

Abstract
Objective:
To assess the prevalence of somatic MTOR mutations in focal cortical dysplasia (FCD) and of germline MTOR mutations in a broad range of epilepsies.
Methods:
We collected 20 blood-brain paired samples from patients with FCD and searched for somatic variants using deep-targeted gene panel sequencing. Germline mutations in MTOR were assessed in a French research cohort of 93 probands with focal epilepsies and in a diagnostic Danish cohort of 245 patients with a broad range of epilepsies. Data sharing among collaborators allowed us to ascertain additional germline variants in MTOR.
Results:
We detected recurrent somatic variants (p.Ser2215Phe, p.Ser2215Tyr, and p.Leu1460Pro) in the MTOR gene in 37% of participants with FCD II and showed histologic evidence for activation of the mTORC1 signaling cascade in brain tissue. We further identified 5 novel de novo germline missense MTOR variants in 6 individuals with a variable phenotype from focal, and less frequently generalized, epilepsies without brain malformations, to macrocephaly, with or without moderate intellectual disability. In addition, an inherited variant was found in a mother–daughter pair with nonlesional autosomal dominant nocturnal frontal lobe epilepsy.
Conclusions:
Our data illustrate the increasingly important role of somatic mutations of the MTOR gene in FCD and germline mutations in the pathogenesis of focal epilepsy syndromes with and without brain malformation or macrocephaly.
A new class of genes encoding components of the mechanistic target of the rapamycin (mTOR) signal transduction pathway has recently been involved in familial focal epilepsies.1 Dishevelled, Egl-10, and Pleckstrin domain-containing protein 5 (DEPDC5) together with nitrogen permease regulator–like 2 (NPRL2) and nitrogen permease regulator–like 3 (NPRL3) form the GTPase-activating protein activity toward Rags complex 1 (GATOR1), an amino acid–sensitive inhibitor of mTORC1.2 Germline mutations in the GATOR1-encoding genes (DEPDC5, NPRL2, and NPRL3) have been reported in numerous familial and sporadic focal epilepsies.3–9 In addition to nonlesional focal epilepsies, GATOR1 mutations have also been recognized in epilepsies associated with developmental cortical malformations such as focal cortical dysplasia (FCD), in particular FCD type IIb (with balloon cells) and IIa (without balloon cells).10–14
Recent studies reported brain somatic missense mutations in the MTOR gene itself in 3 patients with hemimegalencephaly,12 in 12/77 (15%) with FCD II,15 in 6/13 (46%) with FCD IIb,16 in 1 with FCD IIa,17 and in 10 with various developmental disorders from FCD IIa to megalencephaly.18 Evidence for mTORC1 hyperactivity was shown in resected brain tissue after epilepsy surgery.15,16,18
Rare germline heterozygous missense mutations in MTOR have also been reported in a subset of patients with megalencephaly, intellectual disability, and seizures.18–22
In this study, we compared the prevalence and type of MTOR somatic mutations in a cohort of FCD patients, with MTOR germline mutations in cohorts of patients with epilepsy with or without brain malformations.
METHODS
Patients.
As a first step, 3 different cohorts were screened for pathogenic variants in MTOR (figure 1).
Figure 1. Workflow of the study.

FCD = focal cortical dysplasia; MCD = malformation of cortical development.
First, for mosaic mutation detection, we prospectively recruited a cohort of 20 participants with FCD who underwent surgery to treat drug-resistant focal epilepsy. The cohort consisted of 18 children operated at the Fondation Rothschild (Paris, France) between 2015 and 2016 and 2 adult participants operated at the Pitié-Salpêtrière hospital (Paris, France) and at the Swiss Epilepsy Center (Zurich, Switzerland).
Second, for germline mutation detection, we included a previously described French research cohort of 93 unrelated European patients with focal epilepsy, including 55 probands with familial focal epilepsy, 9 sporadic patients with nonlesional focal epilepsy, 14 patients with familial focal epilepsy, and at least one family member with FCD, and 15 sporadic patients with focal epilepsy and FCD.8 In this cohort, 7 individuals were previously shown to have a mutation in one of the GATOR1-encoding genes.8 We also screened a third Danish cohort of 245 unrelated patients with various forms of childhood-onset epilepsies referred for diagnostic gene panel testing. The phenotypic spectrum within this cohort was broad and ranged from benign familial neonatal/infantile epilepsy to generalized epilepsies, focal epilepsies, and severe epileptic encephalopathies.
In addition, we collected 5 previously unreported patients with MTOR mutations through data sharing with Italy and the United States. The Italian cohort consisted of 40 patients with epilepsy, intellectual disability, and malformations of cortical development or morphologic characteristics suggesting altered neuronal proliferation. The US cohort consisted of 338 probands (from 317 families) with developmental delay, intellectual disability, and/or other neurologic symptoms, including epilepsy.
All probands and affected family members underwent detailed clinical examination, including review of the medical files, MRIs, and EEG investigations.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the following local ethical committees: CPP Ile de France II, No ID-RCB/EUDRACT 2015-A00671-48, the Pediatric Ethical Committee of the Tuscany Region, and the Institutional Review Board at the University of Alabama at Birmingham (protocol #X130201001). Written informed consent was obtained from each participant or parents of minors. Parents of participants 10a, 10b, and 12 gave consent for publication of their children's picture. The Danish cohort was recruited in a diagnostic setting, and the patients or parents of minors gave written consent for publications of their findings.
Genetic testing.
Targeted gene panel for detection of somatic variants.
Genomic DNA was extracted from peripheral blood lymphocytes and from fresh frozen resected brain tissue of FCD participants. To search for mosaic mutations, we designed a custom gene panel of 25 genes belonging to the PI3K/AKT/mTOR pathway (SeqCapEZ custom design, Roche NimbleGen) including MTOR and the GATOR1-encoding genes (DEPDC5, NPRL2, and NPRL3).
Deep sequencing was performed on a NextSeq (2 × 75 nt). GenoSplice performed quality control, processing, and detection of the variants. FASTQ sequence files were mapped using hg19 reference with bwa-0.7.12 (mem) and converted to bam with samtools-1-1. Single nucleotide variant and insertion/deletion calling was performed with GATK-2014.3 to call heterozygous germline variants present either in the resected brain or lymphocyte-derived DNA.
Somatic variant calling was performed with samtools-1-1 (mpileup) to detect low frequency variants (i.e., <10%). The pileup was parsed with pileup2base which provides the count of reads for each strand and nucleotides by coordinates. A home-made Perl script was developed to filter variants according to the following criteria: 1/variants not retrieved by GATK; 2/≥ 10 reads with the alternate allele; 3/≥ 1% of allelic frequency; and 4/maximum strand bias value of 0.5.
Picoliter droplet-based digital PCR.
Somatic p.Ser2215Phe and p.Ser2215Tyr mutations were confirmed by digital PCR (dPCR). Genomic DNA was extracted from a second piece of the same tissue block from participants 1–5 and was tested for the presence of specific MTOR mutations. No brain tissue of patient 6 was left. Custom LNA double-dye probes designed to detect both mutant alleles c.6644C>T (p.Ser2215Phe) and c.6644C>A (p.Ser2215Tyr) and wild-type were purchased from Eurogentec. RainDrop Digital PCR system (Raindance Technologies Billerica) was used as previously described23 with the following modifications: dPCR was performed with Taqman Universal Master Mix (life Technologies), 125 nM final concentration of both probes and 250 nM final concentration of primers.
Targeted gene panels for detection of germline variants.
In the French cohort of 93 patients with focal epilepsies, the search for heterozygous variants in genomic blood DNA was accomplished by a previously reported methodology and research epilepsy panel consisting of 76 genes (SeqCapEZ custom design, Roche NimbleGen).8
In the Danish cohort, the genomic blood DNA from 245 patients with various childhood-onset epilepsies was screened using a diagnostic sequencing panel (Sure Select, Agilent Technologies) consisting of 85 epilepsy genes.
The Italian cohort of 40 patients was screened using a gene panel consisting of 15 genes belonging to the PI3K/AKT/mTOR pathway (HaloPlex Target Enrichment System, Agilent Technologies).
MTOR variants were assumed to be pathogenic if they were nonsynonymous missense, seen less than 2 times in control samples (Exome Aggregation Consortium (ExAC) set of ∼61,000 exomes—exac.broadinstitute.org [accessed August 2016]), had arisen de novo in patients without family history of epilepsy, or segregated with the disease in the family. Sanger sequencing was used to confirm all variants.
Exome/genome sequencing for detection of germline variants.
The 338 USA probands were subject to trio-based exome or genome sequencing as part of a Clinical Sequencing Exploratory Research project (conducted at the HudsonAlpha Institute for Biotechnology).24 Whole-exome (patients 10a, 10b, and 11) or genome (patient 12) sequencing was conducted to a median depth of 65X or 30X, respectively. Exome capture was completed using NimbleGen SeqCapEZ Exome version 3. Reads were aligned to reference hg19 using bwa (0.6.2). GATK best practice methods were used to identify variants. Maternity and paternity of each parent were confirmed by whole-exome/genome kinship coefficient estimation with KING. De novo variants were identified as heterozygous variant calls in the proband in which there were at least 10 reads in both parents and the proband, an alternate allele depth of at least 20% in the proband and less than 5% in each parent, and a minor allele frequency <1% in the 1000 Genomes Project and the ExAC. All candidate de novo variants that either affected a protein or had a scaled Combined Annotation Dependent Depletion (CADD) score >10 were manually reviewed.25 In each patient described, no other variants were identified as potentially disease causing. All de novo variants were confirmed by Sanger sequencing to be heterozygous in the probands and absent from both of their parents.
Histopathology and immunostaining.
Histopathologic diagnosis was made according to the classification of the International League Against Epilepsy Diagnostic Methods Commission26 and included 3 FCD I, 9 FCD IIa, 7 FCD IIb, and 1 FCD III. Paraffin-embedded resected brain tissues of patients 1–5 were examined for the presence of cytoarchitectural features of FCD using hematoxylin and eosin (H&E) staining. Brain tissue of patient 6 was not available for further immunostaining. mTORC1 activity was assessed by visualizing the phosphorylation level of its downstream effector, the ribosomal protein S6, a commonly used biomarker in mTORC1 pathway research.27 Sections (10 μm) were probed overnight at 4°C with an antibody against Ser235/236 phosphorylated S6 (#4856, 1:200 dilution; Cell Signaling, Danvers, MA) and visualized using avidin-biotin conjugation (Vectastain ABC Elite; Vector Laboratories, Burlingame, CA). Images were acquired using the Leica DM2500 microscope.
Protein modeling.
Protein modeling was performed using YASARA structure (yasara.org), merging 29 models of multiple PDB files. Molecular dynamic simulations were performed using AMBER031, and structure statistics were calculated with the YASARA2 z score calculation. Eukaryotic linear motif (ELM) predictions were performed on the ELM server 3, and posttranslational modifications were predicted using multiple prediction tools.28–36
RESULTS
Brain somatic mutations in MTOR.
To identify low-level mosaic mutations, we designed a targeted sequencing panel comprising all 57 coding exons and flanking intronic regions of MTOR (NM_004958.3) and the GATOR1-complex encoding genes (DEPDC5, NPRL2, and NPRL3). We achieved a high sequencing coverage of all 4 genes (mean coverage: 2,653×, 2,527×, 2,666×, and 2,342×, respectively). We searched for mosaic variants with allele frequencies ≥1%. We detected neither germline nor mosaic coding pathogenic variants in DEPDC5, NPRL2, or NPRL3 according to our filtering criteria. Brain mosaic mutations in MTOR were found in 2 patients with FCD IIa and 4 patients with FCD IIb (table 1, figure 1), representing 37% (6/16) of all FCD II individuals. A recurrent variant in MTOR (c.6644C>T/p.Ser2215Phe) was identified in patients 1–3 in 138/2,186 (6.31%), 79/2,504 (3.15%), and 28/2,481 (1.13%) reads, respectively. A second recurrent variant (c.6644C>A/p.Ser2215Tyr) affecting the same nucleotide was detected in patients 4–5 in 26/718 (3.62%) and 12/751 (1.6%) reads. Patient 6 with FCD IIb carried 2 MTOR variants located on the same allele: the recurrent variant c.4379T>C/p.Leu1460Pro was detected in 62/2,522 (2.46%) reads and c.4375G>T/p.Ala1459Ser (never reported) was detected in 60/2,493 (2.41%) alleles. To validate these findings, we performed picoliter droplet-based dPCR based on the single-molecule parallel amplification of MTOR wild-type and mutated alleles. dPCR confirmed both the presence and the allelic frequencies of the 2 c.6644C>T/p.Ser2215Phe and c.6644C>A/p.Ser2215Tyr variants in DNA sample replicates from the same tissue block to exclude sequencing artifacts (table 1).
Table 1.
Clinical features of patients with somatic MTOR variants in FCD
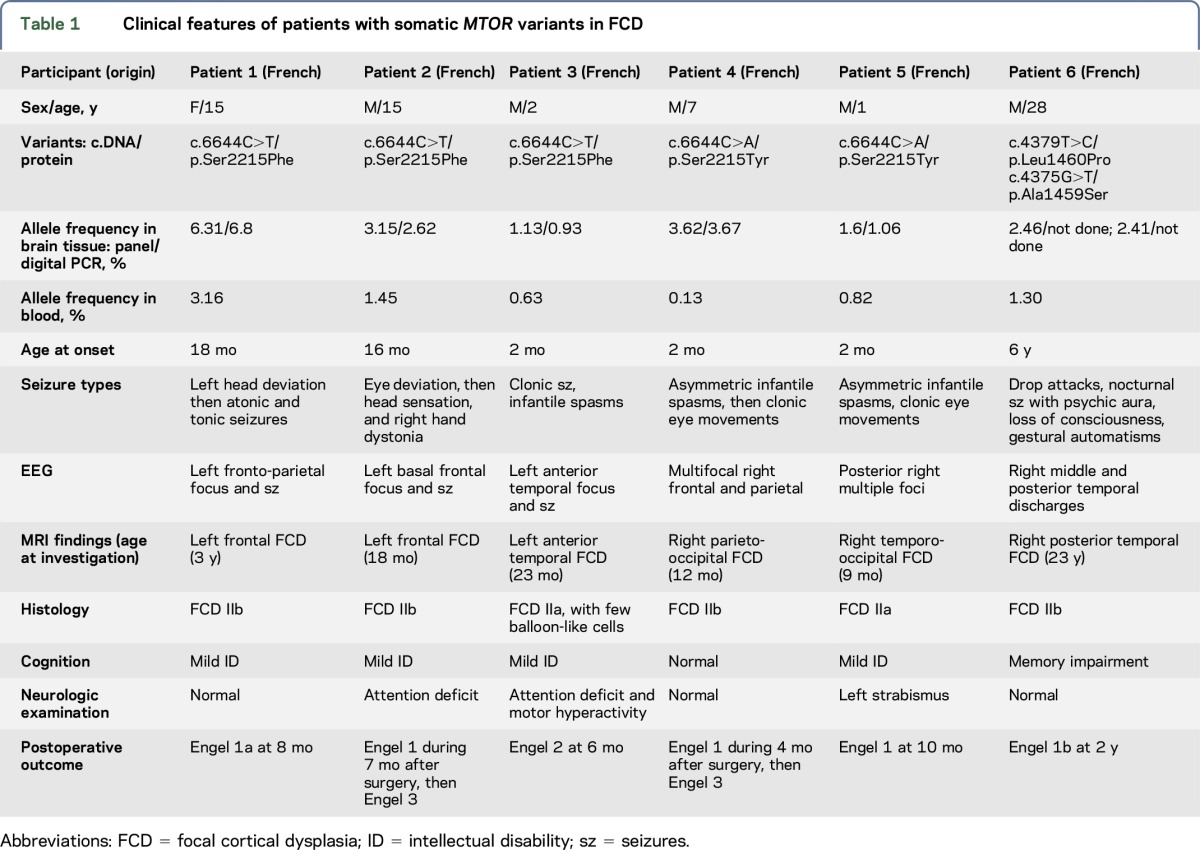
All recurrent mosaic MTOR variants were predicted deleterious by SIFT, disease-causing by Mutation Taster, and possibly damaging by PolyPhen-2, and were absent from the ExAC database. Allele frequencies of the mosaic variants were low in the genomic DNA extracted from the brain tissues (range 1.1%–6.3%) (table 1). We noticed that with the high sensitivity of the deep sequencing, mosaic variants could also be detected in the blood DNA, although with allele frequencies half lower than in the brain DNA (range 0.1%–3%). However, these variants were not present in the blood DNA of patients without detected MTOR brain somatic mutations. The recurrent variants p.Ser2215Phe, p.Ser2215Tyr, and p.Leu1460Pro were considered to be pathogenic as they have already been linked to FCD.15,16,18 These mosaic variants were detected across several studies using distinct technologies and sequencing platforms, excluding that they might correspond to sequencing artifacts.
Germline mutations in MTOR.
In the French cohort of 93 patients with focal epilepsy, we identified only one germline variant (c.4128T>G/p.Asp1376Glu) in a patient with nonlesional focal epilepsy with auditory features. This novel variant was predicted deleterious by all in silico prediction tools and was absent from the ExAC database. Sanger sequencing, however, revealed that the variant was inherited from the asymptomatic father and was thus considered to be a variant of unknown significance.
In the Danish cohort consisting of 245 patients, we detected 2 germline heterozygous variants in MTOR (c.5494G>A/p.Ala1832Thr and c.7501A>G/p.Ile2501Val) corresponding to ∼1% of a diagnostic cohort of patients with childhood-onset epilepsy (figure 1, table 2). Variant c.5494G>A/p.Ala1832Thr occurred de novo in patient 7 with nonlesional occipital lobe epilepsy, macrocephaly, learning difficulties, and attention-deficit/hyperactivity disorder (ADHD). Based on the de novo occurrence, highly evolutionary conservation of the Ala1832 amino acid, and low frequency in the ExAC database (1/110,816 chromosomes), we considered the p.Ala1832Thr variant to be pathogenic. The c.7501A>G/p.Ile2501Val variant, identified in patient 8a, also affects a highly evolutionary conserved amino acid, was predicted to be disease causing by Mutation Taster (but was predicted to be benign by SIFT and PolyPhen-2) and was absent in the ∼61,000 individuals from the ExAC database. The p.Ile2501Val variant was inherited from an affected mother (patient 8b). Both proband and mother had nonlesional nocturnal frontal lobe epilepsy, a phenotype compatible with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). Patients 7 and 8a did not carry additional pathogenic variants in one of the 84 epilepsy genes of the panel, including genes known to be involved in ADNFLE such as CHRNA4, CHRNA2, CHRNB2, DEPDC5, or KCNT1, suggesting that this variant is likely to be pathogenic.
Table 2.
Clinical features of patients with germline MTOR variants
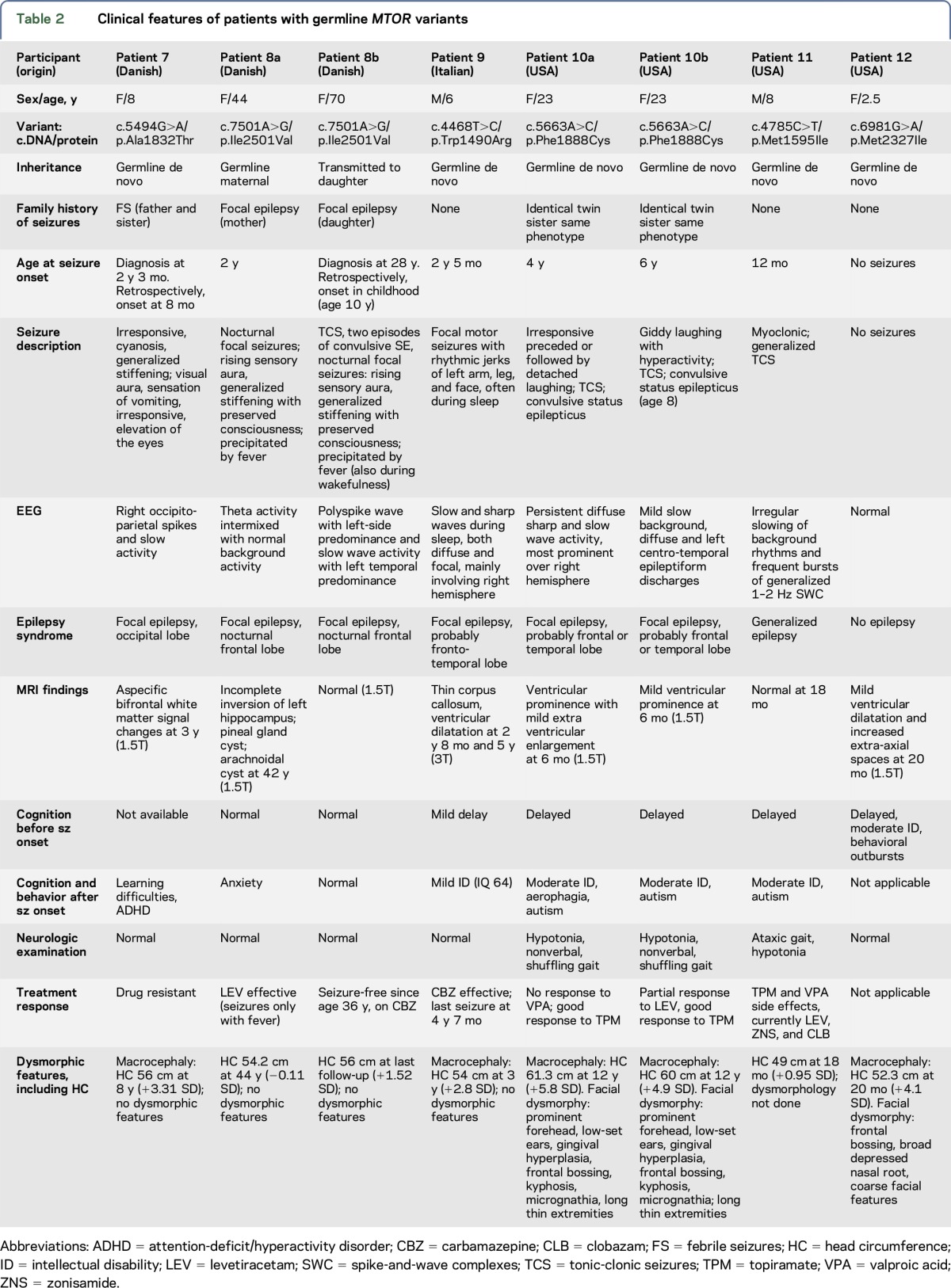
Further germline MTOR mutations were identified through data sharing with collaborators in the United States and Italy. We collected 4 additional de novo germline MTOR mutations as follows: c.4468T>C/p. p.Trp1490Arg (patient 9, Italian cohort); c.5663A>C/p.Phe1888Cys (identical twin pair, patients 10a and 10b, USA cohort); c.4785C>T/p.Met1595Ile (patient 11, USA cohort); and c.6981G>A/p.Met2327Ile (patient 12, USA cohort) (figure 1, table 2). All six variants were predicted to be disease-causing by different in silico prediction methods and were absent from the ExAC database. Sanger confirmation and segregation analysis revealed that all variants occurred de novo, suggesting that they are likely to be pathogenic.
Prediction of pathogenicity.
We assessed the deleteriousness of the MTOR variants using different prediction tools. First, we calculated the CADD scores for all novel germline variants: 15.18 (p.Ile2501Val), 19.76 (p.Ala1832Thr), 26 (p.Met1595Ile), 29.2 (p.Phe1888Cys), and 34 (p.Met2327Ile), as well as the somatic variants: 26.1 (p.Leu1460Pro), 31 (p.Ala1459Ser), 32 (p.Ser2215Tyr), and 33 (p.Ser2215Phe). CADD scores were clearly consistent with them being pathogenic.
Second, we observed in ExAC that the number of missense variants expected in MTOR is 930, while those observed are only 438 (z score = 7.89), indicating that MTOR is intolerant to missense variants. For each variant identified in each cohort, we performed a χ2 test with Yates correction (graphpad.com/quickcalcs/contingency1.cfm) using the ∼61,000 ExAC individuals as a control cohort. All MTOR variants, either novel germline or recurrent somatic, reached extremely statistically significant values (p < 0.0001). In addition, with a 0.34% Residual Variation Intolerance Score (RVIS), MTOR ranks among the top most constrained human genes.37
Furthermore, we asked how the variants may affect the structure and/or function of the mTOR protein. Structural domains are composed of 2 HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1) domains, the FAT (FRAP, ATM, TRRAP) domain, the FKBP12-rapamycin binding domain, the kinase domain, and the FAT C-terminal domain. Most mutations reported here, either somatic or germline, were clustered in and around the FAT and the kinase domains (figure 2). The crystal structure of mTOR has revealed that the FAT domain interacts with the kinase domain38 and DEP domain-containing mTOR-interacting protein (DEPTOR),39 a repressor of mTOR activity. We can speculate that mutations in the FAT domain may alter the conformational structure of protein or inhibit the binding with DEPTOR, maintaining the catalytic site exposed and active.
Figure 2. Domain/structural organization of mTOR protein with the positions of the mutations indicated.

In black: germline mutations; in blue: somatic mutations; in bold: recurrent mutations. Structural domains are composed of HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1) repeats; FAT (FRAP, ATM, TRRAP) domain; FRB (FKBP12-rapamycin binding) domain; kinase domain; and FATC (FAT C-terminal) domain.
We further performed protein modeling of mTOR and showed that most of the variants seen have a high probability of altering protein structure and dynamics through either altered hydrophobic collapse (p.Met1595Ile, p.Met2327Ile, and p.Ile2501Val), helical packing (p.Ala1459Ser, p.Leu1460Pro, and p.Ser2215Phe/Tyr), or surface conserved sites likely involved in protein-protein interactions (p.Trp1490Arg) (table e-1, figure e-1 at Neurology.org/ng). While amino acid 1888 is not as strongly conserved, Phe1888Cys is predicted to elevate the probability for protein lipidation (myristoylation, palmitoylation, and farnesylation), a modification known to alter protein localization, and thus is also likely to deleteriously impact mTOR function. Altogether, these missense variants in MTOR are likely to be pathogenic.
Phenotypic analysis.
Brain mosaic mutations in MTOR were found in 2 patients with FCD IIa and in 4 with FCD IIb (table 1), representing 37% (6/16) of all FCD II individuals. No MTOR variants were found in the 3 patients with FCD I nor in the only patient with FCD III. All patients had characteristic imaging features of FCD (figure 3) and drug-resistant focal epilepsy prior to surgery. Postoperative outcome ranged from Engel class 1 to 3. Histopathology of resected brain tissue showed an FCD IIa with dysmorphic neurons on H&E staining in patients 3 and 5, and an FCD IIb with dysmorphic neurons and balloon cells in patients 1, 2, and 4 (figure 3). We assessed mTORC1 activity by visualizing the phosphorylation level of its downstream effector S6 on Ser235/236 (pS6), a well-recognized marker of mTORC1 activity.27 Hyperactivation of the mTORC1 pathway as shown by S6 intense phosphorylation was observed in dysmorphic neurons of patients with FCD IIa and FCD IIb, in contrast to apparently normal adjacent neurons.
Figure 3. Imaging, histopathology, and mTORC1 pathway activity of patients with focal cortical dysplasia.

(A) MRI with residual lesion after the first and before the second operation because of incomplete initial resection (preoperative MRI unavailable). (D, G, J, M, P) Preoperative brain MRI; arrows indicate the dysplastic lesion. (B, E, H, K, N) Hematoxylin and eosin (H&E) staining; blue arrows indicate balloon cells, and black arrows indicate dysmorphic neurons. (C, F, I, L, O) Ser235/236 phosphorylated S6 (pS6 235/6) immunostaining counterstained with hematoxylin, showing labeling of dysmorphic neurons. Brain tissue of patient 6 was not available for further immunostaining. Scale bar: 20 μm. FCD = focal cortical dysplasia.
Germline mutations were found in patients with a variable phenotypic spectrum (table 2). Seven of the 8 individuals with germline variants had epilepsy. Six had focal epilepsy, and 1 had generalized epilepsy, with age at onset ranging from 8 months to 6 years. The presumptive lobe of seizure onset was variable from frontal, fronto-temporal to occipital. One mother–daughter pair (patients 8a and 8b) had ADNFLE, and 2 patients formed an identical twin pair (patients 10a and 10b) with gelastic seizures. Four patients did not have a family history of epilepsy. Five patients had macrocephaly, and 3 had dysmorphic facial features (figure 4). All patients with germline mutations had either normal imaging or unspecific MRI abnormalities including thin corpus callosum and/or ventricular dilatation (figure 4). Three patients had a normal or near normal development, 1 patient had mild and 4 had moderate intellectual disability. Behavioral and psychiatric disturbances including autistic features and ADHD were reported in 4 patients. No specific dermatological features were observed.
Figure 4. Unspecific MRI findings and facial dysmorphy in patients with germline variants.

(A and B) T2-weighted 3T MRI images of patient 9. Axial image shows dilated lateral ventricles (A), and sagittal image shows thinning of the posterior half of the corpus callosum (B). (C and D) Pictures of identical twin pair 10a and 10b show prominent forehead, low-set ears, gingival hyperplasia, frontal bossing, and micrognathia (MRIs not available for publication). (E–G) Fluid-attenuated inversion recovery (FLAIR) weighted 1.5T MRIs of the brain, and the picture of patient 12. Axial and coronal images shows mild ventricular dilatation. Picture shows macrocephaly with frontal bossing, broad depressed nasal root, and coarse facial features.
DISCUSSION
Brain somatic MTOR mutations have recently been identified in patients with malformations of cortical development, in particular FCD (for review reference 40). In this study, we report recurrent somatic mutations in MTOR in 30% of patients with FCD and 37% of patients considering only patients with FCD II (2 FCD IIa and 4 FCD IIb). We also describe 8 patients with a spectrum of developmental disorders due to germline MTOR mutations. While most patients had focal (and less frequently generalized) epilepsy, mild-to-moderate intellectual disability, and/or dysmorphic features, we extended the phenotypic spectrum associated with germline variants to milder phenotypes than previously reported; none of the patients had a malformation of cortical development identified on MRI, and 3 patients had (near) normal cognitive outcome. At the mildest end of the spectrum, 1 mother–daughter pair had nonlesional treatment-responsive ADNFLE and normal intellectual development. In addition, we report 1 individual with macrocephaly and intellectual disability without seizures. Our results are consistent with previous studies reporting individuals with megalencephaly and seizures due to germline activating mutations in MTOR.18–22
In a previous report, the level of mosaicism of somatic mutations has been suggested to correlate with the extent of the brain malformation.18 In our series of patients with FCD, we could not directly correlate the allele frequencies of somatic variants to the size of the lesion. Given the central and ubiquitous role of the mTORC1 transduction pathway in regulating cell functions, it was surprising to discover heterozygous germline MTOR mutations in the following: (1) patients with purely neurologic symptoms and (2) patients without brain malformations, in contrast to individuals with somatic mutations. Two possible hypotheses can be raised. First, it appears that the amino acids affected by germline mutations are different from the ones affected by somatic mutations. All variants were missense, assumed to enhance the activity of the mTORC1 pathway. However, while somatic mutations may be hyperactivating, and thus cause brain malformations of development, germline mutations could be less activating. A second hypothesis is the possible occurrence during brain development of multiple-hit mosaic mutations (missed with our targeted sequencing strategy) in different genes of the same pathway, explaining the apparition of a focal lesion as seen in FCD. This scenario is known as the multiple-hit or Knudson hypothesis in cancer41 and is supported by our previous report of a patient with FCD with both a somatic and a germline DEPDC5 mutation.11 It is also supported by the finding that brain somatic mutations occur frequently.42 In this FCD cohort, we did not identify an MTOR or a GATOR1 germline mutation in addition to the somatic MTOR variants.
While the somatic MTOR mutations were recurrent, all germline mutations identified in this study were novel. Recurrence of p.Ser2215Phe, p.Ser2215Tyr, and p.Leu1460Pro mutations that result in mTOR activation in multiple FCD cases15,16,18 suggests mutational hotspots. Of interest, the same recurrent mutations were also found in various tumors (cancer.sanger.ac.uk/cancergenome/projects/cosmic).43 Most of these oncogenic mutations, like FCD-causing mutations, cluster near the kinase domain and the FAT domain of mTOR protein. A recent study reported that the hyperactive kinase domain p.Met2327Ile variant confers drug resistance to mTOR second-generation inhibitors suggesting potential issues in the field of precision medicine.44
The role of genes of the GATOR1-mTORC1 pathway (MTOR, DEPDC5, NPRL2, and NPRL3) is being increasingly recognized in focal epilepsies, with and without structural abnormalities.1 Most mutations in GATOR1-encoding genes are loss of function (nonsense, frameshift, splice variants), while all mutations in the MTOR gene are missense mutations predicted to be activating. Both GATOR18,13,14 and MTOR mutations17 have been shown to lead to hyperactivation of the mTORC1 pathway in resected epileptogenic brain tissue. How an increased activity of the intracellular signaling cascade alters network function and ultimately leads to seizures is one of the next challenging questions to be elucidated.
This study confirms the important role of somatic MTOR mutations in the pathogenesis of FCD, and jointly with previous reports shows that germline MTOR mutations may contribute to a broad phenotypic spectrum, ranging from focal epilepsy without MRI-detectable brain abnormalities and normal development, to hemimegalencephaly, intellectual disability, and epilepsy. The treatment effectiveness in these patients with mTORC1 pathway inhibitors needs further evaluation, but meanwhile, screening of MTOR in a broad range of focal epilepsies is warranted.
Supplementary Material
ACKNOWLEDGMENT
The authors thank patients and families for their participation in the study and clinicians, in particular Thomas Dorn from the Swiss Epilepsy Center in Zurich, for referring patients. They thank Eric Noé for technical assistance, Théo Ribierre for critical reading, and Marc Polivka for neuropathology of patients with FCD; and the ICM sequencing platform (Yannick Marie) and the ICM DNA and cell bank (Christelle Dussert, Sylvie Forlani). The digital PCR experiments were performed in the eDIAG platform (UMRS1147, S. Garrigou and P. Nizard). The authors also thank the HudsonAlpha CSER team members, especially Candice Finnila, David Gray, Michelle Amaral, Michelle Thompson, Shirley Simmons, Kelly East, Eddy Lose, Greg Barsh, and Whitley Kelley.
GLOSSARY
- ADHD
attention-deficit/hyperactivity disorder
- ADNFLE
autosomal dominant nocturnal frontal lobe epilepsy
- CADD
Combined Annotation Dependent Depletion
- DEPDC5
Dishevelled, Egl-10, and Pleckstrin domain-containing protein 5
- DEPTOR
domain-containing mTOR-interacting protein
- dPCR
digital PCR
- ELM
eukaryotic linear motif
- ExAC
Exome Aggregation Consortium
- FAT
FRAP, ATM, TRRAP
- FCD
focal cortical dysplasia
- GATOR1
GTPase-activating protein activity toward Rags complex 1
- HEAT
huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1
- H&E
hematoxylin and eosin
- mTOR
mammalian target of rapamycin
- NPRL
nitrogen permease regulator–like
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Rikke S. Møller and Sarah Weckhuysen: analysis and interpretation of genetic and clinical data, and drafting the manuscript. Mathilde Chipaux: acquisition and interpretation of clinical data. Elise Marsan: acquisition and analysis of immunohistochemistry. Valerie Taly: acquisition and analysis of dPCR. E. Martina Bebin: acquisition, interpretation of clinical data, and revising the manuscript for intellectual content. Susan M. Hiatt: acquisition and interpretation of whole-exome/genome data. Jeremy W. Prokop: acquisition and interpretation of protein-modeling data. Kevin M. Bowling: acquisition and interpretation of whole-exome/genome data. Davide Mei and Valerio Conti: analysis of the data. Pierre de la Grange: analysis of genetic data. Sarah Ferrand-Sorbets, Georg Dorfmüller, and Virginie Lambrecq: acquisition and interpretation of clinical data. Line H.G. Larsen: analysis of the data. Eric Leguern: study design. Renzo Guerrini: analysis of the data and revision of the manuscript for intellectual content. Guido Rubboli: acquisition and interpretation of clinical data. Gregory M. Cooper: acquisition, interpretation of whole-exome/genome data, and conception and design of the HudsonAlpha Clinical Sequencing Exploratory Research (CSER) project. Stéphanie Baulac: analysis and interpretation of genetic data, study supervision, drafting the manuscript, and obtaining funding.
STUDY FUNDING
This study was funded by NHGRI grant UM1HG007301, the program “Investissements d'avenir” ANR-10-IAIHU-06 (to S.B., E.L.G., S.W. and V.L.), and the Fondation pour la Recherche Médicale (team FRM DEQ2015033 to S.B. and FDT20160736468 to E.M.). E.M.B. received support from Genomic diagnosis in children with developmental delay HG-12-017 Clinical Sequencing Exploratory Research (UM1) NIH funded-UM1HG007301. This work was supported in part by a grant from the European Union Seventh Framework Program FP7 under the project DESIRE (grant agreement 602531) (to R.G.).
DISCLOSURE
Rikke S. Møller reports no disclosures. Sarah Weckhuysen has received research support from the French program “Investissements d'avenir.” Mathilde Chipaux and Elise Marsan report no disclosures. Valerie Taly has been a consultant for Raindance Technologies (honoraries) and Boehringer Ingelheim; has received travel and speaker honoraria from Raindance Technologies; has served on the editorial board of Biomolecular Detection and Quantification; and holds patents for Surfactant to allow for stabilization of emulsions, and Combinatorial methods for gene library creation for directed evolution applications. E. Martina Bebin has served on scientific advisory boards for Novartis, GW Pharmaceuticals, the FDA Orphan Drug Program, and NIH; has received gifts from Novartis and GW Pharmaceuticals; has served on the editorial board of Pediatric Neurology; has been a consultant for GW Pharmaceuticals; has served on the speakers' bureau of GW Pharmaceuticals; and has received research support from NIH. Susan M. Hiatt has received research support from NHGRI. Jeremy W. Prokop has received research support from NIH. Kevin M. Bowling has received research support from NHGRI. Davide Mei and Valerio Conti have served on the editorial board of Epilepsia. Pierre de la Grange is a cofounder of Genosplice. Sarah Ferrand-Sorbets, Georg Dorfmüller, and Virginie Lambrecq report no disclosures. Line H.G. Larsen is employed by Amplexa Genetics. Eric Leguern has received research support from the program “Investissements d'avenir” (ANR-10-IAIHU-0), INSERM, and Assistance Publique des Hôpitaux de Paris. Renzo Guerrini has served on the scientific advisory boards of Eisai Inc, Novartis, and Zogenix; has served on the editorial boards of Epilepsia, Progress in Epileptic Disorders, Neuropediatrics, Journal of Child Neurology, Seizure, BMC Medical Genetics, Topics in Epilepsy, the Journal of Pediatric Epilepsy, Epileptic Disorders, the European Neurological Journal, Neurology, and the Journal of Embryology & Developmental Biology; receives publishing royalties from Cambridge University Press, Lippincott Williams & Wilkins, John Libbey Eurotext, and Oxford University Press; and has received research support from EPICURE, EC PROGRAM, EU PROGRAM, DESIRE (Development and Epilepsy), Italian Ministry of Health and Tuscany Region, and the Pisa Foundation. Guido Rubboli has served on the editorial boards of Epileptology, Epileptic Disorders, Clinical Cases and Reviews in Epilepsy, and Behavioural Neurology. Gregory M. Cooper has served on the editorial board of PLoS Genetics Genome Research and has received research support from NHGRI. Stéphanie Baulac has received research support from the program “Investissements d'avenir” (ANR-10-IAIHU-06) and the Fondation pour la Recherche Médicale. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Baulac S. mTOR signaling pathway genes in focal epilepsies. Prog Brain Res 2016;226:61–79. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Peled L, Chantranupong L, Cherniack AD, et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340:1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551. [DOI] [PubMed] [Google Scholar]
- 5.Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 2014;82:2101–2106. [DOI] [PubMed] [Google Scholar]
- 6.Ricos MG, Hodgson BL, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol 2016;79:120–131. [DOI] [PubMed] [Google Scholar]
- 7.Korenke GC, Eggert M, Thiele H, Nurnberg P, Sander T, Steinlein OK. Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia 2016;57:e60–e63. [DOI] [PubMed] [Google Scholar]
- 8.Weckhuysen S, Marsan E, Lambrecq V, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 2016;57:994–1003. [DOI] [PubMed] [Google Scholar]
- 9.Striano P, Serioli E, Santulli L, et al. DEPDC5 mutations are not a frequent cause of familial temporal lobe epilepsy. Epilepsia 2015;56:e168–e171. [DOI] [PubMed] [Google Scholar]
- 10.Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 2014;75:782–787. [DOI] [PubMed] [Google Scholar]
- 11.Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol 2015;77:675–683. [DOI] [PubMed] [Google Scholar]
- 12.D'Gama AM, Geng Y, Couto JA, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol 2015;77:720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scerri T, Riseley JR, Gillies G, et al. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol 2015;2:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sim JC, Scerri T, Fanjul-Fernandez M, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol 2016;79:132–137. [DOI] [PubMed] [Google Scholar]
- 15.Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 16.Nakashima M, Saitsu H, Takei N, et al. Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 2015;78:375–386. [DOI] [PubMed] [Google Scholar]
- 17.Leventer RJ, Scerri T, Marsh AP, et al. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology 2015;84:2029–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mirzaa GM, Campbell CD, Solovieff N, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 2016;73:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith L, Saunders C, Dinwiddie D, et al. Exome sequencing reveals de novo germline mutation of the mammalian target of rapamycin (MTOR) in a patient with megalencephaly and intractable seizures. J Genomes Exomes 2013:63–72. [Google Scholar]
- 21.Mroske C, Rasmussen K, Shinde DN, et al. Germline activating MTOR mutation arising through gonadal mosaicism in two brothers with megalencephaly and neurodevelopmental abnormalities. BMC Med Genet 2015;16:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baynam G, Overkov A, Davis M, et al. A germline MTOR mutation in Aboriginal Australian siblings with intellectual disability, dysmorphism, macrocephaly, and small thoraces. Am J Med Genet A 2015;167:1659–1667. [DOI] [PubMed] [Google Scholar]
- 23.Garrigou S, Perkins G, Garlan F, et al. A study of hypermethylated circulating tumor DNA as a universal colorectal cancer biomarker. Clin Chem 2016;62:1129–1139. [DOI] [PubMed] [Google Scholar]
- 24.Green RC, Goddard KA, Jarvik GP, et al. Clinical sequencing exploratory research consortium: accelerating evidence-based practice of genomic medicine. Am J Hum Genet 2016;99:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aronica E, Crino PB. Epilepsy related to developmental tumors and malformations of cortical development. Neurotherapeutics 2014;11:251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 2003;24:1999–2012. [DOI] [PubMed] [Google Scholar]
- 29.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004;4:1633–1649. [DOI] [PubMed] [Google Scholar]
- 30.Krieger E, Joo K, Lee J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 2009;77(suppl 9):114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinkel H, Michael S, Weatheritt RJ, et al. ELM: the database of eukaryotic linear motifs. Nucleic Acids Res 2012;40:D242–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z, Cao J, Ma Q, Gao X, Ren J, Xue Y. GPS-YNO2: computational prediction of tyrosine nitration sites in proteins. Mol Biosyst 2011;7:1197–1204. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, Zheng Y, Li H, et al. GPS-Lipid: a robust tool for the prediction of multiple lipid modification sites. Scientific Rep 2016;6:28249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel 2008;21:639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue Y, Liu Z, Gao X, et al. GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS One 2010;5:e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Q, Xie Y, Zheng Y, et al. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res 2014;42:W325–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrovski S, Gussow AB, Wang Q, et al. The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. PLoS Genet 2015;11:e1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation by the rapamycin-binding domain. Nature 2013;497:217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009;137:873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blumcke I, Sarnat HB. Somatic mutations rather than viral infection classify focal cortical dysplasia type II as mTORopathy. Curr Opin Neurol 2016;29:0. [DOI] [PubMed] [Google Scholar]
- 41.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971;68:820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lodato MA, Woodworth MB, Lee S, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015;350:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaguchi H, Kawazu M, Yasuda T, et al. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci 2015;106:1687–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016;534:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.