

Abstract
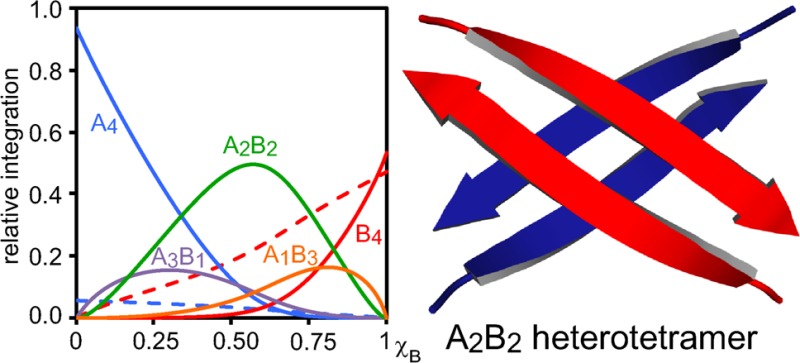
In this paper, we investigate the coassembly of peptides derived from the central and C-terminal regions of the β-amyloid peptide (Aβ). In the preceding paper, J. Am. Chem. Soc.2016, DOI: 10.1021/jacs.6b06000, we established that peptides containing residues 17–23 (LVFFAED) from the central region of Aβ and residues 30–36 (AIIGLMV) from the C-terminal region of Aβ assemble to form homotetramers consisting of two hydrogen-bonded dimers. Here, we mix these tetramer-forming peptides and determine how they coassemble. Incorporation of a single 15N isotopic label into each peptide provides a spectroscopic probe with which to elucidate the coassembly of the peptides by 1H,15N HSQC. Job’s method of continuous variation and nonlinear least-squares fitting reveal that the peptides form a mixture of heterotetramers in 3:1, 2:2, and 1:3 stoichiometries, in addition to the homotetramers. These studies also establish the relative stability of each tetramer and show that the 2:2 heterotetramer predominates. 15N-Edited NOESY shows the 2:2 heterotetramer comprises two different homodimers, rather than two heterodimers. The peptides within the heterotetramer segregate in forming the homodimer subunits, but the two homodimers coassemble in forming the heterotetramer. These studies show that the central and C-terminal regions of Aβ can preferentially segregate within β-sheets and that the resulting segregated β-sheets can further coassemble.
Introduction
Interactions among β-sheets are critical in the aggregation of the β-amyloid peptide (Aβ) to form oligomers and fibrils in Alzheimer’s disease.1 Two regions of the 40- or 42-residue peptide adopt β-sheet structure and promote aggregation: the central region and the C-terminal region.2 The central region comprises the hydrophobic pentapeptide LVFFA (Aβ17–23), and the C-terminal region comprises the hydrophobic undecapeptide AIIGLMVGGVV (Aβ30–40) or the hydrophobic tridecapeptide AIIGLMVGGVVIA (Aβ30–42).
Elucidating the roles of the central and C-terminal regions of Aβ is critical to understanding Aβ aggregation. These two regions assemble differently in the fibrils and in the toxic oligomers that cause synaptic dysfunction and cell death. In Aβ1–40 fibrils, the peptide forms parallel β-sheets, with the central and C-terminal regions laminated together.3,4 In the oligomers, the peptide is thought to form β-hairpins comprising antiparallel β-sheets.5
In
the preceding paper,6 we incorporated
residues from the central and C-terminal regions into macrocyclic
β-sheet peptides 1, and we determined how the peptides
assembled in aqueous solution.6 Peptides 1 consist of a heptapeptide strand, a template strand containing
the unnatural amino acid Hao, and two δ-linked ornithine turn
units.7−9 We incorporated residues LVFFAED (Aβ17–23) and residues AIIGLMV (Aβ30–36) into the
heptapeptide strands of peptides 1a and 1b, respectively. We incorporated isoleucine residues (I8 and I11) into the template strand to promote assembly
and lysine residues (K9 and K10) to maintain
solubility. 1H NMR studies of peptides 1a and 1b show that the peptides assemble to form sandwich-like homotetramers,
consisting of two hydrogen-bonded dimers.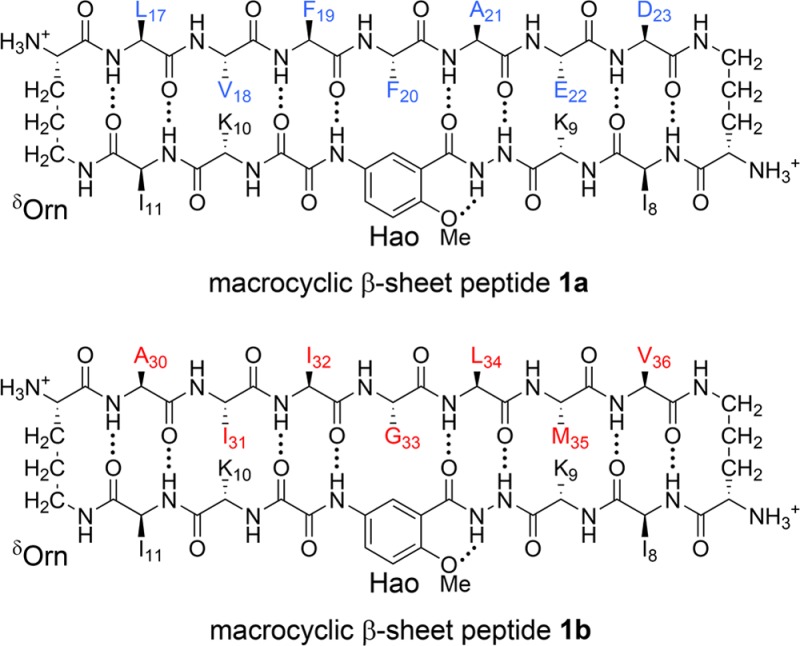
Incorporation of a single isotopic label into peptides 1 facilitated the identification and quantification of the tetramers. The peptides [15N]1a and [15N]1b each contain a single 15N-labeled amino acid in the center of the heptapeptide strand. Peptide [15N]1a contains an 15N label in the F20 residue; peptide [15N]1b contains an 15N label in the G33 residue. 1H,15N HSQC studies show only the resonances associated with the 15N label, reducing each spectrum to two crosspeaks: The 1H,15N HSQC spectrum of peptide [15N]1a shows one crosspeak associated with the monomer and another associated with the homotetramer. The 1H,15N HSQC spectrum of peptide [15N]1b also shows one crosspeak associated with the monomer and another associated with the homotetramer.
In this paper, we ask whether these peptides prefer to coassemble or to segregate.10 To address this question, we mix peptides 1a and 1b and characterize the oligomers that form. 1H NMR studies show that peptides 1a and 1b form a mixture of homotetramers and heterotetramers, but the 1H NMR spectrum of the mixture is largely indecipherable. To characterize the complex mixture of homotetramers and heterotetramers, we use the 15N-labeled peptides [15N]1a and [15N]1b and 1H,15N NMR spectroscopy. 1H,15N HSQC, in conjunction with Job’s method of continuous variation, reveals that the peptides form three heterotetramers in 3:1, 2:2, and 1:3 stoichiometries, in addition to the two homotetramers. The following describes the characterization of these five tetramers and the equilibria among them.
Results and Discussion
Peptides 1a and 1b Coassemble upon Mixing
The 1H NMR spectrum of pure peptide 1a at 8.0 mM predominately shows the homotetramer; the 1H NMR spectrum of pure peptide 1b at 8.0 mM shows the monomer and the homotetramer. In a 1:1 mixture of peptides 1a and 1b at 8.0 mM total concentration, the 1H NMR spectrum shows many new resonances: The resonances from the homotetramer of peptide 1a diminish greatly and the resonances from the homotetramer of peptide 1b nearly disappear. New resonances appear in the spectrum in the aromatic region between 6 and 9 ppm and also in the methyl region below 1 ppm. Several new Hao methoxy (HaoOMe) resonances appear between 4 and 4.5 ppm. The HaoOMe resonance from the homotetramer of peptide 1a diminishes greatly and the HaoOMe resonance from the homotetramer of peptide 1b almost completely disappears. The multitude of new resonances in the spectrum of the 1:1 mixture suggests that several new oligomers form, rather than just one. Figure 1 shows the 1H NMR spectra of pure 1a, pure 1b, and the 1:1 mixture.
Figure 1.

1H NMR spectra of (a) peptide 1a at 8.0 mM, (b) peptide 1b at 8.0 mM, and (c) the 1:1 mixture of peptides 1a and 1b at 8.0 mM total concentration in D2O at 600 MHz and 298 K. Dotted lines illustrate how the resonances from the 1:1 mixture compare with the resonances of pure 1a and pure 1b.
Peptides 1a and 1b Form Heterotetramers
We have previously shown that related macrocyclic β-sheets can assemble to form tetramers.11 In the preceding paper,6 we established that both peptide 1a and peptide 1b form tetramers by measuring the diffusion coefficients (D) with DOSY NMR.6 Here, we use DOSY NMR to determine whether the species that form upon mixing peptides 1a and 1b are also tetramers. The homotetramers of peptides 1a and 1b have diffusion coefficients of about 12 × 10–11 m2/s in D2O at 298 K. The diffusion coefficients of the species that predominate in the 1:1 mixture are comparable, 11.4 × 10–11 m2/s (Table 1), indicating that these species are also tetramers.
Table 1. Diffusion Coefficients (D) of Peptides 1a and 1b in D2O at 298 K.
| MWtetramera (Da) | conc (mM) | D (×10–11m2/s) | oligomer state | |
|---|---|---|---|---|
| 1a | 7068 | 8.0 | 11.8 ± 1.0 | A4 homotetramer |
| 1b | 6572 | 16.0 | 11.9 ± 1.1 | B4 homotetramer |
| 1a + 1b | 8.0b | 11.4 ± 1.1 | heterotetramers |
Molecular weight calculated for the neutral (uncharged) peptide.
Total concentration of the 1:1 mixture of peptides 1a and 1b.
In this paper, we describe the homotetramers and heterotetramers formed by peptides 1a and 1b using the letters A and B. The homotetramers are designated A4 and B4, and the 3:1, 2:2, and 1:3 heterotetramers are designated A3B1, A2B2, and A1B3. Two topological isomers of the A2B2 heterotetramer could form: one consisting of two homodimers (A·A and B·B); the other consisting of two heterodimers (A·B and A·B). Figure 2 illustrates the homotetramers and heterotetramers, where a single β-strand represents either peptide 1a or 1b.
Figure 2.

Cartoons illustrating homotetramers and heterotetramers, in which peptide 1a is represented by a blue arrow and peptide 1b is represented by a red arrow.
The complex mixture of monomers, homotetramers, and heterotetramers can give as many as 16 resonances in the 1H NMR spectrum: two from the A monomer and A4 homotetramer; two from the B monomer and B4 homotetramer, four from the A3B1 heterotetramer, four from the A1B3 heterotetramer, and either two or four from the A2B2 heterotetramer. The A2B2 heterotetramer would give four resonances if both the A·A/B·B and A·B/A·B topological isomers formed, but only two resonances if just one of the two isomers formed.
Elucidation of the A2B2 Topological Isomer
We used peptides [15N]1a and [15N]1b to elucidate the dimers within the A2B2 heterotetramer. In the preceding paper,6 we used these peptides and 15N-edited NOESY to help establish the pairing of the dimers within the A4 and B4 homotetramers.6 Here, we compare the 15N-edited NOESY spectra of pure [15N]1a and pure [15N]1b to that of the 1:1 mixture to determine which A2B2 topological isomer forms (Figure 3). The spectra show that the A2B2 heterotetramer consists of an A·A and a B·B homodimer, and not of two A·B heterodimers (Figure 4).
Figure 3.
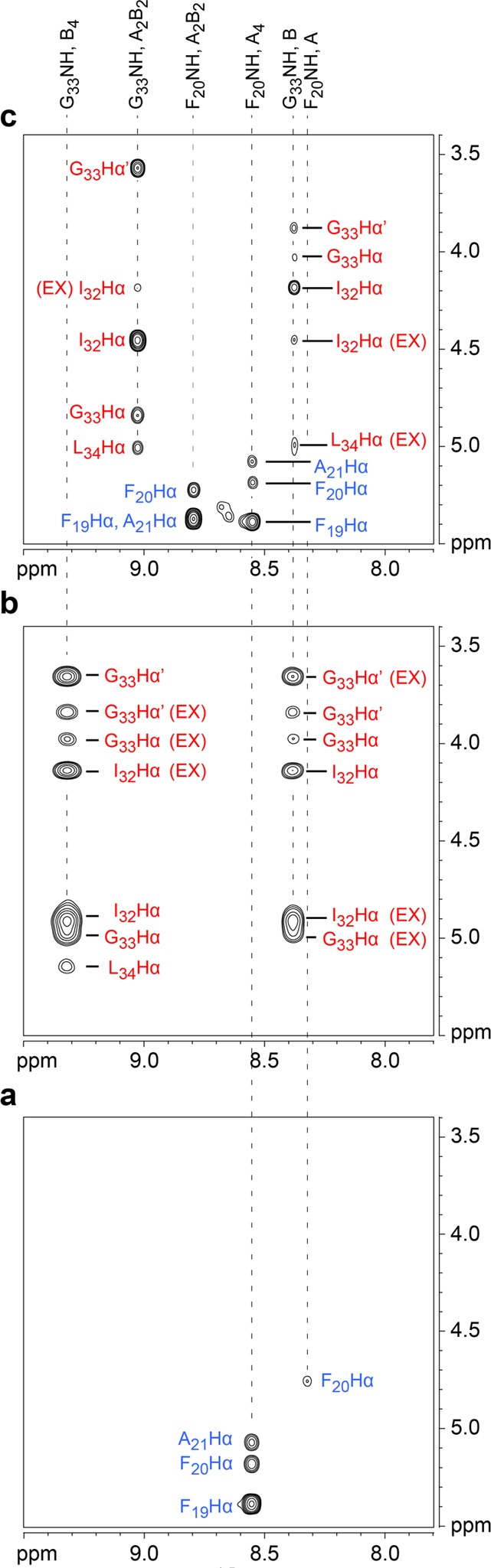
15N-Edited NOESY spectra of (a) peptide [15N]1a at 8.0 mM, (b) peptide [15N]1b at 8.0 mM, and (c) the 1:1 mixture of peptides [15N]1a and [15N]1b at 8.0 mM total concentration in 9:1 H2O/D2O at 600 MHz and 293 K. The G33Hα corresponds to the pro-R α-proton and the G33Hα′ corresponds to the pro-S α-proton. Crosspeaks associated with chemical exchange of peptide 1b between the monomer and the B4 and A2B2 tetramers are labeled EX. Dotted lines illustrate how the crosspeaks from the 1:1 mixture compare with the crosspeaks of pure [15N]1a and pure [15N]1b.
Figure 4.
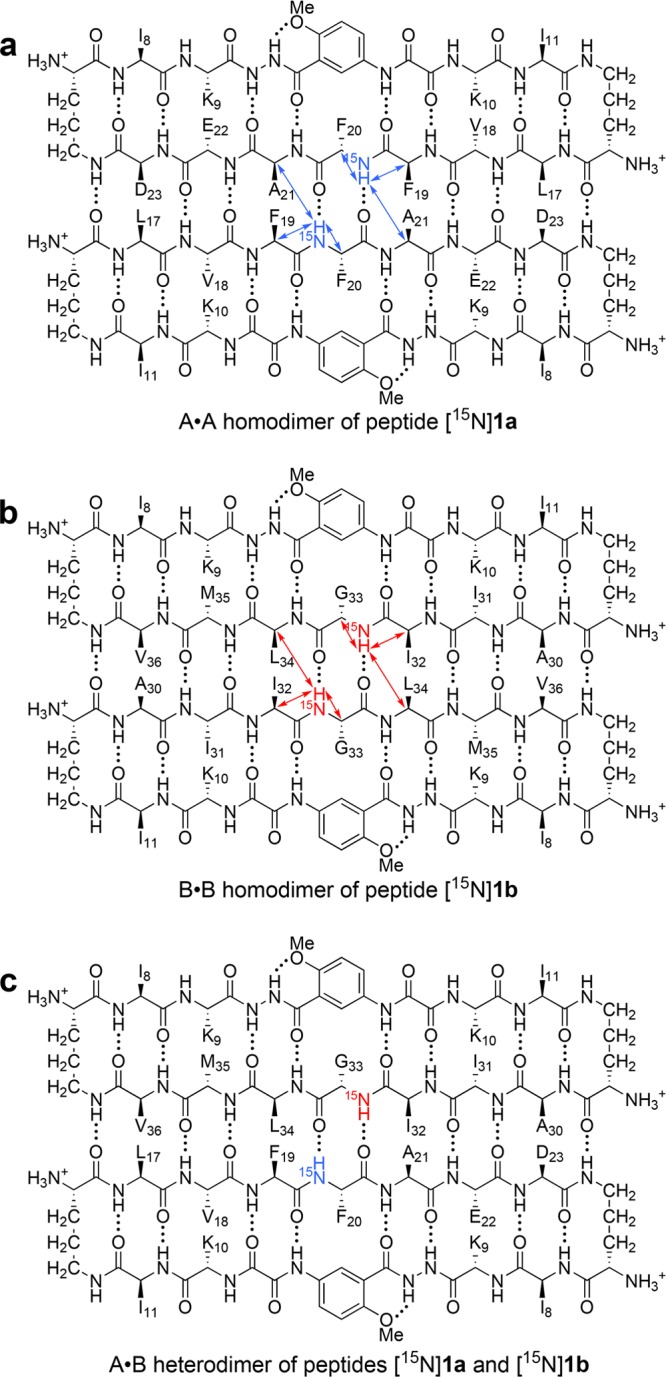
NOEs involving the 15NH protons within the A2B2 heterotetramer. (a) The A·A homodimer with blue arrows illustrating the NOEs observed within the dimers. (b) The B·B homodimer with red arrows illustrating the NOEs observed within the dimers. (c) The A·B heterodimer (not formed).
The 15N-edited NOESY spectrum of the 1:1 mixture of peptides [15N]1a and [15N]1b shows four distinct sets of resonances: two sets associated with the A2B2 heterotetramer; one set associated with the A4 homotetramer; and one set associated with the B monomer (Figure 3c). In addition to the NOEs, the spectrum also shows crosspeaks associated with chemical exchange between the monomer of peptide [15N]1b and the A2B2 heterotetramer.
The A2B2 heterotetramer gives two sets of resonances: one set from the F20NH proton of peptide [15N]1a and the other set from the G33NH proton of peptide [15N]1b. The F20NH proton of peptide [15N]1a gives a strong interresidue NOE to the F19Hα proton and a weaker intraresidue NOE to the F20Hα proton. Figure 4a summarizes these NOEs. An intermolecular NOE between the F20NH and the A21Hα protons is not observed as a separate crosspeak because the F20Hα and the A21Hα resonances overlap.12 The F20NH proton gives an additional NOE to the A21Hβ protons, which corroborates the proximity of these residues (Figure S2). An intermolecular NOE is not observed between the F20NH proton of peptide [15N]1a and the L34Hα proton of peptide [15N]1b (Figure 3c); an intermolecular NOE is also not observed between the F20NH proton of peptide [15N]1a and the G33NH proton of peptide [15N]1b (Figure S3). The absence of these two NOEs indicates that peptide [15N]1a is not part of an A·B heterodimer (Figure 4c).
The G33NH proton of peptide [15N]1b gives an interresidue NOE to the I32Hα proton and intraresidue NOEs to the G33Hα and G33Hα′ protons (Figure 3c). The G33NH proton also gives an intermolecular NOE to the L34Hα proton.12 This NOE confirms that the B·B homodimer forms within the A2B2 heterotetramer and rules out the A·B heterodimer. Figure 4b summarizes these NOEs.13 Collectively, the 15N-edited NOESY studies establish that the A·A/B·B topological isomer that forms exclusively is the A2B2 heterotetramer.
1H,15N HSQC Reveals That Peptides [15N]1a and [15N]1b Form Three Heterotetramers: A3B1, A2B2, and A1B3
We compared the 1H,15N HSQC spectra of pure [15N]1a and pure [15N]1b to that of the 1:1 mixture to show which crosspeaks are associated with heterotetramers.6 The 1H,15N HSQC spectrum of the 1:1 mixture of peptides [15N]1a and [15N]1b at 8.0 mM total concentration shows 10 new crosspeaks (14 crosspeaks in total). The crosspeaks are sharp and distinct, indicating that the tetramers exchange slowly on the NMR time scale. The two crosspeaks designated 1 and 2 come from the monomer and homotetramer of peptide [15N]1a; the two crosspeaks designated 3 and 4 come from the monomer and homotetramer of peptide [15N]1b. The 10 remaining crosspeaks designated 5–14 come from the heterotetramers. Figure 5a shows the 1H,15N HSQC spectrum of the 1:1 mixture of peptides [15N]1a and [15N]1b. Table 2 summarizes the chemical shifts of crosspeaks 1–14.
Figure 5.

1H,15N HSQC spectra of 8.0 mM mixtures in 9:1 H2O/D2O at 600 MHz and 293 K of peptides: (a) [15N]1a and [15N]1b; (b) [15N]1a and 1b; (c) 1a and [15N]1b. The asterisk (*) indicates a crosspeak from a minor unidentified species associated with peptide [15N]1b.
Table 2. Chemical Shifts of Peptides [15N]1a and [15N]1b.
| δ
F20 |
δ G33 |
||||
|---|---|---|---|---|---|
| crosspeak | 1H | 15N | 1H | 15N | species |
| 1 | 8.32 | 122.3 | A monomer | ||
| 2 | 8.56 | 121.3 | A4 homotetramer | ||
| 3 | 8.39 | 112.5 | B monomer | ||
| 4 | 9.33 | 115.8 | B4 homotetramer | ||
| 5 | 8.81 | 124.9 | A2B2 heterotetramer | ||
| 6 | 9.03 | 116.2 | A2B2 heterotetramer | ||
| 7 | 8.69 | 125.7 | A3B1 heterotetramer | ||
| 8 | 8.66 | 121.1 | A3B1 heterotetramer | ||
| 9 | 8.60 | 119.3 | A3B1 heterotetramer | ||
| 10 | 7.94 | 112.8 | A3B1 heterotetramer | ||
| 11 | 8.92 | 120.9 | A1B3 heterotetramer | ||
| 12 | 8.74 | 116.1 | A1B3 heterotetramer | ||
| 13 | 9.25 | 115.8 | A1B3 heterotetramer | ||
| 14 | 9.17 | 115.1 | A1B3 heterotetramer | ||
1H,15N HSQC spectrum was recorded for the 1:1 mixture at 8.0 mM in 9:1 H2O/D2O at 293 K.
The remaining crosspeaks 5–14 come from the A3B1, A2B2, and A1B3 heterotetramers. Crosspeaks 5 and 6 are prominent and strikingly similar in intensity to each other. These two crosspeaks come from the A2B2 heterotetramer. Crosspeaks 7–14 are weaker and are also similar in intensity to each other. These eight crosspeaks are associated with the A3B1 and A1B3 heterotetramers.
We mixed peptides [15N]1a and 1b and also mixed peptides 1a and [15N]1b to assign crosspeaks 7–14 to the respective peptides. Figure 5b,c shows the 1H,15N HSQC spectra of these mixtures of labeled and unlabeled peptides. The 1H,15N HSQC spectrum of peptides [15N]1a and 1b shows that crosspeaks 1, 2, 5, 7, 8, 9, and 11 come from peptide [15N]1a; the 1H,15N HSQC spectrum of peptides 1a and [15N]1b shows that crosspeaks 3, 4, 6, 10, 12, 13, and 14 come from peptide [15N]1b. These spectra confirm that half of the crosspeaks come from peptide 1a and that half of the crosspeaks come from peptide 1b.
Assigning the 1H,15N HSQC Crosspeaks of the A3B1 and A1B3 Heterotetramers
To assign which of the crosspeaks 7–14 come from the A3B1 heterotetramer and which come from the A1B3 heterotetramer, we compared 1H,15N HSQC spectra of 3:1 and 1:3 mixtures of peptides [15N]1a and [15N]1b to that of the 1:1 mixture. In the spectra of the 3:1, 1:1, and 1:3 mixtures, the relative intensities of crosspeaks 1–14 vary, but the chemical shifts do not. The f1 projections of the 1H,15N HSQC spectra conveniently illustrate the relative intensities of the crosspeaks as one-dimensional 15N spectra. Figure 6 shows the f1 projections of pure [15N]1a, the 3:1, 1:1, and 1:3 mixtures, and pure [15N]1b.
Figure 6.

15N spectra from the f1 projections of the 1H,15N HSQC spectra of mixtures of peptides [15N]1a and [15N]1b. Spectra were recorded at 8.0 mM total concentration and varying mole fractions of peptide in 9:1 H2O/D2O at 600 MHz and 293 K. The mole fraction of peptide [15N]1b is designated χB.
The systematic variation of the crosspeaks as a function of the mole fraction χB clearly establishes which crosspeaks are associated with the A3B1 heterotetramer and which are associated with the A1B3 heterotetramer. Crosspeaks 7–10 have maximum relative intensities at χB = 0.25 and come from the A3B1 heterotetramer. Crosspeaks 11–14 have maximum relative intensities at χB = 0.75 and come from the A1B3 heterotetramer. Figure 7 illustrates relative integrations of the crosspeaks versus the mole fraction of peptide [15N]1b, χB.
Figure 7.
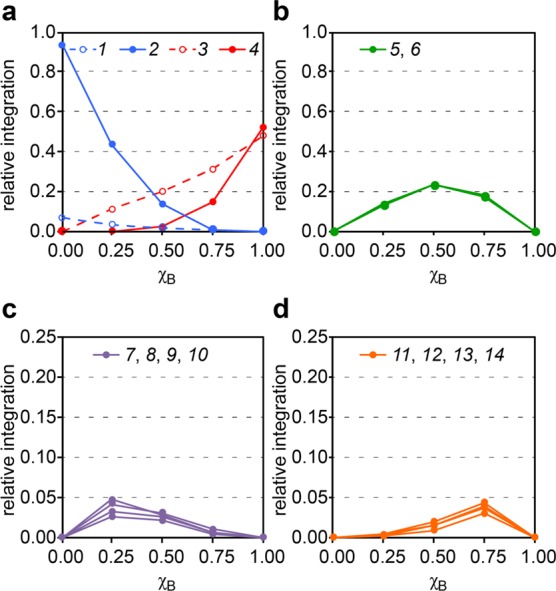
Plot of the relative integrations of crosspeaks 1–14 versus the mole fraction of peptide [15N]1b, χB. The intensities were measured by integrating the crosspeaks in the 1H,15N HSQC spectra of the mixtures of peptides [15N]1a and [15N]1b.
Job’s Method of Continuous Variation
We used Job’s method to determine the relative stabilities of the homotetramers and the heterotetramers of peptides [15N]1a and [15N]1b. Although this method was first introduced to study inorganic complexes, it is useful in all areas of chemistry for studying molecular association.14,15 Job’s method is performed by mixing two compounds “A” and “B” in varying ratios while keeping the total concentration constant. The amount of a complex that forms is then plotted versus the mole fraction to give a plot known as a “Job plot”. The appearance of the Job plot reflects the stoichiometry and relative stability of each complex. The mole fraction at which the maximum amount of the complex forms corresponds with its stoichiometry. For example, an A1B2 heterotrimer would give a maximum in a 1:2 mixture (χB = 0.67).
We applied Job’s method to peptides [15N]1a and [15N]1b, recording 1H,15N HSQC spectra for nine samples at 8.0 mM total concentration.16 We plotted the sum of the relative integrals of the 1H,15N HSQC crosspeaks for each species versus the mole fraction of peptide [15N]1b, χB. For example, we plotted the curve for the A3B1 heterotetramer species using the sum of the relative integrals of crosspeaks 7–10. Figure 8 illustrates the resulting Job plot.
Figure 8.
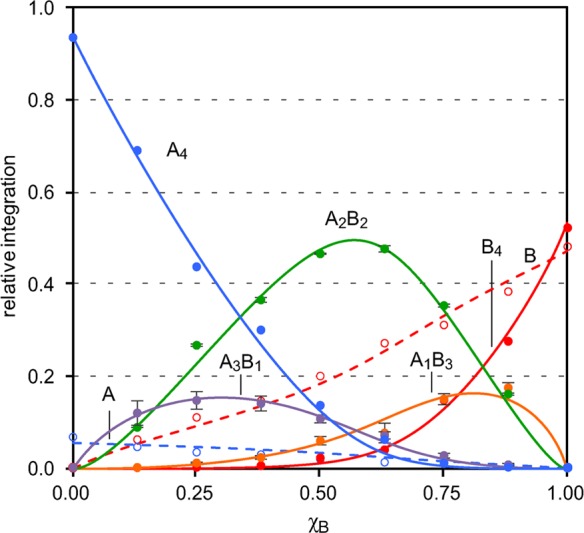
Job plot for peptides [15N]1a and [15N]1b showing the relative integrations of the monomers, homotetramers, and heterotetramers versus the mole fraction of peptide [15N]1b, χB. The curves reflect a monomer–tetramer equilibrium model fitted to the data. The error bars reflect the standard deviations among the individual measurements used to determine the relative integrations of A3B1, A2B2, and A1B3. The relative stabilities determined for each species are ϕ4,4 = 1.00, ϕ4,3 = 0.22, ϕ4,2 = 0.67, ϕ4,1 = 0.12, ϕ4,0 = 0.12, ϕ1,1 = 0.36, and ϕ1,0 = 2.20.
The Job plot shows that the A2B2 heterotetramer predominates over a wide range of mole fractions. At low mole fractions, χB ≤ 0.25, the A4 homotetramer predominates. At high mole fractions, χB ≥ 0.75, the B monomer and B4 heterotetramer predominate. The A2B2 heterotetramer reaches a maximum concentration at a mole fraction χB slightly greater than 0.50. The A3B1 heterotetramer and the A1B3 heterotetramer form to a lesser extent, reaching a maximum concentration at low and high mole fractions χB, respectively.
Simulated Job Plots of Homotetramers and Heterotetramers
We generated simulated Job plots reflecting different homotetramer and heterotetramer stabilities to help interpret the data in Figure 8. We used an implementation developed by Collum and co-workers that readily accommodates homotetramer and heterotetramer equilibria.17−19 We simulated a Job plot for a statistical distribution of homotetramers and heterotetramers and Job plots in which one of the heterotetramers is favored. These plots demonstrate how the relative stabilities of the tetramers affect the shapes of the curves. Figure 9 illustrates the resulting Job plots; the relative integrations of the species are plotted versus the mole fraction χB.
Figure 9.
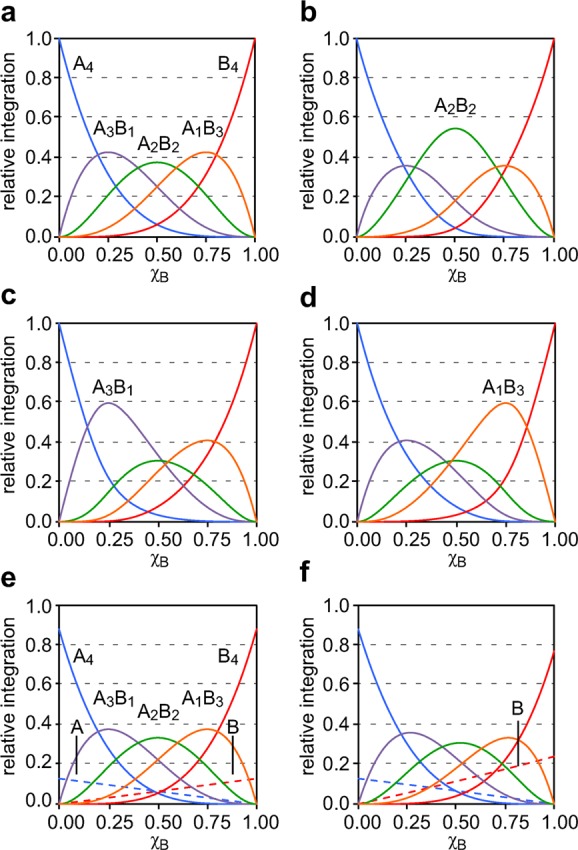
Simulated Job plots that show the relative integrations of the monomers, homotetramers, and heterotetramers versus the mole fraction of B, χB. (a) A statistical distribution of homotetramers and heterotetramers; ϕ4,4 = ϕ4,3 = ϕ4,2 = ϕ4,1 = ϕ4,0 = 1. (b) A2B2 heterotetramer is favored; ϕ4,2 = 2 and ϕ4,4 = ϕ4,3 = ϕ4,1 = ϕ4,0 = 1. (c) A3B1 heterotetramer is favored; ϕ4,3 = 2 and ϕ4,4 = ϕ4,2 = ϕ4,1 = ϕ4,0 = 1. (d) A1B3 heterotetramer is favored; ϕ4,1 = 2 and ϕ4,4 = ϕ4,3 = ϕ4,2 = ϕ4,0 = 1. (e) A statistical distribution of homotetramers and heterotetramers that also includes monomers; ϕ4,4 = ϕ4,3 = ϕ4,2 = ϕ4,1 = ϕ4,0 = 1 and ϕ1,1 = ϕ1,0 = 1. (f) A statistical distribution of homotetramers and heterotetramers that also includes monomers, where the B monomer is favored ϕ4,4 = ϕ4,3 = ϕ4,2 = ϕ4,1 = ϕ4,0 = 1, ϕ1,1 = 1, and ϕ1,0 = 2.
In the implementation by Collum and co-workers, the relative concentrations of the homotetramers and heterotetramers are calculated from equations based on a homotetramer-heterotetramer equilibrium model. The parameters ϕN,n are ascribed to each of the homotetramers and heterotetramers in the equations, where the N and n are integers in which the value of N describes the oligomer size and the value of n describes the number of “A” subunits. The value of each ϕN,n reflects the relative stability of each homotetramer or heterotetramer. The parameters ϕ4,4, ϕ4,3, ϕ4,2, ϕ4,1, and ϕ4,0 describe the relative stabilities of A4, A3B1, A2B2, A1B3, and B4, respectively. When each tetramer is equally stable, all parameters are equal (e.g., ϕ4,4 = ϕ4,3 = ϕ4,2 = ϕ4,1 = ϕ4,0 = 1) and a statistical distribution of homotetramers and heterotetramers forms.
The Job plot of a statistical distribution of homotetramers and heterotetramers is symmetrical, where the maximum of each curve reflects the tetramer stoichiometry. In the 1:1 mixture, the A2B2 heterotetramer predominates, with smaller fractions of the A3B1 and A1B3 heterotetramers in equal amounts, and with traces of the A4 and B4 homotetramers in equal amounts. In the 3:1 mixture, the A3B1 heterotetramer predominates, with smaller fractions of the A4 homotetramer and A2B2 heterotetramer, and with traces of the A1B3 heterotetramer. Similarly, in the 1:3 mixture, the A1B3 heterotetramer predominates, with smaller fractions of the B4 homotetramer and A2B2 heterotetramer, and with traces of the A3B1 heterotetramer. Figure 9a illustrates the Job plot for a statistical distribution of homotetramers and heterotetramers.
The appearance of the Job plot changes if any of the tetramers are favored or disfavored. If the A2B2 tetramer is favored, the A2B2 curve shows a pronounced increase and the A3B1 and A1B3 curves diminish slightly (Figure 9b). If the A3B1 tetramer is favored, the A3B1 curve shows a pronounced increase and the A2B2 curve diminishes slightly (Figure 9c). If the A1B3 tetramer is favored, the A1B3 curve shows a pronounced increase and the A2B2 curve diminishes slightly (Figure 9d).
Analysis of the Job Plot
We modified the implementation by Collum and co-workers to accommodate the equilibrium of the monomers with the homotetramers and heterotetramers. In our implementation, the relative concentrations of the monomers, homotetramers, and heterotetramers are calculated from equations based on a monomer–homotetramer–heterotetramer equilibrium model. The parameters ϕ1,1 and ϕ1,0 reflect the relative stabilities of the monomers A and B. The Job plot of a statistical distribution of homotetramers and heterotetramers that also includes the monomers is similar to the Job plot without monomers, except that the fraction of each tetramer is slightly diminished (Figure 9e). If the equilibrium favors one of the two monomers, a greater fraction of that monomer forms (Figure 9f).
We analyzed the data from our Job’s method experiment by nonlinear least-squares fitting of the model to the data. During the fit, the parameters ϕ4,3, ϕ4,2, ϕ4,1, ϕ4,0, ϕ1,1, and ϕ1,0 were allowed to vary, while the parameter ϕ4,4 remained fixed at 1. Figure 8 illustrates the Job plot with the fitted curves (ϕ4,4 = 1.00, ϕ4,3 = 0.22, ϕ4,2 = 0.67, ϕ4,1 = 0.12, ϕ4,0 = 0.12, ϕ1,1 = 0.36, ϕ1,0 = 2.20).
The model fits the data well. The quality of the fit corroborates that peptides [15N]1a and [15N]1b form a mixture of homotetramers and heterotetramers. The appearance of the resulting plot does not resemble the statistical distribution shown in Figure 9e. The Job plot shows little or no preference for the A2B2 heterotetramer, but it does show suppression of the A3B1 and A1B3 heterotetramers.
The Job’s method of continuous variation study and nonlinear least-squares fitting of the data establish that peptides 1a and 1b prefer to segregate within the heterotetramers. The suppression of the A3B1 and A1B3 heterotetramers shows that the A·B heterodimer subunit is disfavored and that heterotetramers containing an A·B heterodimer subunit are less stable. This finding explains why the A2B2 heterotetramer contains two homodimers rather than two heterodimers. Peptide 1a, which contains Aβ17–23, prefers to pair with itself to form a hydrogen-bonded homodimer; peptide 1b, which contains Aβ30–36, prefers to pair with itself to form a hydrogen-bonded homodimer.
Molecular Models of A2B2 Heterotetramers
We constructed energy-minimized models of A2B2 heterotetramers to help understand the preferential pairing of peptides 1a and 1b to form homodimers. By combining the monomer subunits of the models of the A4 and B4 homotetramers developed in the preceding paper,6 and re-minimizing, we generated two models of the A2B2 heterotetramers: the A·A/B·B topological isomer that was observed, and the A·B/A·B topological isomer that was not. Figure 10 illustrates the resulting models of these two topological isomers.
Figure 10.
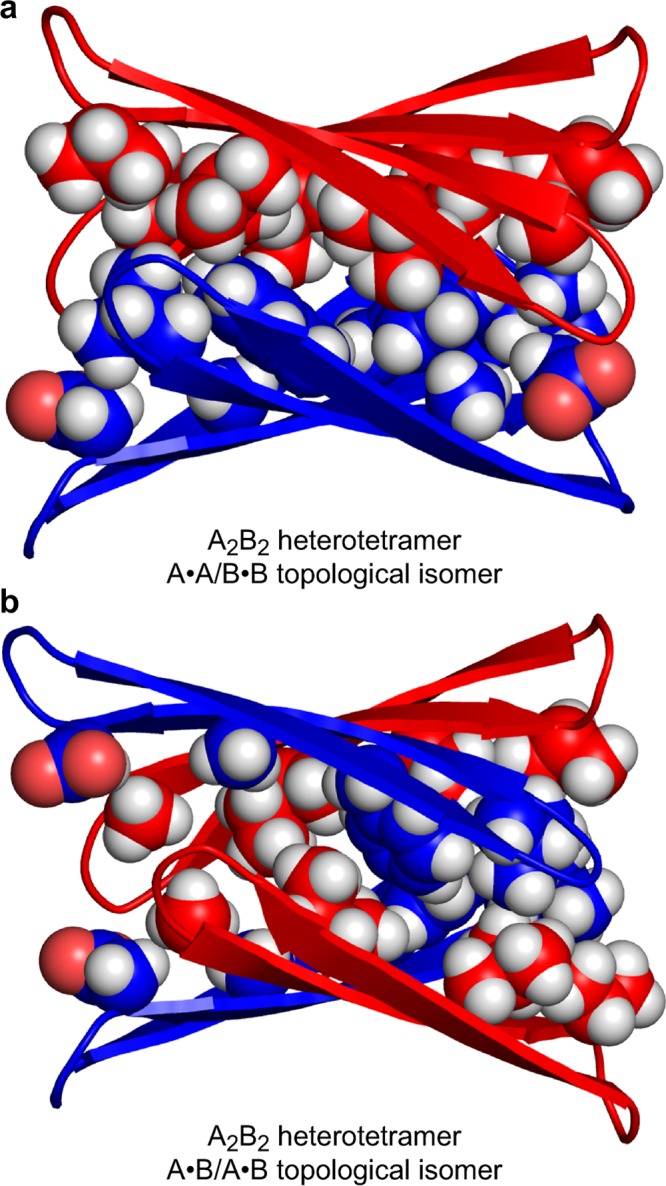
Molecular models of the topological isomers of the A2B2 heterotetramer of peptides 1a and 1b. (a) The A·A/B·B topological isomer. (b) The A·B/A·B topological isomer. Each model is a minimum-energy structure (local minimum) generated with MacroModel using the MMFFs force field with GB/SA water solvation.
The models show that the A2B2 heterotetramers can form sandwich-like structures that are similar to the homotetramers. Both topological isomers consist of two, four-stranded β-sheets that laminate together through hydrophobic packing. The side chains of L17, F19, and A21 from peptide 1a and of A30, I32, L34, and V36 from peptide 1b form hydrophobic surfaces that pack in the hydrophobic core of each heterotetramer. The interface between the A·A and B·B homodimers in the A·A/B·B topological isomer is uniformly packed. In contrast, the interface between the two A·B heterodimers in the A·B/A·B topological isomer is densely packed at one end and lightly packed at the other (Figure 10b).
The A·A homodimer of peptide 1a exhibits a large hydrophobic surface, with intimate contacts between the side chains of L17, F19, and A21. The large F19 and small A21 residues fit together well to help provide a uniformly packed surface (Figure 11a). The B·B homodimer of peptide 1b also exhibits a large hydrophobic surface, with intimate contacts between the side chains of A30, I32, L34, and V36. These residues also provide a uniformly packed surface (Figure 11b). The A·B heterodimer exhibits a hydrophobic surface with intimate contacts between the side chains of L17, F19, and A21 from peptide 1a and the side chains of I32, L34, and V36 from peptide 1b. The side chains do not pack uniformly, but rather the side chains pack densely at one end of the dimer and pack lightly at the other (Figure 11c).
Figure 11.
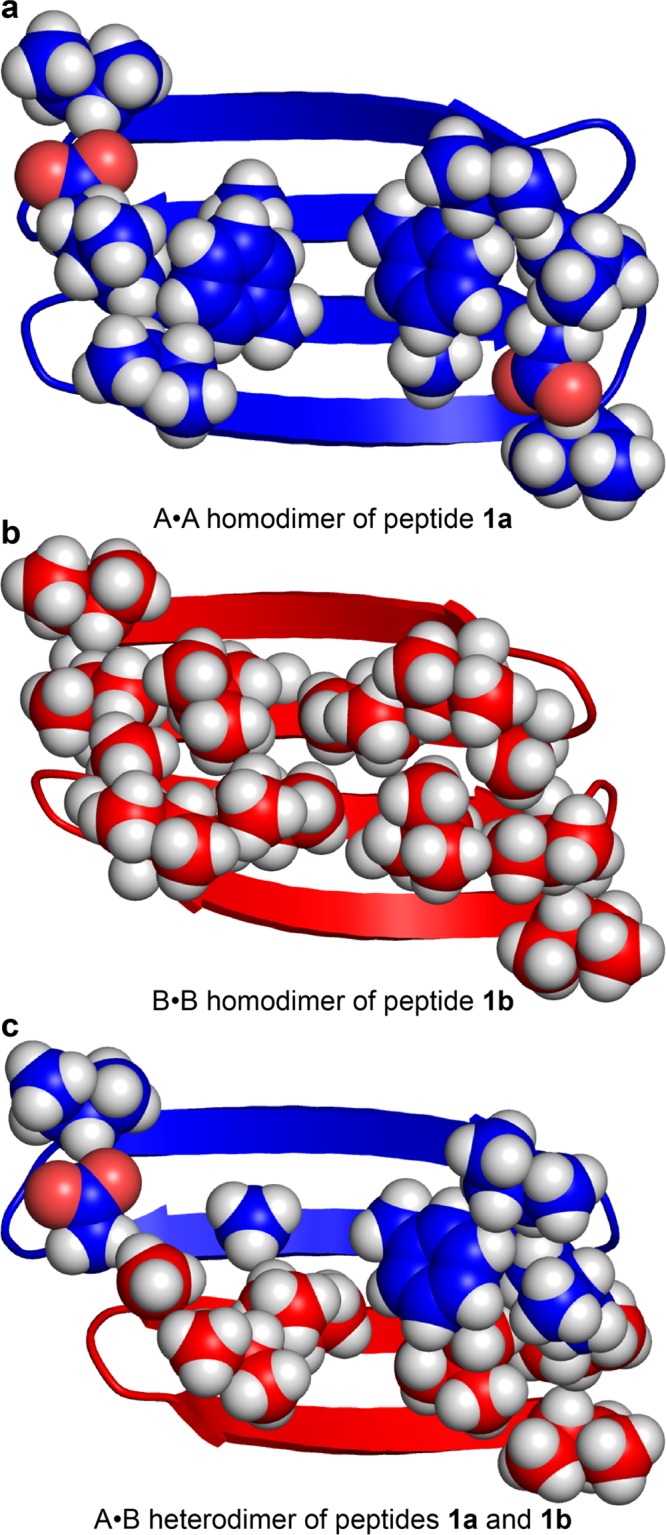
Molecular models of the homodimer and heterodimer subunits of the A2B2 heterotetramers of peptides 1a and 1b.
These molecular models suggest that differences between the homodimers and heterodimers formed by peptides 1a and 1b dictate the observed differences in the A2B2 heterotetramer stability. The uniform packing of the A·A and B·B homodimers appears to drive the formation of the observed A2B2 heterotetramer. The non-uniform packing of the A·B heterodimer appears to suppress the formation of the A3B1 and A1B3 heterotetramers, and also the alternative topological isomer of the A2B2 heterotetramer.
Conclusion
In framing the question behind these studies, we set out to determine whether peptides derived from the central and C-terminal regions of Aβ prefer to coassemble or to segregate. We found that the answer is more nuanced, at least in the context of the model system provided by peptides 1. Peptides 1a and 1b can coassemble, but the resulting heterotetramers reflect a preference to segregate within the dimer subunits. The heterotetramers comprising heterodimers are disfavored, while the heterotetramers comprising homodimers are not. These findings recapitulate the segregation within Aβ1–40 fibrils, in which the central region assembles to form a hydrogen-bonded β-sheet and the C-terminal region assembles to form a hydrogen-bonded β-sheet.3 The two β-sheets coassemble through hydrophobic contacts.
15N-Isotopic labeling, 1H,15N NMR spectroscopy, and Job’s method of continuous variation proved essential in these studies. Incorporation of a single 15N-isotopic label provided a sensitive and non-perturbing spectroscopic probe. 15N-Labeled peptides are readily prepared from commercially available 15N-labeled amino acids using solid-phase peptide synthesis. 1H,15N HSQC facilitated identification of the monomers, homotetramers, and heterotetramers. Job’s method of continuous variation assigned the resonances of each monomer and tetramer and established the relative stability of the tetramers. 15N-Edited NOESY established the identity of the topological isomer of the A2B2 heterotetramer.
These techniques, which proved useful for elucidating the assembly and coassembly of β-sheet peptides, should also be valuable in broader contexts. Peptide and protein assemblies occur widely in coiled coils, helix bundles, and collagen helices, as well as in amyloid oligomers and other β-sheet supramolecular assemblies. We envision that 15N-isotopic labeling in conjunction with 1H,15N NMR spectroscopy and Job’s method will also be valuable for studying these assemblies.
Acknowledgments
We thank the National Institutes of Health for grant support (GM097562). N.L.T. thanks Prof. Melanie J. Cocco and Dr. Philip R. Dennison for assistance with the NMR experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06001.
Figures S1–S5, showing NMR spectroscopic data; materials and methods; mathematical derivation for the monomer–homotetramer–heterotetramer equilibrium model; and procedures for nonlinear least-squares fitting of the Job plot data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Näslund J.; Haroutunian V.; Mohs R.; Davis K. L.; Davies P.; Greengard P.; Buxbaum J. D. JAMA 2000, 283, 1571–1577. 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]; b Haass C.; Selkoe D. J. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]; c Querfurth H. W.; LaFerla F. M. N. Engl. J. Med. 2010, 362, 329–344. 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]; d Knowles T. P.; Vendruscolo M.; Dobson C. M. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Liu R.; McAllister C.; Lyubchenko Y.; Sierks M. R. J. Neurosci. Res. 2004, 75, 162–171. 10.1002/jnr.10859. [DOI] [PubMed] [Google Scholar]
- a Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16742–16747. 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paravastu A. K.; Leapman R. D.; Yau W.-M.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18349–18354. 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tycko R.; Wickner R. B. Acc. Chem. Res. 2013, 46, 1487–1496. 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lu J.-X.; Qiang W.; Yau W.-M.; Schwieters C. D.; Meredith S. C.; Tycko R. Cell 2013, 154, 1257–1268. 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The fibrils formed by Aβ1–42 adopt a more compact structure:; a Xiao Y.; Ma B.; McElheny D.; Parthasarathy S.; Long F.; Hoshi M.; Nussinov R.; Ishii Y. Nat. Struct. Mol. Biol. 2015, 22, 499–505. 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Walti M. A.; Ravotti F.; Arai H.; Glabe C. G.; Wall J. S.; Bockmann A.; Guntert P.; Meier B. H.; Riek R. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4976–E4984. 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Colvin M. T.; Silvers R.; Ni Q. Z.; Can T. V.; Sergeyev I.; Rosay M.; Donovan K. J.; Michael B.; Wall J.; Linse S.; Griffin R. G. J. Am. Chem. Soc. 2016, 138, 9663–9674. 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hoyer W.; Gronwall C.; Jonsson A.; Stahl S.; Hard T. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5099–5104. 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cerf E.; Sarroukh R.; Tamamizu-Kato S.; Breydo L.; Derclaye S.; Dufrene Y. F.; Narayanaswami V.; Goormaghtigh E.; Ruysschaert J. M.; Raussens V. Biochem. J. 2009, 421, 415–423. 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]; c Yu L.; Edalji R.; Harlan J. E.; Holzman T. F.; Lopez A. P.; Labkovsky B.; Hillen H.; Barghorn S.; Ebert U.; Richardson P. L.; Miesbauer L.; Solomon L.; Bartley D.; Walter K.; Johnson R. W.; Hajduk P. J.; Olejniczak E. T. Biochemistry 2009, 48, 1870–1877. 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]; d Sandberg A.; Luheshi L. M.; Söllvander S.; Pereira de Barros T.; Macao B.; Knowles T. P. J.; Biverstål H.; Lendel C.; Ekholm-Petterson F.; Dubnovitsky A.; Lannfelt L.; Dobson C. M.; Härd T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15595–15600. 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lendel C.; Bjerring M.; Dubnovitsky A.; Kelly R. T.; Filippov A.; Antzutkin O. N.; Nielsen N. C.; Hard T. Angew. Chem., Int. Ed. 2014, 53, 12756–12760. 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]; f Spencer R. K.; Li H.; Nowick J. S. J. Am. Chem. Soc. 2014, 136, 5595–5598. 10.1021/ja5017409. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kreutzer A. G.; Hamza I. L.; Spencer R. K.; Nowick J. S. J. Am. Chem. Soc. 2016, 138, 4634–4642. 10.1021/jacs.6b01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truex N. L.; Wang Y.; Nowick J. S. J. Am. Chem. Soc. 2016, 10.1021/jacs.6b06000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cheng P. N.; Liu C.; Zhao M.; Eisenberg D.; Nowick J. S. Nat. Chem. 2012, 4, 927–933. 10.1038/nchem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu C.; Zhao M.; Jiang L.; Cheng P. N.; Park J.; Sawaya M. R.; Pensalfini A.; Gou D.; Berk A. J.; Glabe C. G.; Nowick J.; Eisenberg D. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 20913–20918. 10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Buchanan L. E.; Dunkelberger E. B.; Tran H. Q.; Cheng P. N.; Chiu C. C.; Cao P.; Raleigh D. P.; de Pablo J. J.; Nowick J. S.; Zanni M. T. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19285–19290. 10.1073/pnas.1314481110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowick J. S.; Chung D. M.; Maitra K.; Maitra S.; Stigers K. D.; Sun Y. J. Am. Chem. Soc. 2000, 122, 7654–7661. 10.1021/ja001142w. [DOI] [Google Scholar]
- a Nowick J. S.; Brower J. O. J. Am. Chem. Soc. 2003, 125, 876–877. 10.1021/ja028938a. [DOI] [PubMed] [Google Scholar]; b Woods R. J.; Brower J. O.; Castellanos E.; Hashemzadeh M.; Khakshoor O.; Russu W. A.; Nowick J. S. J. Am. Chem. Soc. 2007, 129, 2548–2558. 10.1021/ja0667965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some related studies of peptide and protein coassembly, see:; a Hammarstrom P.; Schneider F.; Kelly J. W. Science 2001, 293, 2459–2462. 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]; b Schnarr N. A.; Kennan A. J. J. Am. Chem. Soc. 2002, 124, 9779–9783. 10.1021/ja0174940. [DOI] [PubMed] [Google Scholar]; c Hadley E. B.; Testa O. D.; Woolfson D. N.; Gellman S. H. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 530–535. 10.1073/pnas.0709068105. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Xu F.; Zahid S.; Silva T.; Nanda V. J. Am. Chem. Soc. 2011, 133, 15260–15263. 10.1021/ja205597g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fallas J. A.; Hartgerink J. D. Nat. Commun. 2012, 3, 1087. 10.1038/ncomms2084. [DOI] [PubMed] [Google Scholar]; f Thomas F.; Boyle A. L.; Burton A. J.; Woolfson D. N. J. Am. Chem. Soc. 2013, 135, 5161–5166. 10.1021/ja312310g. [DOI] [PubMed] [Google Scholar]; g Negron C.; Keating A. E. J. Am. Chem. Soc. 2014, 136, 16544–16556. 10.1021/ja507847t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Khakshoor O.; Demeler B.; Nowick J. S. J. Am. Chem. Soc. 2007, 129, 5558–5569. 10.1021/ja068511u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pham J. D.; Demeler B.; Nowick J. S. J. Am. Chem. Soc. 2014, 136, 5432–5442. 10.1021/ja500996d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pham J. D.; Spencer R. K.; Chen K. H.; Nowick J. S. J. Am. Chem. Soc. 2014, 136, 12682–12690. 10.1021/ja505713y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We used 1H NMR TOCSY to assign residues associated with these NOEs (Figures S1 and S2).
- The 1H NMR NOESY spectrum of the 1:1 mixture of peptides 1a and 1b shows additional NOEs associated with the stacking of the two homodimers to form a sandwich-like tetramer. The spectrum shows NOEs between the F19 aromatic protons of peptide 1a and the I32 and L34 side-chain protons of peptide 1b (Figure S4a). The spectrum also shows NOEs between the A21 side-chain protons of peptide 1a and the I32 side-chain protons of peptide 1b (Figure S4b). Figure S5 illustrates the stacking of the A·A and B·B homodimers of peptides 1a and 1b consistent with these NOEs.
- Job P. Ann. Chim. Fr. 1928, 9, 113–203. [Google Scholar]
- a Ramanathan P. S. J. Inorg. Nucl. Chem. 1973, 35, 3358–3360. 10.1016/0022-1902(73)80042-9. [DOI] [Google Scholar]; b Ingham K. C. Anal. Biochem. 1975, 68, 660–663. 10.1016/0003-2697(75)90666-1. [DOI] [PubMed] [Google Scholar]; c Huang C. Y. Methods Enzymol. 1982, 87, 509. 10.1016/S0076-6879(82)87029-8. [DOI] [PubMed] [Google Scholar]; d Gil V. M. S.; Oliveira N. C. J. Chem. Educ. 1990, 67, 473–478. 10.1021/ed067p473. [DOI] [Google Scholar]; e Facchiano A.; Ragone R. Anal. Biochem. 2003, 313, 170–172. 10.1016/S0003-2697(02)00562-6. [DOI] [PubMed] [Google Scholar]; f Renny J. S.; Tomasevich L. L.; Tallmadge E. H.; Collum D. B. Angew. Chem., Int. Ed. 2013, 52, 11998–12013. 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 8.0 mM total concentration was chosen to favor tetramers in the mixtures of peptides [15N]1a and [15N]1b.
- a McNeil A. J.; Toombes G. E.; Chandramouli S. V.; Vanasse B. J.; Ayers T. A.; O’Brien M. K.; Lobkovsky E.; Gruner S. M.; Marohn J. A.; Collum D. B. J. Am. Chem. Soc. 2004, 126, 5938–5939. 10.1021/ja049245s. [DOI] [PubMed] [Google Scholar]; b McNeil A. J.; Toombes G. E.; Gruner S. M.; Lobkovsky E.; Collum D. B.; Chandramouli S. V.; Vanasse B. J.; Ayers T. A. J. Am. Chem. Soc. 2004, 126, 16559–16568. 10.1021/ja045144i. [DOI] [PubMed] [Google Scholar]; c Liou L. R.; McNeil A. J.; Ramirez A.; Toombes G. E.; Gruver J. M.; Collum D. B. J. Am. Chem. Soc. 2008, 130, 4859–4868. 10.1021/ja7100642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom B.Statistical Mechanics: A Concise Introduction for Chemists; Cambridge University Press: New York, 2002. [Google Scholar]
- Additional references on quantitative treatment of data from Job’s method of continuous variation:; a Likussar W.; Boltz D. F. Anal. Chem. 1971, 43, 1265–1272. 10.1021/ac60304a006. [DOI] [Google Scholar]; b Bruneau E.; Lavabre D.; Levy G.; Micheau J. C. J. Chem. Educ. 1992, 69, 833–837. 10.1021/ed069p833. [DOI] [Google Scholar]; c Hirose K. J. Inclusion Phenom. Mol. Recognit. Chem. 2001, 39, 193–209. 10.1023/A:1011117412693. [DOI] [Google Scholar]; d Olson E. J.; Bühlmann P. J. Org. Chem. 2011, 76, 8406–8412. 10.1021/jo201624p. [DOI] [PubMed] [Google Scholar]; e Olson E. J.; Bühlmann P. J. Org. Chem. 2014, 79, 830–830. 10.1021/jo402786c. [DOI] [Google Scholar]; f Hirose K. In Analytical Methods in Supramolecular Chemistry, 2nd ed.; Schalley C. A., Ed.; Wiley-VCH: Weinheim, 2012; pp 27–66. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.