

Abstract
NAD+ is required not only for life but for a long life. In this issue, Camacho-Pereira et al. (2016) implicate CD38 in the decline of NAD+ during aging, with implications for combating age-related diseases.
The discovery of nicotinamide adenine dinucleotide (NAD+) as a “cozymase” factor in fermentation has its 110th anniversary this year. Of the two billion people who were alive back in 1906, only 150 people remain. Interestingly, NAD+ itself may be the reason for their longevity. In this issue, Eduardo Chini and colleagues address an open question in biogerontology: why do NAD+ levels fall as we age? They show that the major culprit is an NADase called CD38 whose levels rise during aging. Their results also add to the body of evidence indicating that loss of SIRT3 activity in mitochondria is a cause of age-related metabolic decline (Camacho-Pereira et al., 2016) (Figure 1).
Figure 1. CD38 Regulates Metabolism during Aging through Modulating NAD+ Levels and SIRT3 Activity.
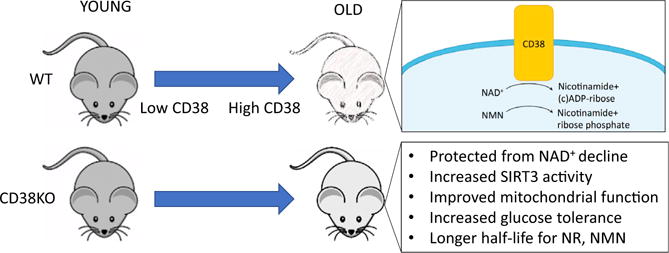
CD38 is a membrane-bound NADase that hydrolyzes NAD+ to nicotinamide and (cyclic-)ADP-ribose. Its protein levels increase during aging, with a corresponding increase in NADase activity and declining NAD+ levels. Mice deficient for CD38 are protected from mitochondrial dysfuntion and diabetes during aging. Many of these effects are mediated through the mitochondrial sirtuin SIRT3. As CD38 also degrades NMN, the CD38 knockout mice also are more sensitive to treatment with NAD+ precursors.
NAD+, and its reduced form NADH, are best known for their roles as coenzymes in redox reactions, linking the catabolic reactions of glycolysis and the TCA cycle to oxidative phosphorylation. In the last two decades, however, another role for NAD+ has been uncovered. Perhaps equally as important (and ancient) is NAD’s role as a signaling molecule. From plants to metazoans, an increase in intracellular levels of NAD+ directs cells to make adjustments to ensure survival, including increasing energy production and utilization, boosting cellular repair, and coordinating circadian rhythms.
NAD+ levels are converted to signals by various enzymes that have evolved to sense NAD+, including the sirtuin deacylases (SIRT1–SIRT7), CtBPs, and poly-ADP-ribose polymerases (PARPs). They can sense NAD+ fluctuations because, unlike the enzymes of glycolysis and the TCA cycle, their dissociation constants for NAD+ are near physiological concentrations. Unfortunately, NAD+ levels steadily decline during aging. By the time a mouse or human is middle aged, levels of NAD+ have fallen to half of youthful levels, with resulting loss of sirtuin and PARP activity. Several studies in recent years have shown that treatment of old mice with PARP inhibitors or precursors to NAD+ can greatly improve health. Observed effects include increased insulin sensitivity, reversal of mitochondrial dysfunction, reduced stem cell senescence, and extension of lifespan (Bai et al., 2011; Gomes et al., 2013; Yoshino et al., 2011; Zhang et al., 2016).
A major question that has remained unanswered is why NAD+ levels decline in the first place. One suggestion was that the synthesis of NAD+ declines with age. Indeed, overexpression of the NAD+ biosynthetic genes NPT1 and PNC1 in yeast extends lifespan by over 50% (Anderson et al., 2002). Other possibilities include increased degradation of NAD+ by hydrolysis or increased NAD+ polymerization to generate poly-ADP-ribose (PAR). Sirtuins degrade NAD+ via a deacylation reaction, but only in a limited fashion. Realistic candidates are the PARPs and the two NADases, CD38 and BST1.
Which brings us to CD38. CD38 is a membrane-bound hydrolase that has been implicated in immune responses and energy metabolism. Mice lacking CD38 or treated with the CD38 inhibitor apigenin have elevated levels of NAD+ and are protected against deleterious effects of a high-fat diet (Escande et al., 2013). In this latest study, the authors show that protein levels of CD38, but not of SIRT1 or PARP1, increased in multiple tissues during aging, along with a corresponding increase in CD38 enzymatic activity (Camacho-Pereira et al., 2016). At 32 months, wild-type mice had about half the NAD+ of a young mouse, but the CD38 knockout showed no decrease.
Mitochondrial dysfunction is a hallmark of metabolic decline during aging. Chini and colleagues found that cells overexpressing CD38 consume less oxygen, have increased lactate levels, and possess irregular mitochondria (Camacho-Pereira et al., 2016). Conversely, mitochondria from the livers of CD38 knockout mice consumed more oxygen and had greater mitochondrial membrane potential. Mitochondrial protein acetylation was decreased in the CD38 knockout mice, indicating that a mitochondrial sirtuin might be involved. Consistent with this, the knockout of SIRT3 in the CD38 knockout background abolished the protective effects of the CD38 knockout alone on mitochondrial respiration and glucose tolerance.
Finally, the authors addressed how CD38 may affect therapies designed to raise NAD+ levels. Currently, the favored approach in mouse and humans is to treat with NAD+ precursors, such as nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN). Interestingly, CD38 not only degrades NAD+ in vivo, but also NMN. When CD38 knockout mice were given injections of NAD+, NMN, or NR (which is converted to NMN), circulating levels of NAD metabolites remained stable after 150 min, long after they began to fall in the wild-type animals. Furthermore, when compared to the wild-type, CD38 knockout mice on a high-fat diet exhibited a much larger improvement in glucose tolerance when given NR. These findings suggest that the efficacy of NAD+ precursors may be enhanced by co-supplementation with CD38 inhibitors, which have been recently identified (Escande et al., 2013; Haffner et al., 2015).
As with all good studies, numerous questions have been raised. It is well established that there are relatively independent pools of NAD+ in the nucleus, cytosol, and mitochondria (Yang et al., 2007). Given that NADase activity has been detected on the plasma membrane and in mitochondria, it will be important to determine how NAD+ levels change in different compartments during aging and how CD38 affects them. Also, what role, if any, does the CD38 homolog BST1, which itself has NADase activity, play in regulating NAD+ levels? And at a more basic level, why do cells have an enzyme to destroy NAD+ and what triggers it to increase during aging? In light of the fact that CD38 expression is regulated by NF-κB (Tirumurugaan et al., 2008) and low-grade inflammation is a characteristic of aging, a possibility is that inflammation is the answer. Either way, the identification of molecules that safely maintain NAD+ levels in humans cannot come soon enough for patients who could benefit and those who hope to celebrate many more anniversaries.
Footnotes
CONFLICTS OF INTEREST
D.A.S. is a consultant and/or inventor on patents licensed to GlaxoSmithKline, Ovascience, Metrobiotech, Caudalie, and Liberty Biosecurity.
References
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, Manchester JK, Gordon JI, Sinclair DA. J Biol Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EM. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, Chini EN. Diabetes. 2013;62:1084–1093. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JSS, Wrann CD, Hubbard BP, et al. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, et al. J Med Chem. 2015;58:3548–3571. doi: 10.1021/jm502009h. [DOI] [PubMed] [Google Scholar]
- Tirumurugaan KG, Kang BN, Panettieri RA, Foster DN, Walseth TF, Kannan MS. Respir Res. 2008;9:26. doi: 10.1186/1465-9921-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. Science. 2016;26:2016. doi: 10.1126/science.aaf2693. Published online April. http://dx.doi.org/10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]