

Abstract
Understanding how to moderate and improve catalytic activity is critical to improving degradable polymer production. Here, di‐ and monozinc catalysts, coordinated by bis(imino)diphenylamido ligands, show remarkable activities and allow determination of the factors controlling performance. In most cases, the dizinc catalysts significantly out‐perform the monozinc analogs. Further, for the best dizinc catalyst, the ligand conformation controls activity: the catalyst with “folded” ligand conformation shows turnover frequency (TOF) values up to 60 000 h−1 (0.1 mol % loading, 298 K, [LA]=1 m), whilst that with a “planar” conformation is much slower, under similar conditions (TOF=30 h−1). Dizinc catalysts also perform very well under immortal conditions, showing improved control, and are able to tolerate loadings as low as 0.002 mol % whilst conserving high activity (TOF=12 500 h−1).
Keywords: dinuclear zinc complexes, homogeneous catalysis, metal–metal cooperativity, ring-opening polymerization, structure–activity relationship
Bimetallic homogeneous catalysts show huge potential in catalysis, with most explanations of observed enhanced performance proposing a synergistic cooperation between the two metal ions.1 This concept is reminiscent of the mode of action of many metalloenzymes.2 Enzymes also moderate activity by adopting particular protein or ligand “conformations”, but the application of this concept to synthetic catalysts is less well developed.3 Pioneering studies of bimetallic catalysts for olefin polymerizations by Marks, and now others, have demonstrated much better activities, selectivities, and control than for mononuclear analogs.4 Bimetallic catalysts have also shown excellent performances in the ring‐opening polymerization (ROP) of lactones,5 and in carbon dioxide/epoxide copolymerizations.6
The ring‐opening polymerization of lactide (LA) is important for production of biodegradable and renewably resourced polylactide (PLA), a material applied in both commodity and medical areas.7 To date, some of the best catalysts are homogeneous zinc complexes, coordinated by electron‐donating ligands, such as β‐diiminate (BDI),8 bis(amino)phenolates9 or pyrazolyol borates.10 Zinc(II) is an attractive choice of metal ion for such catalysts as it is cheap and combines high activity and selectivity with a lack of color, redox chemistry, and toxicity.11
We recently reported a dizinc bis(ethyl) complex coordinated by a bis(imino)diphenylamido macrocycle, [Zn2 LEt(Et)2], as a moderate LA ROP catalyst.12 The catalytic activity was inhibited by the low reactivity of the Zn−Et bonds towards alcohol, a reaction that generates the active dizinc bis(alkoxide) initiator, [Zn2 LEt(OiPr)2] (4). In attempts to overcome this limitation, five new zinc complexes, coordinated by this (LEt)2−, macrocycle, a slightly larger macrocycle, (LPr)2−, or an “open”/acyclic ligand analog, (LOpen)−, and featuring either bis(trimethylsilyl)amido (HMDS) or alkoxide initiating groups, were prepared (catalysts 1–6, Scheme 1). These 22‐ and 24‐membered Schiff‐base macrocycles are attractive as they facilitate formation of bimetallic complexes,13a are synthetically accessible in good yields, provide strong electron donation, and different ring sizes/rigidities are available. The “open” ligand, HLOpen (Figure S1), was prepared in order to provide access to monozinc analogs 3 and 6 (Scheme 1 ) for use as controls.
Scheme 1.
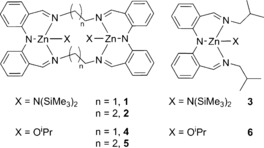
Structures of the family of six zinc complexes studied as rac‐LA ROP catalysts herein: amido complexes [Zn2 LEt(HMDS)2] 1, [Zn2 LPr(HMDS)2] 2, and [ZnLOpen(HMDS)] 3 and the corresponding alkoxides [Zn2 LEt(OiPr)2] 4,[12] [Zn2 LPr(OiPr)2] 5, and [ZnLOpen(OiPr)2] 6.
The three Zn–HMDS complexes 1–3 (Scheme 1) were synthesized, in good yields (>70 %), by the reaction of Zn(HMDS)2 with the appropriate ligand, and were fully characterized, including single‐crystal X‐ray diffraction and multinuclear NMR experiments (Figures S2–11). Complexes 1–3 were all extremely efficient catalysts for rac‐LA ROP. In particular, the dizinc macrocyclic catalysts, 1 and 2, successfully polymerized 1000 equiv. of monomer within one minute, at room temperature, without any exogenous alcohol (Table 1, Figures S12–S20). Catalyst 1 is more than 600 times more active than the bis(ethyl) homologue, [Zn2 LEt(Et)2].12 The activity per zinc center (i.e. corrected for the differing complex nuclearity) of dizinc 2 is three times greater than that of monozinc 3, providing evidence for dinuclear cooperativity. These new catalysts show a linear increase of the molecular weights (MWs), albeit with higher values than predicted, and narrow dispersities throughout the polymerizations. In contrast, other metal amido catalysts have been shown to exhibit reduced control and related behaviors.14 The polymerization kinetics were examined and revealed an unexpected zero‐order rate dependence on lactide (monomer) concentration (Figure 1, LHS). An induction period, typical of metal amido initiators, can also be observed for complexes 1 and 3 until 15–20 % of conversion; after the initiation period, however, both catalysts show clear linear fits to conversion versus time plots (Figure 1 LHS).15 Such rate independence in monomer is very unusual in ROP,15 however Tolman and co‐workers provide a compelling rationale for this in their study of Al catalysts. A Michaelis–Menten mechanistic model is invoked whereby the zeroth order is related to the pre‐equilibrium constant during reversible lactone binding to the metal ion.15c Monomer saturation kinetics are obtained if K m is much smaller than the monomer concentration (where K m is the Michaelis constant related to this fast pre‐equilibrium). The model does not imply a different operative mechanism to the well‐known coordination–insertion process, but rather reflects a different rate‐determining step. In the present case, monomer saturation kinetics are rationalized by the steric hindrance of the HMDS ligand and by the very high rates of initiation and propagation exhibited by complexes 1–3. Indeed, it is notable that within a minute complete monomer conversion occurs, but that this is catalyzed by only a fraction of the available complex (14 % for 1, 40 % for 2 and 64 % for 3), even at low loadings (0.1 mol %). The fractional amount of active catalyst is determined by comparing the higher‐than‐expected MW values with calculated values [Eqs. (S1)–(S3)]. Taking into account the true quantity of active catalyst, 1 is 1.5 times faster than 2 and 6 times more active than monozinc 3. The zinc centers are closer together in 1 than in 2 suggesting that more efficient intermetallic cooperation contributes to its higher activity. The MALDI‐TOF analyses show only peaks due to linear (end‐capped by amide) and cyclic PLA, with evidence of transesterification occurring (Figure S21).
Table 1.
rac‐LA polymerization using zinc–HMDS complexes 1–3.
| Cat [mol %] | Time [s] | Conv [%][a] | M n(exp) [kg⋅mol−1][b] | Ð[b] | M n(cld) [kg⋅mol−1][c] | TOF [h−1][d] |
|---|---|---|---|---|---|---|
| 1 (0.1) | 53 | 60 | 316 | 1.40 | 43 | 20 300 |
| 2 (0.1) | 30 | 73 | 135 | 1.30 | 57 | 45 000 |
| 3 (0.1) | 140 | 56 | 144 | 1.03 | 40 | 14 300 |
| A [e] (0.5) | 4800 | 78 | 9.6 | 1.10 | 11 | 120 |
Polymerization conditions as per Figure 1(LHS). [a] Determined by 1H NMR spectroscopy (CDCl3, 298 K). [b] Determined by SEC, versus polystyrene standards, and corrected by a factor of 0.58.16 [c] Mn(cld)=(conversion/100)×loading/[number of metal centers]×RMM(LA). [d] TOF=(conversion/100)×loading/(time×number of metal centers). [e] A correspond to [Zn2 LEt(Et)2] 2 equiv. of IPA.
Figure 1.
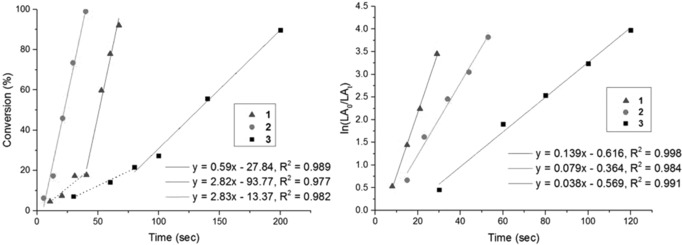
LHS: Plot of rac‐LA conversion (%) versus time (s), using 1 (▲), 2 (•) and 3 (▪) (1000 equiv. of rac‐LA in THF, 25 °C, [rac‐LA]=1 m). Dotted lines represent the induction periods. RHS: Plot of ln([rac‐LA0]/[rac‐LAt]) versus time (s) under immortal conditions, using 1 (▲), 2 (•) and 3 (▪) (1000 equiv. rac‐LA in THF, at 25 °C, with 10 equiv. of IPA: [rac‐LA]=1 m).
In order to understand the hyperactivity observed for this series of zinc–HMDS complexes, 1–3, the analogous zinc alkoxide initiators, 4–6 (Scheme 1), were prepared. The addition of one equiv. of isopropanol (IPA), per zinc, to 1–3, results in complete conversion, within five minutes at 25 °C, to the corresponding alkoxide complexes 4–6, as confirmed by a distinct color change (from red to yellow) and by 1H NMR spectroscopy (Figures S22–24).12 Polymerizations using 4–6 were conducted using in situ prepared complexes, generated by reaction of the HMDS complex with IPA for 30 minutes before monomer addition.
The zinc alkoxides 5 and 6 remain very active, polymerizing 1000 equiv. of rac‐LA in less than two minutes (Table 2 and Table S1, Figures S25–S35). Indeed these di‐ and monozinc catalysts (5 and 6, respectively) show nearly equivalent specific activities (per Zn center). However, under the same conditions, the alkoxide catalyst, 4, is more than 600 times slower than its HMDS analog 1, with only 50 % conversion occurring after 8 h. Further, it is clear that both of the dizinc catalysts, 4 and 5, show a diminution in rate compared to their HMDS analogs (1 and 2).
Table 2.
Selected data for the rac‐LA polymerization using 4–6 and 1–3 under immortal conditions (the complete Table can be found in Table S1).
| Cat [mol %] | Conv [%][a] | M n (exp) [kg⋅mol−1][b] | Ð[b] | M n(cld) [kg⋅mol−1][c] | TOF [h1][d] |
|---|---|---|---|---|---|
| 4 (0.1) | 48 | 50 | 1.10 | 70 | 30 |
| 5 (0.1) | 93 | 53 | 1.04 | 67 | 13 000 |
| 6 (0.1) | 97 | 49 | 1.09 | 70 | 14 000 |
| 1 (0.1) | 97 | 14 | 1.07 | 14 | 60 000 |
| 2 (0.1) | 91 | 13 | 1.03 | 13 | 50 000 |
| 3 (0.1) | 92 | 13 | 1.08 | 13 | 35 000 |
| 2 [e] (0.005) | 80 | 45 | 1.03 | 57 | 16 000 |
| 2 [f] (0.002) | 42 | 48 | 1.02 | 61 | 12 600 |
| A[ g] (0.2) | 97 | 38 | 1.10 | 28 | 600 |
| B [h] (0.1) | 99 | 100 | 1.40 | 142 | 4430 |
| C [i] (0.1) | 63 | 13 | 1.11 | 13 | 18 900 |
| D [j] (0.009) | 83 | 900 | 2.00 | 1344 | 1170 |
Polymerization conditions as per Figure 1 (LHS for 4–6 and RHS for 1–3, immortal conditions) [a] Determined by 1H NMR spectroscopy (CDCl3, 298 K). [b] Determined by SEC versus polystyrene standards, and corrected by a factor of 0.58.16 [c] M n(cld)=(conversion/100)×loading/[number of metal centers]×RMM(LA). [d] TOF=(conversion/100)×loading/(time×number of metal centers). [e] 40 equiv. of IPA per mol of catalyst. [f] 50 equiv. of IPA per mol of catalyst. [g] A=BDIZnOiPr, [rac‐LA]=0.4 m, T=20 °C in CH2Cl2. 8 [h] B=halfsalenZnEt, CH2Cl2.9 [i] C=bis(morpholinomethyl)phenoxyZnEt, 10 equiv. of IPA per mol of catalyst, T=60 °C in toluene.17 [j] D=Sn(Oct)2, T=110 °C in octanoate, neat.18
In direct contrast to the HMDS catalysts 1–3, the alkoxide catalysts 4–6 show the more common first‐order dependence on [LA] concentrations in the kinetic analyses (Figures S26 and S31), emphasizing the importance of the steric hindrance on the Michaelis constant. Carpentier and co‐workers reported similar links between the order in [LA] and the presence of bulky half(salen) ligands at the metal centers.15a As mentioned, 5 and 6 retain impressive activities, with TOF values being 3 times higher than the fastest known zinc catalyst (Table 2: 5, 6, and B) under similar reaction conditions (k obs values, reported in Table S1, being one order of magnitude higher than previously reported).9
The alkoxide catalysts 4–6 all afforded PLA of predictable MW, closely correlated with calculated values, and narrow dispersities—all features of excellent polymerization control. The MALDI‐TOF analyses show a single series corresponding to ester end‐capped PLA and not showing any significant transesterification (Figure S36).
So far, the dizinc–HMDS catalysts 1–3 showed the best rates, whereas the best control was exerted by the alkoxide analogs 4–6. In order to try to combine these beneficial properties, catalysts 1–3 were also tested under immortal conditions. The immortal conditions correspond to the addition of 10 equiv. of IPA (per mole of catalyst) at the same time as the monomer (to prevent the direct formation of only 4–6). Under these conditions, the macrocyclic dizinc catalysts 1 and 2 show outstanding activities, reaching complete conversions within 30 seconds (Table 2 and Table S1, Figures S37–S45). Under such conditions 1 is more than 5500 times more active than 4, whilst 2 is three times more active than 5. Interestingly, the kinetic studies now show a first‐order dependence on the monomer concentration (Figure 1 RHS). TOF values are once again more than 3 times higher than the fastest zinc catalysts reported previously under similar reaction conditions (Table 2: 1, 2, 3 and C).17 Furthermore, the specific activities (per Zn) of 1 and 2 are significantly greater than 3 (mono‐Zn), again highlighting the importance of the zinc–zinc cooperative effect. In all cases, polymerizations are very well controlled, yielding PLA with predictable MW and narrow dispersities. The MALDI‐TOF analyses show PLA with ester and amido end groups, with some transesterification occurring (Figure S46).
Under immortal conditions, 1 and 2 were highly effective at very low loadings: even at just 0.02–0.002 mol % they operated successfully, and the polymerizations progressed rapidly showing linear evolution of MW and narrow dispersities (Table 2 and Table S1, Figures S47–S75). On the other hand, under such industrially relevant catalyst loadings [Industrial scale polymerizations with Sn(Oct)2 are typically performed using loadings between 0.02 and 0.002 mol %],18, 19 monozinc catalyst 3 decomposed, highlighting an issue for many coordination complexes whereby the ligands are subject to protonolysis when used under immortal conditions (Figure S57).20 In the case of the macrocycles, the stability is significantly improved because of the macrocyclic effect and particular coordination modes.
Given the outstanding activities (hyperactivities) observed for some of these complexes, and the dramatic differences in performance, particularly between 1 and 4, it was interesting to correlate this with structural data for the complexes.
The molecular structures of 1–3, obtained from single crystal XRD experiments, all show tetrahedral N4‐coordinated zinc centers (τ 4′=0.79 for 1 and τ 4′=0.84 for 2 and 3).21 Each monoanionic tridentate binding pocket of these ligands coordinates to a zinc ion through the amido and one imine nitrogen atom, forming a planar six‐membered “BDI‐like” chelate ring, whilst the other imine nitrogen atom coordinates out of that plane and the HDMS completes the distorted tetrahedral coordination.22 The aromatic rings in each diphenylamido head unit are therefore dissymmetric.12 The solution structures of 1–3 correspond to the solid‐state structures. The 1H NMR spectrum of 1 (at 193 K) and 2 (at 298 K) in [D8]THF clearly show the two different imine resonances resulting from this asymmetric coordination mode, at 8.71 ppm/7.69 ppm or 8.30/8.26 ppm, respectively (Figures S2 and S6). In contrast, for the monozinc complex of the open ligand, 3, the 1H NMR C6D6 (at 298 K) shows only a single imine signal, presumably due to its significantly higher flexibility enabling fast equilibration between the two imine coordination modes (Figure S9).
Zinc BDI complexes, originally pioneered by Coates and co‐workers and subsequently investigated by many others,8, 23 are some of the most highly active catalysts (Table 2, A). In the present case, the ligands combine a “BDI‐like” chelate with an extra imine donor which enhances the electron donation, and which, in turn, results in faster rates. This observation suggests that increasing the electron density at the active site (metal ion) accelerates the polymerization, perhaps by labilization of the zinc alkoxide bonds.24
Considering the pair of dizinc complexes of the smaller macrocycle: 1 and 4, two distinct ligand conformations are observed which are proposed to contribute to the stark differentiation in activities (Figure 2). The complexes also feature different initiating groups, however initiation effects are not controlling rate because the catalysts show very well‐defined kinetic profiles (albeit quite different ones), exhibit reasonable control, and yield polymers with narrow dispersities. Rather, the radically different performances are mediated by distinct ligand conformations. Complex 1 adopts a “folded” C 2 symmetric structure, in the solid state, with the HMDS groups occupying the same, convex, face of the complex. In cooled solutions (193 K), the NMR data confirms the same folded ligand conformation, further supported by multinuclear and ROESY experiments (Figure S76–S77). VT NMR experiments show that the folded structure is retained until at least 253 K. From 263—293 K, signal coalescence occurs, due to a relatively slow equilibration between two conformers, and at temperatures above 303 K averaged signals are observed. The dynamic process is proposed to involve the rotation of the two phenyl rings around the central (amido) nitrogen of the diphenylamine head unit, with concomitant reversal of the short versus long Zn−Nimine bonds (Figures S78–S80). During equilibration, the folded structure is maintained, but the flat “Zn‐BDI‐like” chelate flips from one side of the molecule to the other. The folded structure is proposed to be retained at all temperatures, stabilized by the π–π interactions between the phenyl rings (Figure S79). An Eyring analysis showed the equilibrium thermodynamic parameters (ΔG ≠=15 kcal) were in close agreement with the theoretical barrier obtained from DFT calculations of the process (ΔG ≠=17 kcal, Figure S91, Table S2).
Figure 2.
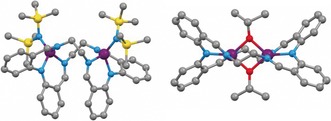
Molecular structures of 1 and 4,12 which are coordinated by the same (LEt)2− macrocycle but show distinct ligand conformations (heteroatoms color assignment: purple: zinc; blue: nitrogen; yellow: silicon; red: oxygen).
In contrast to the folded ligand structure observed, under the polymerization conditions, for 1, the molecular structure of 4 shows a different, “planar”, ligand conformation.12 The structure of 4 has D 2 symmetry and features two κ2 alkoxide groups, bridging between the zinc centers (Figure S81).12 Thus, the “BDI‐like” zinc chelate seen in 1 is not present in 4, and the pair of single‐atom bridging alkoxides massively reduces the conformational flexibility of the macrocycle. The 1H NMR spectrum of 4, in [D8]THF at 298 K, shows resonances in accordance with a D 2 symmetry, and confirms the zinc coordination modes (Figure S82). It is proposed that the higher activity of 1 results from its folded shape whereby the zinc‐active sites are pointing out on the convex face of the complex, facilitating both monomer insertion and coordination after the initiation steps, the latter of which becomes limiting under monomer saturation conditions.
The precise nature of the active‐site structure(s) when using 1 under immortal conditions is a more complex problem. It seems unlikely that a mixture of 1 and 4 is present, as the relative polymerization rate of 4 is so much lower than 1 (1 is 5833 times faster). Given that complete conversion occurs in only 29 seconds, under immortal conditions, there would not be sufficient time for 4 to function independently (c.f. 4 takes 8 h to reach just 50 % conversion under the same conditions). Nonetheless, alkoxide end groups, as well as amide groups, are observed in the MALDI‐TOF spectrum (Figure S46) and the control exerted is excellent (N.B. the alkoxide groups do not form under the mass spectrometry conditions and are real products of the catalysis). The data could be rationalized if the rate of initiation from 1 is approximately equal to the rate of chain exchange with alcohol and if an intermediate alkoxide complex forms which matches the activity of 1, but is orders of magnitude faster than 4. There is preliminary evidence for such a putative dizinc alkoxide species from LT‐NMR data (183 K), which show unassigned low intensity intermediate alkoxide signals during alcoholysis to form 4 (Figure S83). It is possible to infer the formation of complex 4 at the end of polymerizations run under immortal conditions as the addition of a second portion of monomer at this stage results in extremely slow rates of polymerization, consistent with its much lower activity (Figure S84–S89). It is tentatively proposed that a highly active alkoxide complex, featuring a folded ligand conformation, is partly responsible for the outstanding activity under immortal conditions. The intermediate alkoxide rearranges to the thermodynamically more stable bridging species, 4, within one minute under the reaction conditions, but in many cases polymerization is already complete by this time (Scheme S90). Accordingly, the rate of the initiation (k i) and the rate of the co‐ligand exchange are proposed to be of the same magnitude but are greater than the isomerization rate (k ex≈k i>k iso). Unfortunately, the lifetime of the intermediate is still too transient for more detailed analysis using common spectroscopic techniques.
In conclusion, dizinc complexes of strongly donating diphenylamine‐based [2+2] imine macrocycles, show better rates than the previous record‐holding zinc catalysts17 for lactide ring‐opening polymerizations. They also significantly out‐perform the monozinc analogs, showing rates which are up to 6 times higher per active site, indicating a cooperative interaction between the two zinc ions, tuned by the ancillary ligand. Further, the best activities result from the dizinc complexes in which the ligand adopts a folded conformation, which combines strong electron donation with short intermetallic distances and open coordination sites at the metal centers. The best catalysts are also able to operate under immortal conditions, that is, in the presence of exogenous alcohol, and can tolerate loadings as low as 0.002 mol % (vs. monomer) which is highly industrially relevant and puts them at the forefront in the field. These dizinc catalysts are significantly more stable to alcohol than the monozinc analogs. As a whole, the results demonstrate some of the critical features for the ancillary ligand and highlight its importance in promoting activity, selectivity, and stability. It is envisaged that both the absolute rates obtained and the insights into ligand conformation structural control will stimulate the development of catalysts showing improved performances in both ROP and other, related, polymerization processes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The EPSRC (grant numbers EP/K035274/1, EP/K014070/1, EP/K014668/1, EP/L017393/1), Climate KIC (studentship to A.T.), University of Otago (including Ph.D. scholarship to H.J.D.), MacDiarmid Institute for Advanced Materials and Nanotechnology (NZ), are acknowledged for research funding. Dr. Jennifer Garden and Dr. Anish Cyriac are thanked for technical assistance during the kinetic experiments. We thank Michael Crawford (Dunedin) for creating the front cover image from a concept provided by S.B. We thank Imperial College HPC for the computing resources.
A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker, C. K. Williams, Angew. Chem. Int. Ed. 2016, 55, 8680.
Contributor Information
Prof. Sally Brooker, Email: sbrooker@chemistry.otago.ac.nz.
Prof. Charlotte K. Williams, Email: c.k.williams@imperial.ac.uk.
References
- 1.
- 1a. Matsunaga S., Shibasaki M., Chem. Commun. 2014, 50, 1044–1057; [DOI] [PubMed] [Google Scholar]
- 1b. Liu Y., Ren W.-M., He K.-K., Lu X.-B., Nat. Commun. 2014, 5, 5687; [DOI] [PubMed] [Google Scholar]
- 1c. Bratko I., Gomez M., Dalton Trans. 2013, 42, 10664–10681; [DOI] [PubMed] [Google Scholar]
- 1d. Armstrong D. R., Garden J. A., Kennedy A. R., Mulvey R. E., Robertson S. D., Angew. Chem. Int. Ed. 2013, 52, 7190–7193; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7331–7334; [Google Scholar]
- 1e. Radlauer M. R., Buckley A. K., Henling L. M., Agapie T., J. Am. Chem. Soc. 2013, 135, 3784–3787; [DOI] [PubMed] [Google Scholar]
- 1f. Park J., Hong S., Chem. Soc. Rev. 2012, 41, 6931–6943; [DOI] [PubMed] [Google Scholar]
- 1g. Nakano K., Hashimoto S., Nozaki K., Chem. Sci. 2010, 1, 369–373. [Google Scholar]
- 2.
- 2a. Mitić N., Smith S. J., Neves A., Guddat L. W., Gahan L. R., Schenk G., Chem. Rev. 2006, 106, 3338–3363; [DOI] [PubMed] [Google Scholar]
- 2b. Jedrzejas M. J., Setlow P., Chem. Rev. 2001, 101, 607–618. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Arbaoui A., Redshaw C., Hughes D. L., Chem. Commun. 2008, 4717–4719; [DOI] [PubMed] [Google Scholar]
- 3b. Straub B. F., Adv. Synth. Catal. 2007, 349, 204–214; [Google Scholar]
- 3c. Ackermann J., Meyer F., Kaifer E., Pritzkow H., Chem. Eur. J. 2002, 8, 247–258. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Boulho C., Zijlstra H. S., Harder S., Eur. J. Inorg. Chem. 2015, 2132–2138; [Google Scholar]
- 4b. Radlauer M. R., Day M. W., Agapie T., J. Am. Chem. Soc. 2012, 134, 1478–1481; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Delferro M., Marks T. J., Chem. Rev. 2011, 111, 2450–2485; [DOI] [PubMed] [Google Scholar]
- 4d. Li H., Marks T. J., Proc. Natl. Acad. Sci. USA 2006, 103, 15295–15302; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Li L., Metz M. V., Li H., Chen M.-C., Marks T. J., Liable-Sands L., Rheingold A. L., J. Am. Chem. Soc. 2002, 124, 12725–12741. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Tseng H.-C., Chen H.-Y., Huang Y.-T., Lu W.-Y., Chang Y.-L., Chiang M. Y., Lai Y.-C., Chen H.-Y., Inorg. Chem. 2016, 55, 1642–1650; [DOI] [PubMed] [Google Scholar]
- 5b. Chen L., Li W., Yuan D., Zhang Y., Shen Q., Yao Y., Inorg. Chem. 2015, 54, 4699–4708; [DOI] [PubMed] [Google Scholar]
- 5c. Normand M., Roisnel T., Carpentier J. F., Kirillov E., Chem. Commun. 2013, 49, 11692–11694; [DOI] [PubMed] [Google Scholar]
- 5d. Chen H.-Y., Liu M.-Y., Sutar A. K., Lin C.-C., Inorg. Chem. 2010, 49, 665–674; [DOI] [PubMed] [Google Scholar]
- 5e. Sarazin Y., Howard R. H., Hughes D. L., Humphrey S. M., Bochmann M., Dalton Trans. 2006, 340–350; [DOI] [PubMed] [Google Scholar]
- 5f. Bukhaltsev E., Frish L., Cohen Y., Vigalok A., Org. Lett. 2005, 7, 5123–5126. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Kissling S., Lehenmeier M. W., Altenbuchner P. T., Kronast A., Reiter M., Deglmann P., Seemann U. B., Rieger B., Chem. Commun. 2015, 51, 4579–4582; [DOI] [PubMed] [Google Scholar]
- 6b. Garden J. A., Saini P. K., Williams C. K., J. Am. Chem. Soc. 2015, 137, 15078–15081; [DOI] [PubMed] [Google Scholar]
- 6c. Thevenon A., Garden J. A., White A. J. P., Williams C. K., Inorg. Chem. 2015, 54, 11906–11915. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Wang X., Thevenon A., Brosmer J. L., Yu I., Khan S. I., Mehrkhodavandi P., Diaconescu P. L., J. Am. Chem. Soc. 2014, 136, 11264–11267; [DOI] [PubMed] [Google Scholar]
- 7b. Bakewell C., White A. J. P., Long N. J., Williams C. K., Angew. Chem. Int. Ed. 2014, 53, 9226–9230; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9380–9384; [Google Scholar]
- 7c. Wheaton C. A., Hayes P. G., Ireland B. J., Dalton Trans. 2009, 4832–4846; [DOI] [PubMed] [Google Scholar]
- 7d. Platel R. H., Hodgson L. M., Williams C. K., Polym. Rev. 2008, 48, 11–63; [Google Scholar]
- 7e. Dechy-Cabaret O., Martin-Vaca B., Bourissou D., Chem. Rev. 2004, 104, 6147–6176. [DOI] [PubMed] [Google Scholar]
- 8. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238. [DOI] [PubMed] [Google Scholar]
- 9. Williams C. K., Breyfogle L. E., Choi S. K., Nam W., Young V. G., Hillmyer M. A., Tolman W. B., J. Am. Chem. Soc. 2003, 125, 11350–11359. [DOI] [PubMed] [Google Scholar]
- 10. Chisholm M. H., Eilerts N. W., Huffman J. C., Iyer S. S., Pacold M., Phomphrai K., J. Am. Chem. Soc. 2000, 122, 11845–11854. [Google Scholar]
- 11. Inkinen S., Hakkarainen M., Albertsson A.-C., Södergård A., Biomacromolecules 2011, 12, 523–532. [DOI] [PubMed] [Google Scholar]
- 12. Romain C., Bennington M. S., White A. J. P., Williams C. K., Brooker S., Inorg. Chem. 2015, 11842–11851. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Cameron S. A., Brooker S., Inorg. Chem. 2011, 50, 3697–3706; [DOI] [PubMed] [Google Scholar]
- 13b. Black D., Rothnie N., Aust. J. Chem. 1983, 36, 2395–2406; [Google Scholar]
- 13c. Black D. St. C., Rothnie N. E., Tetrahedron Lett. 1978, 19, 2835–2836. [Google Scholar]
- 14.
- 14a. Ajellal N., Carpentier J.-F., Guillaume C., Guillaume S. M., Helou M., Poirier V., Sarazin Y., Trifonov A., Dalton Trans. 2010, 39, 8363–8376; [DOI] [PubMed] [Google Scholar]
- 14b. Dove A. P., Gibson V. C., Marshall E. L., White A. J. P., Williams D. J., Dalton Trans. 2004, 570–578. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Normand M., Dorcet V., Kirillov E., Carpentier J.-F., Organometallics 2013, 32, 1694–1709; [Google Scholar]
- 15b. Huang Y., Wang W., Lin C.-C., Blake M. P., Clark L., Schwarz A. D., Mountford P., Dalton Trans. 2013, 42, 9313–9324; [DOI] [PubMed] [Google Scholar]
- 15c. Ding K., Miranda M. O., Moscato-Goodpaster B., Ajellal N., Breyfogle L. E., Hermes E. D., Schaller C. P., Roe S. E., Cramer C. J., Hillmyer M. A., Tolman W. B., Macromolecules 2012, 45, 5387–5396. [Google Scholar]
- 16. Kowalski A., Duda A., Penczek S., Macromolecules 1998, 31, 2114–2122. [Google Scholar]
- 17. Poirier V., Roisnel T., Carpentier J.-F., Sarazin Y., Dalton Trans. 2009, 9820–9827. [DOI] [PubMed] [Google Scholar]
- 18. Nijenhuis A. J., Grijpma D. W., Pennings A. J., Macromolecules 1992, 25, 6419–6424. [Google Scholar]
- 19.P. J. Sicco de Vos, Research Gate 2015.
- 20. Poirier V., Roisnel T., Sinbandhit S., Bochmann M., Carpentier J.-F., Sarazin Y., Chem. Eur. J. 2012, 18, 2998–3013. [DOI] [PubMed] [Google Scholar]
- 21. Okuniewski A., Rosiak D., Chojnacki J., Becker B., Polyhedron 2015, 90, 47–57. [Google Scholar]
- 22. Cheng M., Moore D. R., Reczek J. J., Chamberlain B. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 8738–8749. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Whitehorne T. J. J., Vabre B., Schaper F., Dalton Trans. 2014, 43, 6339–6352; [DOI] [PubMed] [Google Scholar]
- 23b. Abbina S., Du G., ACS Macro Lett. 2014, 3, 689–692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Du G., Wei Y., Zhang W., Dong Y., Lin Z., He H., Zhang S., Li X., Dalton Trans. 2013, 42, 1278–1286; [DOI] [PubMed] [Google Scholar]
- 23d. Drouin F., Oguadinma P. O., Whitehorne T. J. J., PruO'homme R. E., Schaper F., Organometallics 2010, 29, 2139–2147; [Google Scholar]
- 23e. Chisholm M. H., Gallucci J. C., Phomphrai K., Inorg. Chem. 2005, 44, 8004–8010; [DOI] [PubMed] [Google Scholar]
- 23f. Chen H.-Y., Huang B.-H., Lin C.-C., Macromolecules 2005, 38, 5400–5405; [Google Scholar]
- 23g. Chisholm M. H., Gallucci J., Phomphrai K., Inorg. Chem. 2002, 41, 2785–2794; [DOI] [PubMed] [Google Scholar]
- 23h. Chisholm M. H., Huffman J. C., Phomphrai K., J. Chem. Soc. Dalton Trans. 2001, 222–224. [Google Scholar]
- 24.
- 24a. Wang X., Brosmer J. L., Thevenon A., Diaconescu P. L., Organometallics 2015, 34, 4700–4706; [Google Scholar]
- 24b. Bakewell C., Cao T.-P.-A., Long N., Le Goff X. F., Auffrant A., Williams C. K., J. Am. Chem. Soc. 2012, 134, 20577–20580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary