sanger DNA sequencing. Hence, the motivation for this study was to obtain a more complete data set of the cccDNA population after CRISPR-Cas9 cleavage using next generation sequencing (NGS). Moreover, we sought to determine the frequency of G:A transitions indicative of cytosine deamination in normal and cytokine-treated cells and devise an experimental strategy that would permit us to determine whether cccDNA can be edited in HepG2 cells. Our results were consistent with our original observations indicating that single nucleotide insertions and deletions dominated all other mutations in cccDNA following Cas9 cleavage. They also revealed that approximately 7% of edited DNA contained in frame deletions indicating that a single CRISPR target on the HBV genome might not always be sufficient to inactivate cccDNA. Furthermore they indicated that G:A transitions accumulate at a much lower frequency compared with Cas9-induced mutations. Evidence for editing of cccDNA following APOBEC deamination of cytosine residues could not be obtained. However, our results revealed that virion DNA derived from the HepG2–derived cell lines HepAD3811 and 2.2.1512 commonly used for the production of HBV for infection studies already exhibits G:A transitions.
Results
Spectrum and frequency of CRISPR/Cas9 mutations on cccDNA
We have previously reported cleavage and repair of HBV cccDNA by Cas9.3 This study revealed that the majority of cleavage events lead to a single nucleotide insertion or deletion. Larger deletion could also be observed albeit at a lower frequency. However, exact numbers could not be determined due to the limited number of cloned PCR-derived HBV fragments available for nucleotide sequence analysis.
To determine the nature and frequency of Cas9 mutations on cccDNA in a more comprehensive fashion and to identify sgRNAs for subsequent experiments to investigate APOBEC-mediated editing of HBV DNA (see below), we designed two sgRNAs, HBx2 and HBx4, targeting HBx and the overlapping polymerase gene (Figure 1a,b). Previous studies demonstrated that HBx is required for the transcription of viral RNAs from cccDNA, permitting us to measure Cas9 activity indirectly with immunofluorescence (IF) microscopy using an HBcAg antibody.3,13,14,15
Figure 1.

Spectrum of CRISPR/Cas9 cleavage. (a) The figure shows a physical map of HBV with the four open reading frames and viral RNAs. Also indicated are the positions of enhancers I and II (EN1, EN2). For additional details, see ref. 35. (b) Target sequences of sgRNAs HBx4 and HBx2 in reference to the polymerase (pol) and HBx (X) genes. Note, for HBx4, the complementary (plus strand) sequence is shown. (c) HepG2/NTCP/Cas9 cells expressing HBx2 or HBx4 were infected with HBV, treated with IFNα for 5 days and processed for IF analysis with HBcAg specific antibody C1-5 10 days post infection. HBcAg positive cells were counted with an automated microscope. UI; uninfected, CO; Control, d; doxocycline, I, IFNα; P values for CO1/HBx2,d and HBx/HBx2,d were 0.0003 and 0.0001, respectively. (d) The figure depicts a pie chart representing the frequencies of reads with specific mutations, as well as wildtype (wt). The data used for the chart are shown in Table 1. del*; frequency of one and two nucleotide-long deletions spanning positions -1 to -6 with respect to the PAM sequence. Deletion at position -4 is shown separately (del C), ins*; frequencies of A,G,T insertions at position -4., ins C; insertion of a C residue at position -4.
HepG2/NTCP cells expressing the single guide RNA (sgRNAs) under the control of the tetracycline promoter exhibited a ca. 8- to 10-fold reduction in the number of HBcAg expressing cells compared with control cells (Figure 1c). To determine the nature of the mutations introduced into cccDNA, we extracted DNA from approximately 100,000 infected, HBx2 expressing cells with the Hirt procedure that permits selective extraction of cccDNA.10 We then PCR amplified the purified DNA with primers spanning the cohesive overlap region of the viral genome (PCR1, Figure 3a). The selection of the primer favors amplification of cccDNA over the more abundant rcDNA that is to some extent still present in the Hirt extracts.3,7 The PCR products were then prepared for NGS. The results from three separate infections, based on more than one million sequence reads per experiment, demonstrated that approximately 90% of the reads carried mutations within the target sequence for the HBx2 sgRNA (Table 1). This result correlated well with the observed 8- to 10-fold reduction in HBcAg expression in cells expressing HBx2 (Figure 1c). Over 60% of the sequencing reads exhibited a single nucleotide deletion at position -4 with respect to the protospacer adjacent motif (PAM) sequence. About 8% of reads carried an insertion of a C residue at position -4. It should be noted that Cas9 preferentially cleaves at position -3 from the PAM site, which indicates that the terminal G residue of the HBx2 guide RNA sequence served as the first nucleotide of the PAM region (Figure 1b).16 About 10% of the reads carried a single nucleotide insertion at this site and ca. 16% exhibited deletions ranging from 2 to 6 nucleotides in length. Notably, the variation among samples of the three infections was minimal (Table 1).
Table 1. Cas9 mutations in cccDNA.
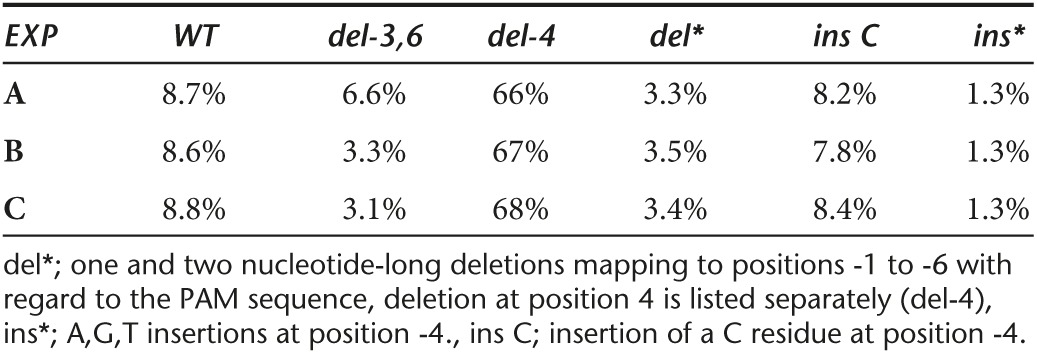
A more detailed analysis of the mutations created by Cas9 followed by nonhomologous end joining (NHEJ) revealed that deletions of 3 and 6 nucleotides which maintain the open reading frame of HBx amounted to almost 7% of the population (Table 1 and Figure 1d). Assuming that HBx mutants with short in frame deletions remain functional, the results indicated that a single sgRNA might be insufficient to completely inactivate cccDNA. The reason why ca. 8% of cccDNA remained wildtype is not known (see discussion), but could be explained by difference in the Cas9 or sgRNA expression levels among cells used for HBV infections. It is also possible that protein-free rcDNA produced directly from virion DNA during infection could have contributed to the fraction of wildtype DNA, because it can be co-purified with cccDNA.17,18
Evidence for editing of HBV DNA
In a previous report we demonstrated that interferon alpha (IFNα) did not significantly inhibit infection of HepG2/NTCP cells with HBV.3 We confirmed those results with additional experiments using IFNα, as well as IFNγ and tumor necrosis factor α (TNFα). As expected, IFNα induced translocation of STAT1 from the cytoplasm to the cell nucleus and lead to the phosphorylation of STAT1 (Figure 2b,c). Similarly, IFNγ induced nuclear accumulation of STAT1. Consistent with a previous study,19 we observed that IFNα induced transcription of APOBEC3G and to a lesser extent APOBEC3A. APOBEC 3B was not induced by the cytokine (Supplementary Figure S1). Finally, we noted that addition of IFNα within 2 days of HBV infections caused cell death indicating that, under these conditions, the cytokine activated a cytotoxic innate immune response in HepG2/NTCP cells (results not shown, see discussion). Treatment of HBV infected cells with either IFNα, IFNγ, or TNFα 5 days post infection did not exhibit any obvious cytotoxicity and did not reduce the efficiency of HBV infections (Figure 2d).
Figure 2.

Effect of cytokines on HBV infection. (a) The figure depicts the method used for infection of HepG2/NTCP cells with HBV. Addition of IFNα at day 2 postinfection (p.i.) leads to toxicity (see text). (b) The figure shows images captured from HepG2 cells treated with IFNα (2000 IU/ml) and IFNγ (20 g/ml) and processed for IF with a STAT1-specific antibody. (c) Western blot of whole cell extracts from HepG2 cells incubated for 2 (lanes 2) and 4 (lanes 3) days in 2% dimethyl sulfoxide (DMSO) with IFNα (2000 IU/ml) developed with STAT1 and phospho STAT1 (pSTAT1) antibodies, respectively. (d) Analysis of HBcAg expression as described in the legend to Figure 1c. Differences among the four samples (CO, IFNα, IFNγ and TNFα were obviously not significant (P > 0.1). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
To determine whether IFNα could induce deamination of cytosine residues in HBV infected HepG2/NTCP cells, i.e., through activation of APOBEC proteins, we isolated HBV cccDNA from infected cells with the Hirt procedure (Figure 2a). cccDNA was then PCR amplified with primer pairs spanning the cohesive overlap region as above (PCR1, Figure 3a). Deamination of cytosine results in an increase of the A:T / G:C ratio in rcDNA and in cccDNA derived from rcDNA. As a consequence, APOBEC-edited HBV DNA has a lower melting temperature compared with wildtype DNA. To detect edited HBV DNA, we used the “3D-PCR” method to selectively amplify A:T rich HBV DNA present in a G:C rich region overlapping HBx (3D-PCR, Figure 3a).20 The input DNA for the 3D-PCR reaction was derived from the PCR1 reaction described above. Hence, 3D-PCR is a “nested” PCR reaction. We optimized the conditions for the 3D-PCR reaction by reducing the melting temperature stepwise from 95 °C to 85 °C. With a melting temperature of 87 °C, PCR reactions with serially diluted DNA from the PCR1 reactions of IFNα-treated cells yielded products of expected size that were absent from the same reactions with PCR1 DNA produced with untreated HBV infected cells (Figure 3b). We then cloned and sequenced the DNA from the differentially amplified DNA fragments. DNA sequencing revealed clones with extensive G:A transitions consistent with cytosine deamination on the minus strands of HBV DNA (Figure 4). Within a 180 nucleotide-long segment spanning pos. 2753–2932, 29 and 32 out of 50 dG residues were changed to dA. Remarkably, in clone MT3Db every G residue was changed to dA between position 2806 and 2932, essentially as reported previously.7
Figure 3.

3D-PCR of cccDNA. (a) The figure shows a map of the HBV genome spanning the region targeted for PCR amplification of cccDNA (Hirt cccDNA, PCR1), virion DNA (Virion rcDNA, vPCR1) and 3D-PCR using either PCR1 or vPCR1 as a source. The position of the 5′ end of minus stand DNA is indicated with a solid circle. The position of the 5′ end of the target sequence for sgRNA HBx2 is shown. (b) PCR reactions from Hirt DNA extracted from control cells (CO), cells treated with IFNα at day 2 and 5 p.i., respectively, and uninfected cells (UI). Mitochondrial DNA (MITO) was PCR amplified to normalize the HBV-specific products from the PCR1 reaction used for 3D-PCR. A fraction (1/40) of the PCR1 reaction was 10-fold serially diluted for the 3D-PCR reaction. The arrow points to the PCR product that was cloned and sequenced. Td, denaturation (melting) temperature.
Figure 4.

Sequence analysis of 3D-PCR-derived clones. Nucleotide sequence of two clones (MT3Da, MT3Db) obtained from the 3D-PCR reaction (Figure 3b). G:A transitions are indicated in bold black type face and marked with an asterisk. AYW, wild type HBVayw.
G:A editing of HBV DNA is a rare event
A potential problem with the 3D-PCR method is that the conditions for selective amplification of A:T rich DNA result in the production of chimeric DNA fragments with very high A:T content. In other words, the cloned DNA fragments with massive G:A transitions, i.e., like MT3Db, could be the product of DNA recombination events known to occur during PCR reactions (see discussion). Moreover, 3D-PCR does not provide any information about the frequency of G:A mutations. Hence, to validate the 3D-PCR results, we used NGS of PCR amplified HBV DNA (PCR1, Figure 3a) from HepG2/NTCP infected cells incubated with and without IFNα. NGS would also permit us to directly compare the efficiency of G:A editing with that of Cas9-induced mutations. Therefore, we used sgRNA HBx2 expressing cells for these experiments. Consistent with the previous experiment, we observed 3D-PCR reactions from IFNα treated cells at lower concentrations of input DNA (PCR1) compared with untreated cells (Figure 5a). Surprisingly, NGS of PCR1 fragments derived from IFNα treated and control cells revealed that G:A transitions were present independently of whether the cells were treated with the cytokine or not (Figure 5b,c). G:A transitions were present at much lower frequencies compared to Cas9 editing. As with the previous experiment, more than 90% of the reads carried mutations in the CRISPR-Cas9 target site for HBx2 (Figure 5c, note, only reads with 1 nucleotide deletions are shown). In contrast, the frequencies of reads with G:A mutations was over 15,000 to 100,000-fold lower compared to Cas9-inudced mutations. Moreover, an analysis of the 80 nucleotide-long reads spanning pos. 2816–2895 did not reveal any evidence for extensive editing as observed with 3D-PCR, where, as described above, in some clones almost every dG residue was changed to dA (Figure 5b, compare R2a-R5a with R18a* derived from MT3Db in Figure 4).
Figure 5.

NGS-based analysis of cccDNA. (a) The figure shows the 3D-PCR results from a second experiment with HBV infected HepG2/NTCP/Cas9 cells expressing sgRNA HBx2 (HBx2, dox) treated and not treated with IFNα (IFN, 2000 IU/ml). The products of the PCR1 reaction (not 3D-PCR! Figure 3a) were processed for NGS. (b) The figure shows selected nucleotide sequences obtained with NGS from PCR1 of HBV infected HepG2 cells treated with IFNα (panel a, Hbx2, IFN (d5-9)). The sequence spans positions 2816 to 2895 on the HBVayw genome. For illustrative purposes, the figure shows R18a* derived from clone MT3Db (Figure 4). The right column indicates the frequency of each sequence. R2a to R5a refer to selected sequence reads with 2, 3 4 and 5 G:A substitutions, respectively. (c) Same as B, except that PCR1 was derived from DNA marked “HBx2, IFN, dox” in panel a (see text). (d) NGS results obtained from vPCR1 (Figure 3a) with virion DNA purified from the supernatant of HepAD38 cells (a complete list with sequencing reads is available upon request).
These unexpected results suggested that extensive G:A editing might be an artifact created by the 3D-PCR reactions. Importantly, they also indicated that editing was not primarily a consequence of IFNα treatment, but might occur in untreated cells as well and/or might have already been present in HBV particles derived from HepAD38 cells used for infections of HepG2/NTCP cells. Why 3D-PCR was more efficient with DNA from IFNα treated cells compared with controls could not be explained by our results (Figure 5a, see discussion).
Virion DNA produced in different HepG2-derived cell lines carries G:A transitions
The observation that untreated cells contained G:A edited HBV DNA prompted us to analyze DNA extracted from virions produced in HepAD38 cells used for infection of HepG2/NTCP cells. Purified virion DNA was PCR amplified (vPCR1, Figure 3a) and processed for NGS. Analysis of the 80 nucleotide-long reads spanning pos. 2816–2895 revealed that virion DNA indeed exhibited G:A transitions, as observed with Hirt DNA derived from infected HepG2/NTCP cells (Figure 5d). The maximal number of G:A transitions observed in the 80 nucleotide-long fragment spanning position 2816 to 2895 was 6 of a total of 24 possible G:A transitions. The frequencies of reads with G:A mutations was generally a few fold higher in DNA from virions compared with Hirt DNA from HBV infected HepG2/NTCP cells (compare Figure 5b,d).
To determine whether G:A editing was a property unique to HepAD38 cells and to perform a parallel analysis of virion-derived DNA with NGS and 3D-PCR, we analyzed HBV virion DNA harvested from the supernatant of the HBV producer cell line HepG2 2.2.15.12 Virion DNA was amplified as before (vPCR1, Figure 3a) and used for 3D-PCR and NGS, respectively. Analysis of 3D-PCR in the 80 nucleotide-long fragment described above yielded clones with as many as 17 G:A transitions (H23D, Figure 6a). In contrast, with NGS, we observed at most 5 of 24 possible G:A transitions per sequencing read. We calculated the frequency of reads with 0–5 G:A transitions. Data points could be fitted to an exponential trendline (y = 0.17 × e(−1.12x), R2 = 0.99), Figure 6b), indicating that reads with more than 5G:A transitions might be very rare. Based on this trendline, the expected frequency of 80 nucleotide-long fragments with 17 G:A transitions, as observed with 3D-PCR, would be about 1 in one billion (9 × 10−10). We made similar observations when we used intracellular rcDNA extracted from HepAD38 cells in lieu of virion DNA (results not shown).
Figure 6.

NGS-based analysis of virion DNA. (a) The figure shows an 80 nucleotide-long sequence derived from wildtype (AYW), 3D-PCR (H23D), and NGS (H25a) from virus produced by HepG2.2.15 cells (see text). H25a refers to selected reads obtained with NGS. (b) The graph shows the frequency of occurrence of zero to five G:A transitions in the region shown in panel a.
Our results thus supported our hypothesis that 3D-PCR yields DNA with significantly higher G:A transitions than are present in input DNA, presumably as a consequence of DNA recombination during the PCR reaction (see discussion). They also demonstrated that G:A editing of virion DNA occurs in at least two HepG2-derived HBV producer cell lines.
APOBEC editing in the PAM sequence abrogates Cas9 editing
Having established that input HBV DNA is edited, we thought to determine whether G:A transitions in the PAM sequence of HBx2 would interfere with Cas9 cleavage. We also reasoned that if Cas9-mediated cleavage could be observed in conjunction with G:A transitions in the PAM sequence, it could provide evidence that cccDNA were the target of CRISPR/Cas9 as it has been concluded previously.7 We analyzed sequence reads obtained from PCR1 of control and IFNα treated HepG2 cells described above (Figure 5). Specifically, we searched for sequences with at least three G:A transitions in the 5′-GGGG-3 motif at the 3′ end of the HBx2 sgRNA target sequence (Figure 7a). We identified 26 and 16 reads, respectively, none of which exhibited the characteristic one nucleotide insertion or deletion at position -4 (Figure 7b). We estimated that about 23 (out of 26) or 14 (out of 16) PAM-site mutations would have Cas9-mediated mutations, provided that cccDNA were the sole target for G:A editing. This estimate is based on our observation that almost 90% of HBx2 target sites exhibited Cas9 mediated mutations. Hence, our results suggested that G:A transitions occurred prior to cccDNA formation, consistent with our previous results, revealing that intracellular rcDNA and virion DNA already contain G:A transitions.
Figure 7.

APOBEC editing and mutations in the PAM sequence. (a) The figure shows the nucleotide sequence used to search for DNA reads with HBx2 mediated 1 nt insertions or deletions and G:A transitions in the PAM sequence (capitalized G residues, see text for details). (b) The figure shows the results of the analysis obtained with reads from HBx2 and HBx2 and IFNα treated cells described in the legend to Figure 5a.
Discussion
The results from this study described for the first time the complete repertoire of cccDNA genomes present in a liver-derived cell line expressing the CRISPR/Cas9 system targeting a locus on HBV DNA. An important finding is that approximately 7% of cleaved cccDNA genomes are repaired in a fashion that yields in frame mutants that might not abrogate function of the targeted gene, in our case HBx (Figure 1d). Hence, multiple sgRNAs targeting different loci on the HBV genome might be required to inactivate cccDNA. Assuming that 10% of hepatocytes in a liver are infected (ca. 5 × 1010 hepatocytes) each carrying four copies of cccDNA, at least 10 loci would have to be targeted to inactivate each cccDNA molecule (2 × 1011 × 0.0710 = 0.56). Therefore, a more practical approach would be to target a sequence motif on the HBV genome known to be required for gene function, such as the active site of the DNA polymerase or RNaseH domains of the pol gene. The terminal protein region of the pol gene where tyrosine 68 is known to be essential for protein priming would be another possibility.21 It is also conceivable that in frame deletions in HBx as observed in our study might hamper the function of this protein to permit efficient transcription from cccDNA,3,13,15 a possibility we have not not yet examined.
While CRISPR/Cas9 is the most efficient method known to date for inactivation of genes, it requires therapeutic administration of at least two components enabling expression of Cas9 and sgRNA in the same cell.16,22 In contrast, editing and inactivation of genes through activation of innate immune pathways could, in theory, be accomplished through oral administration of small molecules. In this regard, activation of APOBEC proteins has been proposed as a potential method to inactivate and even destroy cccDNA.7,23 We have directly compared APOBEC editing of HBV DNA with CRISPR/Cas9 cleavage and found that it is very inefficient compared with CRISPR/Cas9. The results showed that the frequency of cytosine deamination is at least 15,000- to 100,000-fold lower than observed with Cas9. Importantly, they revealed that individual HBV DNA genomes carry only a small number of G:A transitions, which are in most, if not all, cases already present on input virion DNA. Importantly, we found that the 3D-PCR method, often used to measure APOBEC-mediated editing of HBV genomes, yields erroneous results that greatly exaggerate the extent of editing. Hence, our study revealed that 3D-PCR is an unreliable predictor for both, the frequency and in particular the extent of cytosine deamination (Figures 456). The reason lies in the method that selects for amplification of DNA strands with a reduced Tm and hence, high A:T content. The resulting artifact is most likely caused by re-annealing and extension of incomplete PCR products following the denaturation step, leading to a stepwise increase in the number of chimeric products with increasing A:T content. In vitro chimera formation during PCR reactions has been observed previously when input DNA consisted of two or more alleles of the gene targeted for PCR amplification.24,25,26 A curious observation derived from our study is that IFNα treatment did increase the yield of DNA products from the 3D-PCR reaction (Figures 3b and 5a), although NGS did not reveal any significant difference in G:A hypermutations between control and IFNα treated samples (Figure 5b). Perhaps very small differences in the A:T content within the population of input DNA for 3D-PCR accounts for this difference.
A limitation of our study is that it was performed in HepG2 cells, which might not adequately mimic conditions in hepatocytes in the liver. While information about the activity of CRISPR/Cas9 in humans is not yet available, considering that the system depends only on two components, Cas9 and sgRNA and then on the endogenous NHEJ pathway, we believe that our observations demonstrating efficient cleavage of cccDNA are valid also under conditions in patients with CHB. As indicated above, this conclusion is based on the premise that Cas9 and sgRNA can be delivered efficiently to the infected liver. Similarly, it is possible that HepG2 cells, or other hepatocyte cultures do not adequately reproduce conditions for APOBEC-mediated deamination of HBV DNA in infected hepatocytes. However, our results showed that IFNα as well as IFNγ induce STAT1 phosphorylation and translocation into the nucleus, evidence for activation of an innate immune response (Figure 2b,c). In addition, we observed a cytotoxic effect of IFNα when added within 2 days of HBV infections. While the reason for this toxicity is not known, the observation provided further evidence for activation of an innate immune response in HepG2 cells by the cytokine. Finally, we found that APOBEC3G was induced by IFNα as reported previously with HepG2 cells, primary hepatocyte cultures and even in vivo (Supplementary Figure S1).19 In spite of these reservations, it is notable that our results correlate well with reports from in vivo studies. For example, hypermutated genomes could be recovered from some sera of chronically infected HBV patients at a frequency of ca. 10−4, suggesting that deaminated HBV DNA is produced in a very small fraction of HBV-infected hepatocytes.27 A more precise estimate for the fraction of G to A edited HBV genomes was derived from an NGS analysis of virion DNA obtained from CHB patients.28 Median hypermutation frequency in virion DNA from HBeAg-positive patients ranged from 0 to 10−4 depending on the 100 nucleotide long segment analyzed. Notably, editing occurred preferentially in the region of the HBV genome that is single stranded, consistent with the known substrate preference of APOBEC proteins and the prevailing view consistent with our observations that editing occurs on rc, rather than cccDNA.29 Much higher frequencies of editing have so far only been reported in viral DNA from patients with advanced cirrhosis, a condition marked by high cytokine levels.30 Hence deamination of HBV DNA is generally a rare event in infected patients with minimal or no consequences for viral persistence. For this reason it is not surprising that HBV does not encode a protein that inhibits APOBEC activity, like HIV. The latter encodes Vif that causes degradation of APOBEC proteins by the proteasome.31,32 Moreover, all evidence points to a mechanism where deamination of HBV DNA occurs during minus strand DNA synthesis in core particles present in the cytoplasm of infected cells. Our study is in agreement with this consensus because we could not find any evidence for editing of cccDNA following Cas9 cleavage (Figure 7). Lastly, although the Hirt procedure for DNA isolation and the selection of primers flanking the cohesive overlap region were used for PCR amplification of cccDNA, rather than rcDNA (PCR1, Figure 3a), we are aware of the fact that some rcDNA could still have been co-amplified with cccDNA. Hence, given the low frequency of the observed G:A transitions observed with DNA extracted from HBV infected HepG2/NTCP, it is plausible that these mutations were derived from PCR amplified rcDNA, not cccDNA.
Even if editing would occur to some extent on cccDNA, given the low number of deaminated residues present on cccDNA, it is almost certain that removal of the uracil base by uracil-DNA glycosylase is followed by a DNA repair mechanism, not destruction of cccDNA, essentially as observed with chromosomal DNA.8 The present and our recent study demonstrated that cccDNA is efficiently repaired following Cas9 cleavage by the DNA repair machinery, like chromosomal DNA.3 In other words, there is no solid evidence to date for selective recognition of cccDNA by cellular DNA modifying enzymes that would destroy it. Thus, more than 15 years after the proposal that cccDNA could be cleared from infected hepatocytes by a cytokine-mediated non-cytolytic event,4 we still have no insights into a possible mechanism that could explain such an event. For this reason, it appears that exogenous nucleases, such as Cas9, are the sole means known to date to functionally inactivate cccDNA and hopefully cure CHB.
Materials and Methods
Cells and virus infections. HepG2/NTCP cells were produced by infection of HepG2 cells with lentivirus NT-GFP as described earlier3; GFP expressing cells were enriched with the help of FACS. NTCP/Cas9 cells were derived from HepG2/NTCP cells following infection with lentivirus vector pCW-Cas9 (refs. 3,33) and selection with puromycin (1 μg/ml) as described previously.3 Infection of HepG2 cells with HBV occurred in the presence of 4% polyethylene glycol 8000 in complete serum-free medium (DMEM/F12, pyruvate, non-essential amino acids, and penicillin/streptomycin) containing 2% DMSO with an estimated 50 genome equivalents per cell. Medium was replaced with complete medium containing 10% fetal calf serum and 2% DMSO 24 hours after infection. HBV was concentrated 100-fold from the culture medium of HepAD38 or 2.2.15 cells with 6% polyethylene glycol 800 and resuspended in serum-free OptiMEM medium (GIBCO, Waltham, MA). HBcAg antibody C1-5 was obtained from Santa Cruz Biotechnology (Dallas, TX). STAT1 and pSTAT1 antibodies were purchased Bethyl Laboratories (Montgomery, TX) and Cell Signaling Technology Danvers, MA) respectively.
Vectors. HBx2 and HBx4 sgRNAs were cloned into lentivirus vector pLX-SG1 as described previously in Seeger and Sohn.3
Isolation of cccDNA and virion rcDNA. HBV cccDNA was isolated from infected cells by the Hirt procedure.10 Briefly, cells from a well of a 6-well plate were lysed in 1.0 ml of SDS lysis buffer (50 mmol/l Tris-HCl pH7.5, 10 mmol/l EDTA, 0.5% SDS) for 10 minutes at 37 °C. Two hundred μl of a solution of 2.5 mol/l KCl (0.5.M KCl final) was added and incubated at room temperature for 30 minutes to precipitate proteins and chromosomal DNA. After centrifugation at 10,000× g for 10 minutes, the Hirt supernatant was extracted twice with phenol, and once with butanol:isopropanol (7:3). DNA was precipitated with two volumes of ethanol at room temperature for 2 hours and resuspended in 30 μl Tris-EDTA (10:1). HBV DNA was isolated from concentrated virus produced in HepAD38 (ref. 11)and HepG2-2.2.15 cells12 essentially as described by Yang et al.34 for cytoplasmic rcDNA. Briefly, virus particles were lysed in 0.5-ml core lysis buffer (50 mmol/l Tri-HCl pH 7.5, 1 mmol/l EDTA, 1% NP40) for 10 minutes on ice. After removal of cell debris, the cleared supernatant was incubated in a buffer containing 0.5% SDS, 10 mmol/l EDTA, 100 mmol/l NaCl, and 400 μg/ml pronase for 1 hour. at 37 °C. DNA was extracted with phenol and butanol:isopropanol and then precipitated with ethanol at −20 °C for 2 hours. After centrifugation, the pellet containing virion rcDNA was resuspended in Tris-EDTA (10:1).
Immunofluorescence. For immunostaining, cells were fixed in 96-well plates with 4% paraformaldehyde for 10 minutes and processed for immunofluorescence with HBcAg monoclonal antibody C1-5 (Santa Cruz Biotechnology (Dallas, TX). The fraction of HBcAg positive cells was determined with an ImageXpress Micro automated microscope (Molecular Devices, Sunnyvale, CA). Images from 16 preset positions at ×10 magnification with two channels (DAPI, Cy5) were collected from each 96 well. Images were analyzed with MetaXpress imaging and analysis software using the multi-wavelength cell scoring module.
PCR reactions. Primers for PCR1 were 5′-2338-GCCTATTGATTGGAAAGTATGT-2359-3 and 5′-195-AGCTGAGGCGGTATCTA-179-3′, for vPCR1 5′-2743-ATGGCTGCTARGCTGTGCTGCCAA-2766-3′ and 5′-15-GTGAAAAAGTTGCATGGTGCTG-3176-3′; for 3D-PCR 5′-2743-ATGGCTGCTARGCTGTGCTGCCAA-2766-3′ and 5′-2955-AAGTGCACACGGTYYGGCAGAT-2934-3′. To increase the efficiency of the PCR reaction, the isolated cccDNA was linearized with EcoRI.
Statistical analysis. Unpaired, one-tailed t-tests (Prism 5) were used for statistical analyses of the results shown in Figures 1c and 2d.
SUPPLEMENTARY MATERIAL Figure S1. Induction of APOBEC genes following IFNα treatment of HepG2 cells.
Acknowledgments
We acknowledge the help of Yan Zhou in processing the NGS data. We thank Jianming Hu and William Mason for helpful comments to the manuscript. We acknowledge services provided by the following FCCC facilities: High throughput, Imaging, DNA sequencing, Genomics and Statistics. C.S. acknowledges support from NIH grants AI103514 and AI119433 and the Commonwealth of Pennsylvania.
Supplementary Material
References
- Seeger, C and Mason, WS (2000). Hepatitis B virus biology. Microbiol Mol Biol Rev 64: 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger, C and Mason, WS (2015). Molecular biology of hepatitis B virus infection. Virology 479–480: 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger, C and Sohn, JA (2014). Targeting Hepatitis B Virus With CRISPR/Cas9. Mol Ther Nucleic Acids 3: e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti, LG, Rochford, R, Chung, J, Shapiro, M, Purcell, R and Chisari, FV (1999). Viral clearance without destruction of infected cells during acute HBV infection. Science 284: 825–829. [DOI] [PubMed] [Google Scholar]
- Murray, JM, Wieland, SF, Purcell, RH and Chisari, FV (2005). Dynamics of hepatitis B virus clearance in chimpanzees. Proc Natl Acad Sci USA 102: 17780–17785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, WS, Xu, C, Low, HC, Saputelli, J, Aldrich, CE, Scougall, C et al. (2009). The amount of hepatocyte turnover that occurred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J Virol 83: 1778–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora, J, Xia, Y, Reisinger, F, Zhang, K, Stadler, D, Cheng, X et al. (2014). Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343: 1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama, T and Wilson, DM 3rd (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12: 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari, FV, Mason, WS and Seeger, C (2014). Virology. Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science 344: 1237. [DOI] [PubMed] [Google Scholar]
- Hirt, B (1967). Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26: 365–369. [DOI] [PubMed] [Google Scholar]
- Ladner, SK, Otto, MJ, Barker, CS, Zaifert, K, Wang, GH, Guo, JT et al. (1997). Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41: 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells, MA, Chen, ML and Acs, G (1987). Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA 84: 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora, J, Arzberger, S, Durantel, D, Belloni, L, Strubin, M, Levrero, M et al. (2011). Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 55: 996–1003. [DOI] [PubMed] [Google Scholar]
- Belloni, L, Pollicino, T, De Nicola, F, Guerrieri, F, Raffa, G, Fanciulli, M et al. (2009). Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA 106: 19975–19979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breugel, PC, Robert, EI, Mueller, H, Decorsière, A, Zoulim, F, Hantz, O et al. (2012). Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology 56: 2116–2124. [DOI] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, W and Hu, J (2007). Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81: 6164–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H, Jiang, D, Zhou, T, Cuconati, A, Block, TM and Guo, JT (2007). Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81: 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y, Marusawa, H, Seno, H, Matsumoto, Y, Ueda, Y, Kodama, Y et al. (2006). Anti-viral protein APOBEC3G is induced by interferon-alpha stimulation in human hepatocytes. Biochem Biophys Res Commun 341: 314–319. [DOI] [PubMed] [Google Scholar]
- Suspène, R, Henry, M, Guillot, S, Wain-Hobson, S and Vartanian, JP (2005). Recovery of APOBEC3-edited human immunodeficiency virus G->A hypermutants by differential DNA denaturation PCR. J Gen Virol 86: 125–129. [DOI] [PubMed] [Google Scholar]
- Zoulim, F and Seeger, C (1994). Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol 68: 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA and Charpentier, E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Y, Stadler, D, Lucifora, J, Reisinger, F, Webb, D, Hösel, M et al. (2016). Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 150: 194–205. [DOI] [PubMed] [Google Scholar]
- Judo, MS, Wedel, AB and Wilson, C (1998). Stimulation and suppression of PCR-mediated recombination. Nucleic Acids Res 26: 1819–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhans, A, Vartanian, JP and Wain-Hobson, S (1990). DNA recombination during PCR. Nucleic Acids Res 18: 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn, R, Cedroni, M, Haselkorn, T, Grover, C and Wendel, JF (2002). PCR-mediated recombination in amplification products derived from polyploid cotton. Theor Appl Genet 104: 482–489. [DOI] [PubMed] [Google Scholar]
- Suspène, R, Guétard, D, Henry, M, Sommer, P, Wain-Hobson, S and Vartanian, JP (2005). Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA 102: 8321–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggel, B, Münk, C, Däumer, M, Hauck, K, Häussinger, D, Lengauer, T et al. (2013). Full genome ultra-deep pyrosequencing associates G-to-A hypermutation of the hepatitis B virus genome with the natural progression of hepatitis B. J Viral Hepat 20: 882–889. [DOI] [PubMed] [Google Scholar]
- Harris, RS and Dudley, JP (2015). APOBECs and virus restriction. Virology 479–480: 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian, JP, Henry, M, Marchio, A, Suspène, R, Aynaud, MM, Guétard, D et al. (2010). Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog 6: e1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy, AM, Gaddis, NC and Malim, MH (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9: 1404–1407. [DOI] [PubMed] [Google Scholar]
- Marin, M, Rose, KM, Kozak, SL and Kabat, D (2003). HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9: 1398–1403. [DOI] [PubMed] [Google Scholar]
- Wang, T, Wei, JJ, Sabatini, DM and Lander, ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W, Mason, WS and Summers, J (1996). Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J Virol 70: 4567–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Zoulim F, Mason WS (2013). Hepadnaviruses. In: Knipe, DM, Howley, P, (eds.). Fields VirologyLippincott, Williams and Wilkins: Philadelphia. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.