

Abstract
Purpose
In contrast to Hodgkin lymphoma and systemic anaplastic large-cell lymphoma, CD30 expression of malignant lymphocytes in mycosis fungoides (MF) and Sézary syndrome (SS) is quite variable. Clinical activity and safety of brentuximab vedotin, a CD30 targeting antibody-drug conjugate, was evaluated in MF and SS. Tissue and blood biomarkers of clinical response were explored.
Patients and Methods
In this phase II study, patients with MF or SS with negligible to 100% CD30 expression levels were treated with brentuximab vedotin (1.8 mg/kg) every 3 weeks for a maximum of sixteen doses. The primary end point was overall global response rate. Secondary end points included correlation of tissue CD30 expression level with clinical response, time to response, duration of response, progression-free and event-free survivals, and safety.
Results
Of the 32 patients enrolled and treated, 30 patients had available efficacy evaluations. Objective global response was observed in 21 (70%) of 30 patients (90% CI, 53% to 83%). CD30 expression assessed by immunohistochemistry was highly variable, with a median CD30max of 13% (range, 0% to 100%). Those with <5% CD30 expression had a lower likelihood of global response than did those with 5% or greater CD30 expression (P < .005). CD163 positive tumor-associated macrophages, many of which coexpress CD30, were abundant in tissue. Peripheral neuropathy was the most common adverse event.
Conclusion
Brentuximab vedotin demonstrated significant clinical activity in treatment-refractory or advanced MF or SS with a wide range of CD30 expression levels. Additional biomarker studies may help optimize rational design of combination therapies with brentuximab vedotin.
INTRODUCTION
Mycosis fungoides (MF) and Sézary syndrome (SS) are the most common subtypes of cutaneous T-cell lymphoma (CTCL).1–3 Patients with early stage disease have an excellent prognosis; however, survival in patients with advanced disease is compromised, especially when adverse prognostic factors are present, such as folliculotropic, large-cell transformed, or extracutaneous disease. Current therapies rarely provide durable responses, and the only potentially curative therapy is allogeneic hematopoietic stem-cell transplantation. There is a great need for new therapies to treat MF/SS in patients with advanced or refractory disease.
Brentuximab vedotin (BV, SGN-35) is an anti-CD30 monoclonal antibody conjugated with monomethyl auristatin E by a protease-cleavable linker.4,5 BV has shown dramatic efficacy in the treatment of Hodgkin lymphoma and systemic anaplastic large-cell lymphoma,6,7 both of which uniformly express CD30 as assessed by immunohistochemistry (IHC) with use of BerH2 antibody. However, response rates are lower in systemic lymphomas with variable CD30 expression.8,9 Malignant T cells in MF/SS skin lesions have median CD30 expression levels of 10% to 15% of total mononuclear infiltrate, as assessed by routine IHC, with greater CD30 levels in those with large-cell transformation (LCT).10,11
The goals of this phase II study were to explore the clinical activity of BV in patients with MF/SS with any CD30 expression level (including a negligible level), establish the safety profile in an MF/SS population, and assess for potential biomarkers of clinical response.
PATIENTS AND METHODS
Study Design
This was a phase II, investigator-initiated, multi-institution study (study No. LYMNHL0089 and SU-06212011-7946). CD30 expression levels were grouped as low (<10%, Group A), intermediate (10% to 50%, Group B), and high (>50%, Group C) to ensure enrollment of patients across a wide range of expression (Appendix Figure A1, online only). A minimum of two skin biopsies (of different lesion morphology and site) were obtained for each patient at baseline, and the maximum level of CD30 expression (CD30max) among all biopsies was used for group A, B, or C designation.
The primary end point was the overall response rate of BV in this patient population. Secondary objectives were to explore potential biomarkers of response or resistance including tissue CD30 expression level, tumor microenvironment, and soluble CD30 (sCD30). Utility of multispectral image (MSI) analysis to assess CD30 expression was evaluated in collaboration with the University of Wisconsin. Additional secondary end points included time to response (TTR), duration of response (DOR), progression-free survival (PFS), event-free survival (EFS), and safety.
All patients were enrolled and treated at the primary institution (Stanford University). Memorial Sloan Kettering Cancer Center served as an independent review site for clinical and pathologic evaluations. This study was approved by the institutional review board at Stanford University and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient.
Patient Population and Treatment
Patients with MF or SS, stages IB through IVB,3 who had at least one systemic therapy failure were eligible. Subjects with any levels of CD30 expression (0% to 100%) in the skin or other compartments were included. An age of ≥ 18 years and an Eastern Cooperative Oncology Group performance status of 0 to 2 were required. Additional inclusion criteria were an absolute neutrophil count of ≥ 1,000/μL, a platelet count of ≥ 50,000/μL, a serum creatinine level of ≤ 2× the upper limit of normal, and ALT and AST levels of ≤ 3× the upper limit of normal. Topical or systemic corticosteroids were not allowed for treatment of MF or SS symptoms. All patients were treated with as many as eight cycles of BV (1.8 mg/kg) administered every 3 weeks (Appendix Figure A1). Patients with continued clinical improvement were allowed to have a maximum of eight additional cycles; those with complete response (CR) were allowed to have two more cycles.
Efficacy and Safety Assessments
The skin was the primary efficacy assessment; however, in patients with extracutaneous disease, all involved compartments were assessed and a global response incorporating nonskin compartments was determined.12 Clinical response documentation required confirmation of response at the subsequent study visit. Skin disease burden was measured with the modified Severity-Weighted Assessment Tool (mSWAT)12 at screening, before each dose of BV, at the end of treatment, and every 2 to 3 months until progression. Patients with lymph-node or visceral disease had whole body positron emission tomography-computed tomography at the time of screening, at the end of cycles 2, 5, 8, 11, and 14, and at the end of treatment, and patients with blood involvement had Sézary flow cytometry at the same time points as the skin assessments.
Adverse events were recorded in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.12a Given the neurotoxicity associated with BV, a detailed neurologic evaluation was performed at the time of screening, at 6 months, and at the end of treatment. For patients who had grade 2 peripheral neuropathy, a formal neurologic evaluation was updated every two cycles or every 2 months (off study).
Tissue and Blood Biomarker Analysis
Tissue IHC for biomarker assessment.
Biopsies were fixed in 4% formalin and embedded in paraffin. Commercially available antibodies were used to assess the following markers with use of standard dilutions, antigen retrieval, and chromogen development methods: CD30 (BerH2, DAKO, Carpinteria, CA), CD8 (clone C8/144B, DAKO), CD20 (clone L26, DAKO), CD163 (clone 10D6, Leica, Buffalo Grove, IL), PD-1 (clone NAT105, Cell Marque, Rocklin, CA), FoxP3 (clone 236A/E7, eBioscience, San Diego, CA). IHC was graded according to the percentage of total mononuclear cell infiltrate that was positive .
Serum sCD30 level.
sCD30 was detected in human serum with use of a Luminex-based sandwich fluoroimmunoassay (Life Technologies, Carlsbad, CA). Luminex beads conjugated with an antihuman sCD30 monoclonal antibody were incubated with samples, and then a phycoerythrin-conjugated antihuman sCD30 monoclonal antibody was added. Beads were analyzed with a Bio-Rad Bio-Plex 100 reader (Bio-Rad Laboratories, Hercules, CA).
MSI acquisition and analysis.
All images and quantitative IHC data were generated with the Nuance system (PerkinElmer, Waltham, MA), equipped with Nuance (version 3.0.2) multispectral imaging software and inForm (version 1.4) advanced image analysis software. A spectral library with characteristic wavelength emission curves of hematoxylin, Fast Red, and diaminobenzidine was built by sampling spectra from tissue sections stained with single dyes. For visualization of the signals in the MSIs, the pseudo colors red (CD30), green (CD8 and CD163), and blue (hematoxylin) were used. To quantitate CD30 expression, 161 to 238 mononuclear cells per biopsy were manually designated as regions of interest. Using the spectral library as reference, the system measured the amount of signal within each region of interest, converting spectral data into optical density (OD) units. To assess for coexpression of CD30 with CD8 or CD163, MSIs of double-stained slides were acquired as above in selected biopsies. The percentage of double- and single-positive cells was determined using inForm.
T-Cell Receptor Clonality by High-Throughput Sequencing
Genomic DNA was prepared from formalin-fixed, paraffin-embedded tissue with a QIAamp DNA Mini Kit (QIAGEN, Valencia, CA). A multiplex PCR system was used to amplify rearranged T-cell receptor beta (TCRB) and/or gamma (TCRG) loci and high-throughput sequencing (HTS) performed with Illumina GA2 system or HiSeq platform (Illumina, San Diego, CA) as previously described to generate reads that cover the entire complementarity-determining region 3 (CDR3).13 The dominant CDR3 sequence of tumor cells was determined from the baseline samples.
Statistical Analysis
A sample size of 30 patients would provide 80% power to reject a 35% response rate, (the general response rate of the US Food and Drug Administration's recently approved systemic agents),14–16 if the true (alternative) response rate were 60% or higher using binomial test at a one-sided significance level of 5%. A 90% two-sided exact CI is reported. There were no predefined subpopulations for statistical analysis.
DOR, PFS, EFS, and duration of neuropathy were analyzed with use of the Kaplan-Meier method. DOR was calculated from date of response to progressive disease (PD) and PFS from date of first BV dose to the time of PD or death.11a,15 For DOR and PFS, patients were censored at the last follow-up visit if any new treatment was initiated before documentation of PD. EFS was calculated from the date of the first BV dose to the time of toxicity-related termination, PD, next significant therapy (eg, the use of a systemic agent or total skin electron beam therapy), or death by any cause (whichever occurred first). For biomarker analyses comparing response with nonresponse, Wilcoxon rank-sum and Fisher's exact tests were applied, as appropriate.
RESULTS
Patient Characteristics
Thirty-two patients were enrolled (Table 1); all were included in the safety analysis. Two patients without any response assessment data were excluded from the efficacy analyses. Most patients had advanced disease (stage IIB or higher, 88%) with adverse prognostic features of folliculotropic mycosis fungoides (FMF) or LCT in 29 (91%) of 32. The median number of prior systemic therapies was 3 (range, 1 to 13 therapies); most of these patients had prior cytotoxic agents, one with prior allogeneic stem-cell transplantation. Fourteen (44%) of the 32 patients had a low baseline maximum level of CD30 expression (CD30max; Group A), and only four had >50% CD30max (Group C).
Table 1.
Patient Baseline Demographics, Clinical Characteristics, and Clinical Response
| Characteristics | All Patients, N = 32, n (%) | Evaluable for Response, n = 30 |
ORR,* n (%) | ||||
|---|---|---|---|---|---|---|---|
| CR | PR | SD | PD | NE | |||
| Sex | |||||||
| Male | 19 (59) | 0 | 13 | 1 | 4 | 1 | 13 of 18 (72) |
| Female | 13 (41) | 1 | 7 | 3 | 1 | 1 | 8 of 12 (67) |
| Age, years, median (range) | 62 (20-87) | 78 | 60 (38-87) | 60 (20-82) | 64 (57-77) | 60 (50-70) | |
| Clinical stage | |||||||
| All | 32 (100) | 1 | 20 | 4 | 5 | 2 | 21 of 30 (70) |
| IB | 4 (13) | 0 | 3 | 1 | 0 | 0 | 3 of 4 (75) |
| IIB | 18 (56) | 0 | 14 | 2 | 2 | 0 | 14 of 18 (78) |
| IV/SS† | 10 (31) | 1 | 3 | 1 | 3 | 2 | 4 of 8 (50) |
| Adverse prognostic factors | |||||||
| LCT or FMF | 29 (90) | 1 | 19 | 3 | 5 | 1 | 20 of 28 (71) |
| LCT | 16 (50) | 1 | 9 | 2 | 3 | 1 | 10 of 15 (67) |
| FMF | 8 (25) | 0 | 7 | 1 | 0 | 0 | 7 of 8 (88) |
| LCT + FMF | 5 (16) | 0 | 3 | 0 | 2 | 0 | 3 of 5 (60) |
| No. of prior systemic therapies | |||||||
| < 3 | 15 (47) | 0 | 8 | 2 | 4 | 1 | 8 of 14 (57) |
| ≥ 3 | 17 (53) | 1 | 12 | 2 | 1 | 1 | 13 of 16 (81) |
| CD30 grouping at screening | |||||||
| A (< 10%) | 14 (44) | 0 | 7 | 4 | 2 | 1 | 7 of 13 (54) |
| B (10% to 50%) | 14 (44) | 0 | 11 | 0 | 3 | 0 | 11 of 14 (79) |
| C (> 50%) | 4 (13) | 1 | 2 | 0 | 0 | 1 | 3 of 3 (100) |
Abbreviations: CR, complete response; FMF, folliculotropic mycosis fungoides; LCT, large-cell transformation; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; SD, stable disease; SS, Sézary syndrome.
Objective clinical response was observed in 21 (70%) of the 30 efficacy-evaluable patients
Of 10 stage IV patients, three patients had SS with one CR, one PR, and one PD; one patient was stage IVB who had PR.
Clinical Activity
Objective clinical response was observed in 21 (70%) of the 30 efficacy-evaluable patients (90% CI, 53% to 83%; Table 1; Fig 1). Responses were observed across all clinical stages and in patients with FMF and/or LCT regardless of number or type of prior systemic therapies. The majority of patients had improvement in the skin, with a maximum mSWAT change from the time of screening (mSWATmax) median score reduction of 73% (range, 100% to −54%, Fig 2A). Although only one patient had documented global CR, seven patients had a >90% reduction in their mSWAT scores. Patient age, sex, clinical stage, baseline disease burden (as assessed on the mSWAT), or FMF and/or LCT status did not correlate with clinical response.
Fig 1.
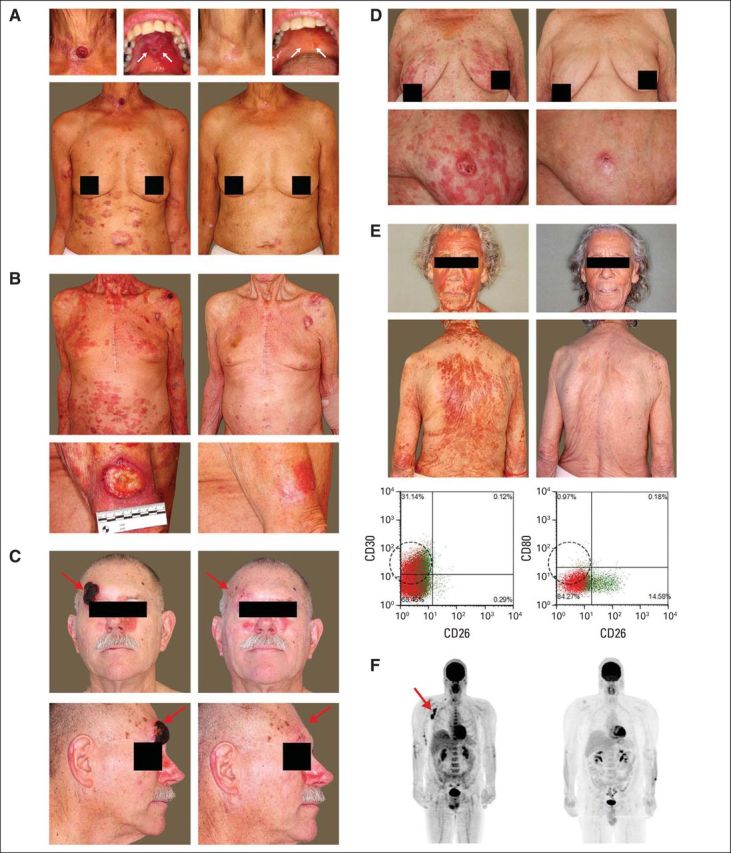
Clinical presentation of the patients at baseline and with brentuximab vedotin treatment. (A) 66-year-old woman with mycosis fungoides (MF) stage IVB with oral disease; the patient had a maximum reduction in modified Severity-Weighted Assessment Tool score from time of screening (mSWATmax) of 86%. (B) 87-year-old man with MF stage IIB with ulcerative tumor on the arm; the patient had an mSWATmax reduction of 92%. (C) 66-year-old man with MF stage IIB with forehead tumor; the patient had an mSWATmax reduction of 97%. (D) 78-year-old woman with Sézary syndrome (SS) stage IVA1; the patient had an mSWATmax reduction of 100% (global complete response). (E) 80-year-old woman with SS stage IVA1; the patient had an mSWATmax reduction of 89%; flow cytometry showing reduction of CD4+/CD26-/CD30+ Sézary population after treatment. (F) 45-year-old man with MF IVA2 with reduction in lymph-node disease after treatment, as shown in the positron emission tomography-computed tomography scan.
Fig 2.
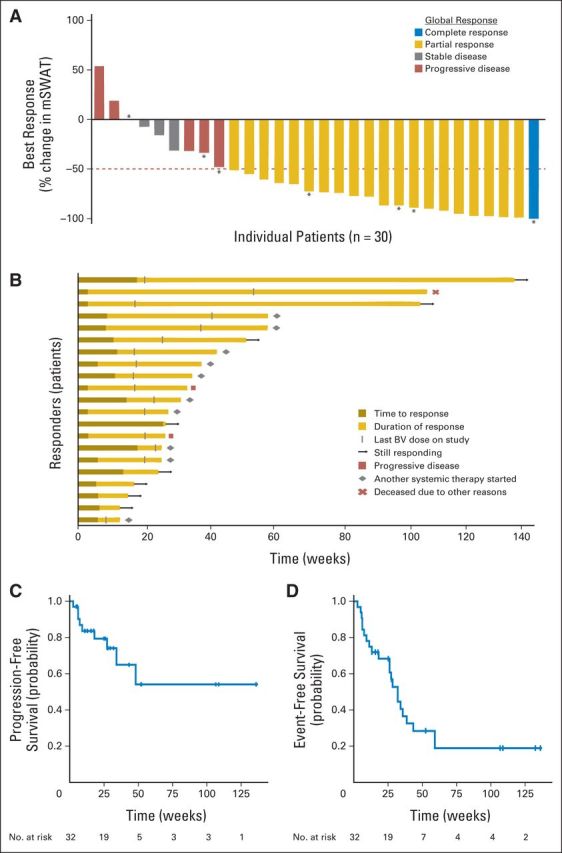
Primary study end-point analyses. (A) Maximum change in modified Severity Weighted Assessment Tool (mSWAT) score (skin disease burden) compared with baseline in all patients treated with brentuximab vedotin (n = 30). Eight patients had more than 90% reduction in their mSWAT score, including one complete response (stage IV/SS) and seven near complete responses, including two stage IB (patch/plaque only) and five stage IIB (patch-plaque and tumor). Clinically meaningful responses were seen in both tumor and patch-plaque lesions. Horizontal line at −50% indicates the threshold for defining partial response in the skin. (*) Patients with stage IV (extracutaneous) disease. Overall global response was determined by composite of skin, lymph node, viscera, and blood compartment responses.15 (B) Time course in patients who had global clinical responses to brentuximab vedotin (n = 21). (C) Progression-free survival (n = 32). (D) Event-free survival (n = 32), with event defined as toxicity-related termination, progressive disease, death, or start of another systemic therapy. BV, brentuximab vedotin.
At the time of analysis, the median observation time for all subjects (n = 32) was 71.7 weeks (range 13.0 to 156.6 weeks) and five patients remained on therapy. The median TTR was 6.6 weeks (range, 3.0 to 27.0 weeks). Figure 2B depicts the time course and current status of the 21 responders. At 6 and 12 months, Kaplan-Meier estimates showed that 90% and 79% of responses, respectively, were continuing; 79% and 54% of responders, respectively, were progression-free; and 61% and 28% of responders, respectively, were event-free (Fig 2C, 3D). Twelve patients initiated another round of systemic therapy without attaining PD.
Fig 3.

Maximum change in modified Severity Weighted Assessment Tool (mSWAT) by tissue CD30max (maximum level of CD30 expression) assessed by immunohistochemistry, stratified by disease stage and global clinical response. Data support a correlation between skin CD30max and depth of skin response (reduction in mSWAT; P = .0031).The dotted line depicts CD30max of 5%. Patients with CD30max of < 5% had a significantly lower likelihood of clinical response (P = .0046).
Biomarkers of Clinical Response
By tissue IHC evaluation, the median CD30max of all patients was 13% (range, 0% to 100%; six patients had a CD30max of <5%). Although clinical responses were observed across all expression levels (groups A, B, and C), the median CD30max was higher in responders (global response CR/partial response, 15%), versus nonresponders (global response stable disease/PD, 3.0%; P = .037; Fig 3). The correlation of response with CD30max was greatest in stage IIB (skin tumors, P = .0072). All eight stage IIB patients with a CD30max of 10% or higher responded to BV, and 3 of 8 had a >90% mSWAT reduction. Patients with a CD30max of <5% had a significantly lower likelihood of clinical response (17% v 83%, P = .0046). Furthermore, our data support a correlation between skin CD30max and depth of skin response (reduction in mSWAT; P = .0031; Fig 3). There was no significant correlation between tissue CD30max and TTR, DOR, PFS, or EFS.
Skin microenvironment biomarkers studied included tumor-infiltrating CD8+ T cells, B cells (CD20+), macrophages (CD163+), Foxp3+ T cells, or PD-1+ T cells. Of these, CD163+ macrophages were most abundant with median of 40% (range 5% to 80%) of total mononuclear infiltrate in pretreatment samples assessed. However none were associated with response in a univariable analysis, (P = .38, .25, .98, .29, .36, respectively).
Twenty-two of 32 patients had baseline sCD30 assessed. Median sCD30 was 86 ng/mL (range, 28 to 955 ng/mL; n = 14) in responders and 137 ng/mL (range, 26 to 282 ng/mL; n = 8) in nonresponders (P = .92).
MSI Analysis of CD30 and Coexpression With CD163 or CD8
Seven (54%) of 13 evaluable patients in the low CD30max group (Group A) demonstrated objective clinical response, including one patient with negligible baseline CD30max by routine IHC (Fig 4). We sought to measure low levels of CD30 by the MSI technique. Whereas light microscopy relies on arbitrary scoring, MSI allows for computerized scoring and quantification of biomarkers. The online Appendix demonstrates the superior sensitivity of MSI analysis over routine IHC. This technique allowed for the unmixing or separation of chromogens, even if they spatially overlap. The unmixed images were used to quantitate each chromogen individually, which allows for greater sensitivity and specificity of assessing CD30 expression.17 Cells with ODs above isotype control (background levels) were considered CD30 positive. By viewing individual cells with known OD values in their original light microscopy mode, we identified a threshold value of approximately 0.1 OD per cell, designated as the “visibility threshold,” below which the signal was read as positive by MSI but was undetectable by light microscopy (Fig 4). The MSI analysis demonstrated quantifiable CD30 staining in 19 (95%) of 20 samples interpreted as negligible CD30 by routine IHC.
Fig 4.
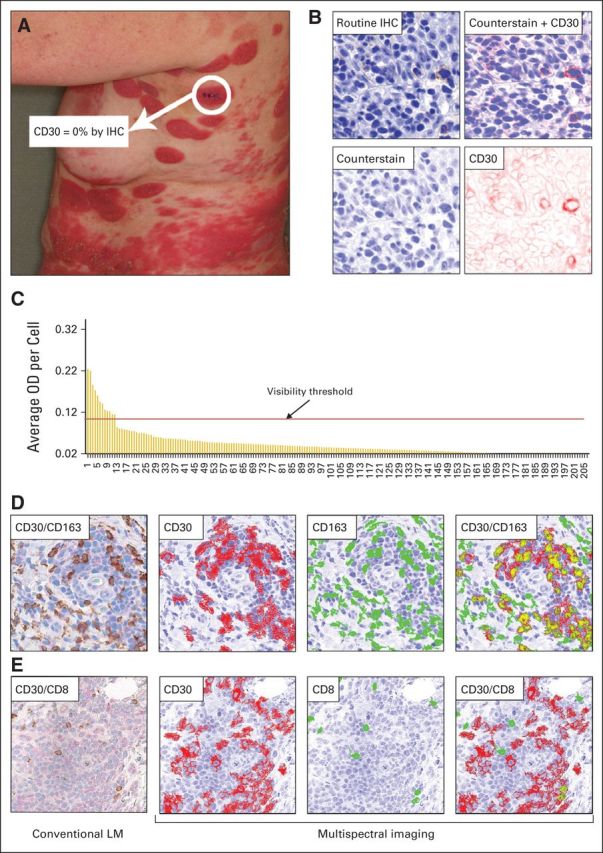
Multispectral imaging (MSI) analysis of CD30 expression. (A) Clinical photograph of a patient at the time of screening. Immunohistochemistry (IHC) stain of the highlighted biopsy site showed CD30 level of 0% as assessed by conventional light microscopy (LM). (B) MSI series of a representative CD30 immunostained area from the same biopsy. The MSI system converts the standard light microscopic image into a pseudo-colored composite: the counterstain is shown in the pseudo color blue, and the CD30 is represented by the pseudo color red. The composite image can be unmixed into single components, counterstain and CD30. The CD30 panel highlights the CD30 stain in pseudo color red. (C) Histogram showing optical density (OD) values per cell in biopsy. Each bar represents the average OD value of an individual cell (n = 207 cells). Nonspecific background OD (0.02, including two stable disease) was subtracted from all values, and any bar therefore signifies specific CD30 staining. The visibility threshold indicates the OD below which CD30 stain was undetectable by visual inspection with conventional LM but positive by MSI analysis. (D) CD30 and CD163 double-stained lesional skin: conventional LM image of CD30 (light pink) and CD163 (brown) double stain. Here, the areas that exceed the nonspecific background OD for CD30 are shown in red, thus the color red depicts CD30 positivity. CD163+ cells are shown in green. The areas of spatial colocalization of both stains are highlighted in gold, demonstrating the extent of CD30 and CD163 coexpression in this lesion. (E) CD30 and CD8 double-stained lesional skin from the same subject: conventional LM image, showing CD30 (light pink) and CD8 (brown). Corresponding MSI of CD30+ (red) and CD8+ (green) cells and the coexpression of both markers (gold) in this area.
To distinguish expression of CD30 by neoplastic T cells versus tumor-infiltrating cytotoxic T cells or macrophages, we performed double staining for coexpression of CD30 and CD8 (cytotoxic T cells) or CD163 (macrophages) and assessed with routine IHC and MSI analysis. The MSI technique can differentiate and unmix up to 14 different stains per pixel, whereas the human eye cannot easily distinguish stains with similar colors nor can it deconvolute spatially colocalized chromogens. The online Appendix demonstrates significant CD30-CD8 and CD30-CD163 coexpression not appreciated by routine IHC. Of six patient samples, each double stained for CD30-CD8 and CD30-CD163 and coexpression ranged from 0.2% to 14.8% (median 1.7%) and 1.0% to 40.9% (median 9.7%) of cells by MSI analysis, respectively.
TCR Clonality Assessment of New Tumors With Therapy
We analyzed seven patients with new tumor lesions with TCR HTS to assess for a new dominant TCR clone. Six patients had identifiable TCR-β clone before BV, and the HTS TCR profile of their new tumors showed identical dominant TCR CDR3 sequence as the pretreatment clone, including five of six tumor samples with loss of CD30 expression.
Safety and Tolerability
The median number of BV doses was six (range, 1 to 16 doses). The most common adverse events related to BV were peripheral neuropathy (66%), fatigue (47%), nausea (28%), alopecia (22%), and neutropenia (19%; Table 2). Grade 3/4 related adverse events were observed in 10 patients; the most common were neutropenia (n = 4) and skin eruption (n = 3). Three treatment-related serious adverse events were reported, including one event each of confusion, acute renal failure, and neuropathy.
Table 2.
Most Common Drug-Related Adverse Events (experienced in > three of 32 patients)
| Adverse Event | All Grades |
Grade 1 |
Grade 2 |
Grade 3/4 (severe adverse event) |
||||
|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | |
| Peripheral neuropathy | 21 | 66 | 9 | 28 | 11 | 34 | 1 | 3 |
| Fatigue | 15 | 47 | 11 | 34 | 4 | 13 | 0 | 0 |
| Nausea | 9 | 28 | 9 | 28 | 0 | 0 | 0 | 0 |
| Alopecia | 7 | 22 | 6 | 19 | 1 | 3 | 0 | 0 |
| Neutropenia | 6 | 19 | 1 | 3 | 1 | 3 | 4 | 13 |
| Anorexia | 6 | 19 | 5 | 16 | 1 | 3 | 0 | 0 |
| Skin eruption | 4 | 13 | 0 | 0 | 1 | 3 | 3 | 9 |
| Dyspepsia | 4 | 13 | 4 | 13 | 0 | 0 | 0 | 0 |
| Diarrhea | 3 | 9 | 2 | 6 | 1 | 3 | 0 | 0 |
| Skin infection | 3 | 9 | 2 | 6 | 1 | 3 | 0 | 0 |
| Weight loss | 3 | 9 | 1 | 3 | 2 | 6 | 0 | 0 |
Ten patients had dose delay and/or reduction (to 1.2 mg/kg), and six patients (19%) were terminated from the study prematurely due to toxicities. Peripheral neuropathy was the most common cause of dose modification or toxicity-related early termination. Most peripheral neuropathy was a combined sensory-motor neuropathy. Twelve of 21 patients with peripheral neuropathy had grade ≥2 peripheral neuropathy. One with grade 4 peripheral neuropathy died of pneumonia as a complication of the neuropathy. Median time to any peripheral neuropathy was 13 weeks (range, 3.0 to 38.6 weeks) and, to grade 2 peripheral neuropathy, 20.8 weeks (range, 15.0 to 46.0 weeks). By Kaplan-Meier calculation, the median time to improvement of peripheral neuropathy was 49.0 weeks (20.4 to 70.1 weeks), with 59% showing improvement or resolution by 12 months and 86% by 24 months.
DISCUSSION
This study shows significant clinical activity of BV in patients with MF and SS who have CD30 expression in skin (by routine IHC) from a nondetectable level to 100% of the mononuclear cell infiltrate. The primary end point of the study was met with an overall response rate of 70% (90% CI, 53% to 83%). Although we observed only one CR, seven patients (23%) had skin improvement of > 90%.
The majority of our patients had advanced clinical stage with adverse prognostic factors, such as LCT and/or FMF; thus, the overall response rate of 70% with acceptable DOR, PFS, and EFS data reflects the potential for addressing unmet need in these patients with aggressive, refractory disease profile. There were no statistically significant differences in response rates among clinical stages, and greater sample size would be needed to make additional interpretation of clinical effectiveness of BV in the extracutaneous compartments.
Although, objective clinical responses were observed in all CD30 enrollment groups, the median CD30max was higher in responders than in nonresponders (P < .05). In particular, if the CD30max was low (<5%), a value that is often considered as “negative” by pathologists,18,19 the likelihood of response was significantly lower (P < .005). We did not observe a significant association of CD30 expression levels with DOR, PFS, or EFS.
Standard IHC to detect tissue CD30 expression using BerH2 antibody and glass slide examination by the human eye has intrinsic limits of sensitivity and specificity with regard to low levels of CD30 staining and the discrimination of malignant cells versus infiltrating nonmalignant cells. When MSI analysis was used to assess for CD30 expression in lesions with negligible or nondetectable CD30 by routine IHC, significant low-level CD30 was visualized and quantitated, possibly accounting for the clinical activity seen in these patients.
We assessed the potential role of the tumor-infiltrating cells that have been implicated in hematolymphoid malignancies, including cytotoxic T cells (CD8+), B cells (CD20+), macrophages (CD163+), regulatory T cells (FoxP3+), and checkpoint regulator (PD-1+).20–29 Of these microenvironment factors evaluated, CD163+ M2-type tumor-associated macrophages were quite abundant (median, 40% of infiltrate). M2-type macrophages secrete T-helper type-2 cytokines (interleukin-4, interleukin-13, and transforming growth factor-β), thus can contribute to an immune-suppressive tumor environment by down-regulating M1 and T-helper type-1 cells and promote tumor-cell survival.24 These M2 macrophages have been linked to disease progression in CTCL30 and, in a xenografted CTCL murine model, depletion of M2-like tumor-associated macrophages delayed tumor development.31 In classic Hodgkin lymphoma, tumor-associated macrophages correlated with resistance to chemotherapy as well as to poor survival outcome.23,32 MSI evaluation of the double-stained lesional tissues showed that a significant proportion of these M2 tumor-associated macrophages also coexpress CD30. BV may target these macrophages in addition to the malignant T cells and disrupt their tumor-promoting function. Moreover, it is possible that these CD30 coexpressing macrophages may offer an additional source of monomethyl auristatin E to the nearby malignant T cells and augment tumor cell death. These data suggest a potential role of the microenvironment in influencing the clinical outcome with BV therapy.
Brentuximab vedotin was well tolerated in this CTCL population. As anticipated, peripheral neuropathy was a commonly observed toxicity (66% of 32 patients). Our patients were consistently evaluated by a neurologist, which may have increased the detection of neuropathy. Not all peripheral neuropathy was reversible and one patient had rapidly progressive grade 4 peripheral neuropathy. Thus, the potential for peripheral neuropathy must be carefully assessed and monitored while the patient is receiving therapy. All other adverse events were manageable and reversible.
The encouraging clinical responses in a population known to have variable CD30 expression levels along with acceptable toxicity profile in patients who have advanced and/or refractory disease with adverse prognostic features support that BV is a promising agent that warrants further clinical study either as monotherapy or in combination with other treatments for MF and SS. The ongoing phase 3 clinical trial comparing BV with methotrexate or oral bexarotene should add insight on the efficacy and safety of BV. We utilized MSI to enhance the detection of CD30 and to show coexpression of CD30 by tumor-infiltrating cells. These findings suggest that further studies of tissue biomarkers are warranted and may help optimize management strategies with BV in CTCL.
Appendix
Fig A1.

Study design. CR, complete response; EOT, end of treatment; IHC, immunohistochemistry; MF, mycosis fungoides; PD, progressive disease; PR, partial response; SS, Sézary syndrome.
Footnotes
Supported by Seattle Genetics and the Haas Family Foundation.
Presented in part at the 54th Annual Meeting of the American Society of Hematology, December 8-11, 2012, Atlanta, GA, the 56th Annual Meeting of the American Society of Hematology, December 6-9, 2014, San Francisco, CA, and the 72nd Annual Meeting of the Society for Investigative Dermatology, May 9-12, 2012, Raleigh, NC.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT01396070.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Youn H. Kim, Steven M. Horwitz
Collection and assembly of data: Youn H. Kim, Mahkam Tavallaee, Katrin A. Salva, Sima Rozati, Seema Nagpal, Michael Krathen, Sunil Reddy, Richard T. Hoppe, Annie Nguyen-Lin, Wen-Kai Weng, Randall Armstrong, Melissa Pulitzer
Data analysis and interpretation: Youn H. Kim, Mahkam Tavallaee, Uma Sundram, Gary S. Wood, Shufeng Li, Michael Krathen, Richard T. Hoppe, Annie Nguyen-Lin, Wen-Kai Weng, Melissa Pulitzer, Ranjana H. Advani, Steven M. Horwitz
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Youn H. Kim
Consulting or Advisory Role: Actelion Pharmaceuticals, Millennium Pharmaceuticals, Eisai, Kyowa-Hakko Kirin, Seattle Genetics, Celgene, Galderma, miRagen
Research Funding: Seattle Genetics, Kyowa-Hakko Kirin, Millennium Pharmaceuticals, Eisai, Merck, TetraLogic Pharmaceuticals
Travel, Accommodations, Expenses: Seattle Genetics, Celgene, Actelion Pharmaceuticals, Galderma
Mahkam Tavallaee
No relationship to disclose
Uma Sundram
No relationship to disclose
Katrin A. Salva
No relationship to disclose
Gary S. Wood
No relationship to disclose
Shufeng Li
No relationship to disclose
Sima Rozati
No relationship to disclose
Seema Nagpal
Consulting or Advisory Role: Nektar, Novartis, Novocure
Research Funding: Nektar
Michael Krathen
Consulting or Advisory Role: Actelion Pharmaceuticals, Millenium Pharmaceuticals
Sunil Reddy
No relationship to disclose
Richard T. Hoppe
Stock or Other Ownership: Johnson & Johnson, Pfizer
Honoraria: Clarient, Takeda Pharmaceuticals
Consulting or Advisory Role: Clarient
Travel, Accommodations, Expenses: Takeda Pharmaceuticals
Annie Nguyen-Lin
No relationship to disclose
Wen-Kai Weng
No relationship to disclose
Randall Armstrong
No relationship to disclose
Melissa Pulitzer
No relationship to disclose
Ranjana H. Advani
Honoraria: Millennium Takeda, General Electric/Clarient
Research Funding: Millennium Takeda, Seattle Genetics, Genentech/Roche, Allos Therapeutics, Pharmacyclics, Janssen Pharmaceuticals, Celgene, Idera Pharmaceuticals, Agensys, Regeneron
Travel, Accommodations, Expenses: Seattle Genetics
Steven M. Horwitz
Consulting or Advisory Role: Celgene, Amgen, Bristol-Myers Squibb, Janssen Pharmaceuticals, Millennium Pharmaceuticals, Seattle Genetics, Kiowa Hakko Kirin, ADC Therapeutics, Immunogen, Spectrum Pharmaceuticals
Research Funding: Celgene, Millennium Pharmaceuticals, Infinity, Kiowa Hakko Kirin, Seattle Genetics, Spectrum Pharmaceuticals
Travel, Accommodations, Expenses: Infinity Pharmaceuticals, RAND Corporation
REFERENCES
- 1.Swerdlow SH, Campo E, Harris NL, et al., editors. Lyon: IARC; 2008. 4th Edition of the WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. [Google Scholar]
- 2.Horwitz SM, Olsen EA, Duvic M, et al. Review of the treatment of mycosis fungoides and Sézary syndrome: A stage-based approach. J Natl Compr Canc Netw. 2008;6:436–442. doi: 10.6004/jnccn.2008.0033. [DOI] [PubMed] [Google Scholar]
- 3.Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC) Blood. 2007;110:1713–1722. doi: 10.1182/blood-2007-03-055749. [DOI] [PubMed] [Google Scholar]
- 4.Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 5.Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–897. doi: 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- 6.Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. 2012;30:2183–2189. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol. 2012;30:2190–2196. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123:3095–3100. doi: 10.1182/blood-2013-12-542142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartlett NL, Sharman JP, Oki Y, et al. A phase 2 study of brentuximab vedotin in patients with relapsed or refractory CD30-positve non-Hodgkin lymphomas: Interim results in patients with DLBCL and other B-cell lymphomas. Blood. 2013;122(suppl):21. abstr 848. [PubMed] [Google Scholar]
- 10.Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860–1868. doi: 10.1097/PAS.0b013e3181bf677d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benner MF, Jansen PM, Vermeer MH, et al. Prognostic factors in transformed mycosis fungoides: A retrospective analysis of 100 cases. Blood. 2012;119:1643–1649. doi: 10.1182/blood-2011-08-376319. [DOI] [PubMed] [Google Scholar]
- 11a.Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: A consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598–2607. doi: 10.1200/JCO.2010.32.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 12a.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40.
- 13.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 15.Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: A consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598–2607. doi: 10.1200/JCO.2010.32.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weng W-K, Armstrong R, Arai S, et al. Minimal residual disease monitoring with high-throughput sequencing of T cell receptors in cutaneous T cell lymphoma. Sci Transl Med. 2013;5:ra171. doi: 10.1126/scitranslmed.3007420. [DOI] [PubMed] [Google Scholar]
- 17.Mansfield JR, Hoyt C, Levenson RM. Visualization of microscopy-based spectral imaging data from multi-label tissue sections. Curr Protoc Mol Biol. 14:14. doi: 10.1002/0471142727.mb1419s84. Unit 14 19, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Sundram U, Harvell JD, Rouse RV, et al. Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod Pathol. 2003;16:802–810. doi: 10.1097/01.MP.0000081726.49886.CF. [DOI] [PubMed] [Google Scholar]
- 19.Ogura M, Ishida T, Hatake K, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol. 2014;32:1157–1163. doi: 10.1200/JCO.2013.52.0924. [DOI] [PubMed] [Google Scholar]
- 20.Hoppe RT, Medeiros LJ, Warnke RA, et al. CD8-positive tumor-infiltrating lymphocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol. 1995;32:448–453. doi: 10.1016/0190-9622(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 21.Vermeer MH, van Doorn R, Dukers D, et al. CD8+ T cells in cutaneous T-cell lymphoma: Expression of cytotoxic proteins, Fas ligand, and killing inhibitory receptors and their relationship with clinical behavior. J Clin Oncol. 2001;19:4322–4329. doi: 10.1200/JCO.2001.19.23.4322. [DOI] [PubMed] [Google Scholar]
- 22.Ni X, Hazarika P, Zhang C, et al. Fas ligand expression by neoplastic T lymphocytes mediates elimination of CD8+ cytotoxic T lymphocytes in mycosis fungoides: A potential mechanism of tumor immune escape? Clin Cancer Res. 2001;7:2682–2692. [PubMed] [Google Scholar]
- 23.Chetaille B, Bertucci F, Finetti P, et al. Molecular profiling of classical Hodgkin lymphoma tissues uncovers variations in the tumor microenvironment and correlations with EBV infection and outcome. Blood. 2009;113:2765–3775. doi: 10.1182/blood-2008-07-168096. [DOI] [PubMed] [Google Scholar]
- 24.Tiemessen MM, Mitchell TJ, Hendry L, et al. Lack of suppressive CD4+CD25+FOXP3+ T cells in advanced stages of primary cutaneous T-cell lymphoma. J Invest Dermatol. 2006;126:2217–2223. doi: 10.1038/sj.jid.5700371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nam SJ, Go H, Paik JH, et al. An increase of M2 macrophages predicts poor prognosis in patients with diffuse large B-cell lymphoma with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone. Leuk Lymphoma. 2014;55:2466–2476. doi: 10.3109/10428194.2013.879713. [DOI] [PubMed] [Google Scholar]
- 27.Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108:2957–2964. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 28.Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J Clin Oncol. 2010;28:4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jullié ML, Carlotti M, Vivot A, Jr, et al. CD20 antigen may be expressed by reactive or lymphomatous cells of transformed mycosis fungoides: Diagnostic and prognostic impact. Am J Surg Pathol. 2013;37:1845–1854. doi: 10.1097/PAS.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 30.Sugaya M, Miyagaki T, Ohmatsu H, et al. Association of the numbers of CD163(+) cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. J Dermatol Sci. 2012;68:45–51. doi: 10.1016/j.jdermsci.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Schulte BC, Zhou Y, et al. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J Invest Dermatol. 2014;134:2814–2822. doi: 10.1038/jid.2014.206. [DOI] [PubMed] [Google Scholar]
- 32.Tan KL, Scott DW, Hong F, et al. Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: A correlative study from the E2496 Intergroup trial. Blood. 2012;120:3280–3287. doi: 10.1182/blood-2012-04-421057. [DOI] [PMC free article] [PubMed] [Google Scholar]