

Abstract
Hepatocellular carcinoma (HCC) is a cancer lacking effective therapies. Several measures have been proposed to treat HCCs, such as senescence induction, mitotic inhibition, and cell death promotion. However, data from other cancers suggest that single use of these approaches may not be effective. Here, by genetic targeting of Survivin, an inhibitor of apoptosis protein (IAP) that plays dual roles in mitosis and cell survival, we identified a tumor necrosis factor alpha (TNFα)‐mediated synergistic lethal effect between senescence and apoptosis sensitization in malignant HCCs. Survivin deficiency results in mitosis defect‐associated senescence in HCC cells, which triggers local inflammation and increased TNFα. Survivin inactivation also sensitizes HCC cells to TNFα‐triggered cell death, which leads to marked HCC regression. Based on these findings, we designed a combination treatment using mitosis inhibitor and proapoptosis compounds. This treatment recapitulates the therapeutic effect of Survivin deletion and effectively eliminates HCCs, thus representing a potential strategy for HCC therapy. Conclusion: Survivin ablation dramatically suppresses human and mouse HCCs by triggering senescence‐associated TNFα and sensitizing HCC cells to TNFα‐induced cell death. Combined use of mitotic inhibitor and second mitochondrial‐derived activator of caspases mimetic can induce senescence‐associated TNFα and enhance TNFα‐induced cell death and synergistically eliminate HCC. (Hepatology 2016;64:1105‐1120)
Abbreviations
- CPC
chromosomal passenger complex
- DEN
diethylnitrosamine
- DDC
3,5‐diethoxycarbonyl‐1,4‐dihydro‐collidine
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- IAP
inhibitor of apoptosis proteins
- mRNA
messenger RNA
- PCR
polymerase chain reaction
- PDX
patient‐derived xenograft
- PLK1
polo‐like kinase 1
- qPCR
quantitative PCR
- SA‐β‐gal
senescence‐associated β‐galactosidase activity
- Ptges
prostaglandin E synthase
- SAHF
senescence‐associated heterochromatin foci
- siRNAs
small interfering RNAs
- SMAC
second mitochondrial‐derived activator of caspases
- TNFα
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Hepatocellular carcinoma (HCC) is the fifth‐most common cancer and the third cause of cancer death worldwide.1 The only approved drug for HCC treatment is sorafenib, which prolongs overall survival by around 3 months.2 There is an urgent need to develop more‐effective therapies. A notable feature of HCC is that it usually develops in inflammatory conditions, for example, viral infection and metabolic disease‐associated steatohepatitis.3, 4 One of the key players in regulating cancer‐associated immune responses is cellular senescence.5, 6
Cellular senescence, a state of irreversible cell‐cycle arrest, is considered a tumor‐suppressive mechanism.7, 8 Cellular senescence has been documented as a pivotal barrier for initiation and progression of HCC.6, 9 Moreover, senescent HCC‐associated stromal cells secrete extracellular‐matrix‐degrading enzymes, which are believed to enhance immune surveillance and facilitate HCC resolution.10 In these scenarios, both innate immune responses and T‐cell‐mediated adaptive immunity are involved.6, 9
Approaches to induce efficient senescence in vivo have been actively investigated.8 Recent studies showed that inhibiting the chromatin passenger complex (CPC) function caused cellular senescence.11 CPC complex controls chromatin alignment during mitosis, the inhibition of which leads to mitosis arrest and proliferation halt.12 Polo‐like kinase 1 (PLK1) and Aurora B kinase are two key kinases in regulating CPC functions.13 There are several highly selective compounds targeting against PLK1 or Aurora B. Both types of inhibitors caused remarkable mitosis arrest and senescence. Surprisingly, the outcomes of several clinical trials for these compounds were disappointing, making some debates on the therapeutic value for these mitotic inhibitors.14, 15, 16 To that end, it was proposed to exploit these compounds in combination with other drugs, mainly chemotherapeutic drugs15, 17; however, the underlying rationale for such use was not fully clarified.
Both senescence‐based or mitosis arrest‐based anticancer treatment induce a cytostatic status, but do not directly eliminate cell‐cycle‐arrested cells.18, 19 A desirable therapy would not only induce cytostasis, but also eliminate these cancer cells efficiently. It is proposed to take advantage of HCC‐associated inflammation. For example, senescence and infiltration of immune cells trigger locally increased concentrations of cytokines in HCC tissues. Some of these inflammatory factors, such as tumor necrosis factor alpha (TNFα), are cell death inducers.20 The difficulty of this approach is that HCC cells are well protected from cell death by antiapoptotic proteins, including members of the family of inhibitor of apoptosis proteins (IAPs). It is thus tempting to develop a combinational strategy to eliminate cytostatic cancer cells by unleashing the death‐inducing effects of inflammatory cytokines.
Survivin is the smallest member of the family of IAPs and is highly expressed in precancerous liver lesions and in malignant HCC cells.21 Besides its functions in cell death, Survivin is also a major component of the CPC complex and controls mitosis.12 Previous studies have proposed that Survivin could be a therapeutic target for cancer treatment. However, development of Survivin inhibitors has so far been unsuccessful.22, 23 Further delineation of the oncogenic properties of Survivin may provide insights for the development of novel anticancer strategies that bypass direct targeting of Survivin. In that respect, we have previously shown that Survivin promotes survival of HCC initiating cells by regulation of activator protein 1 and sirtuin 6.21 However, it remains unknown whether Survivin controls HCC malignancy at late stages and, if so, how the molecular mechanism could be translated into a potential therapeutic strategy.
In this study, we found a near‐complete HCC repression using genetic deletion of Survivin. A careful analysis of Survivin deletion in HCC led to an unexpected finding of a synergistic effect between mitosis defect‐induced senescence and apoptosis sensitization, mediated by TNFα, on eliminating HCC cells. Survivin deletion causes mitosis defect and senescence, which further induces inflammation and TNFα expression locally. Remarkably, because of the hypersensitivity of Survivin‐deficient HCC cells to TNFα‐triggered cell death, Survivin‐deficient HCC cells undergo extensive cell death, thereby leading to drastic HCC regression. By taking advantage of these findings, we additionally designed and validated a new HCC therapeutic strategy by combination use of mitotic inhibitor and second mitochondrial‐derived activator of caspases (SMAC) mimetic to induce mitosis arrest‐associated senescence and enhance TNFα‐induced cell death, respectively.
Materials and Methods
PRIMARY CULTURED HCC CELLS AND PATIENT‐DERIVED XENOGRAFT
Human HCC samples used for primary HCC cell culture and patient‐derived xenograft (PDX) transplantation were collected from Eastern Hepatobiliary Surgery Hospital, Second Military Medical University (Shanghai, China). All procedures of human sample collection were approved by the Ethical Committee of Eastern Hepatobiliary Surgery Hospital.
For primary HCC cell culture, HCC tissues were snipped followed by collagenase digestion. HCC cells were cultured in RPMI 1640 medium with 10% fetal calf serum on collagen‐coated dishes. For PDX transplantation, HCC tissues were cut into pieces and subcutaneously transplanted into nod‐scid mice and then transplanted into athymic nude mice where tumors were allowed to grow.
MICE AND LIVER TUMORIGENESIS PROTOCOL
Mx‐cre; Survivin f/f (svvΔ li*) and Alb‐cre; Survivin f/f mice were generated by crossing svv f/f mice to Alb‐cre mice and Mx‐cre mice. Genotyping was performed by polymerase chain reaction (PCR) of tail genomic DNA. To induce HCCs in mice, a single dose of diethylnitrosamine (DEN; 25 mg per kilogram of body weight; Sigma‐Aldrich, St. Louis, MO) was injected into 2‐week‐old male mice intraperitoneally and mice were euthanized at the age of 10 months. Genotyping primers are listed in the Supporting Tables. Mice were kept under specific pathogen‐free conditions at the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. All mouse experiments were approved by the Institutional Animal Care and Use Committee of the Institute of Biochemistry and Cell Biology and performed in accord with this committee's guidelines.
MICROARRAY ANALYSIS
Total RNAs were hybridized to whole‐mouse gene expression microarray (Agilent Technologies, Inc., Lexington, MA) in accord with the manufacturer's instructions. Data were normalized using Gene‐Spring (Agilent). Original data are available in the NCBI Gene Expression Omnibus (GSE64804).
STATISTICAL ANALYSIS
For most statistical analyses, the unpaired Student t test was applied for calculating statistical probability. For survival analyses, the Kaplan‐Meier log‐rank test was applied. Statistical calculations were performed using the Statistical Program for Social Sciences (SPSS) software (IBM Corp., Armonk, NY). For all statistics, data from at least three independent samples or repeated experiments were used.
Results
SURVIVIN DELETION INDUCES REGRESSION OF MALIGNANT HCCs
We previously showed that Survivin promotes survival of HCC‐initiating cells.21 However, genetic evidence is still lacking on whether Survivin controls HCC progression and malignancy. We generated Mx‐cre, svv f/f mice by crossing mice carrying floxed Survivin alleles (svv f/f) and mice carrying Mx1 promoter‐controlled Cre recombinase (Mx‐cre). Poly(IC) treatment induces Cre expression in hepatocytes and hematopoietic cells in Mx‐cre, svv f/f mice.24 To specifically delete Survivin in hepatocytes, Mx‐cre, svv f/f mice were irradiated and transplanted with bone marrows from svv f/f siblings before poly(IC) treatment (svv Δli*). In line with our previous report, the morphology and function of hepatocytes remained normal in svv Δli* mice.24
HCCs were induced with DEN in svv f/f and Mx‐cre, svv f/f mice at the age of 2 weeks. Both svv f/f and Mx‐cre, svv f/f mice then received bone marrow transplantation at the age of 2 months and injected with poly(IC) to delete Survivin either at the age of 5 (Fig. 1A) or 8 months (Fig. 1D). Poly(IC) treatment efficiently deleted Survivin in livers and HCCs in Mx‐cre, svv f/f mice, but not in bone marrow cells or tumor‐infiltrated Kupffer cells and T cells (Supporting Fig. S1A,B). In both experimental settings, the formation of HCCs was comparable between svv f/f and Mx‐cre, svv f/f mice before Survivin deletion (Supporitng Fig. S1C,D). At the age of 10 months, mice were sacrificed and HCCs were quantified. Control svv f/f mice developed a large number of HCCs (Fig. 1B,C). By contrast, HCCs were almost undetectable in svv Δli* mice with Survivin deletion at the age of 5 months (Fig. 1B,C), and HCCs were markedly reduced in svv Δli* mice with Survivin deletion at the age of 8 months (Fig. 1E,F). Serum alanine aminotransferase, aspartate aminotransferase, and total bilirubin levels were decreased in svv Δli* mice (Supporting Fig. S1E,F), suggesting improved liver function. Moreover, cancer‐bearing svv Δli* mice survived much longer than svv f/f mice (Supporting Fig. S1G).
Figure 1.
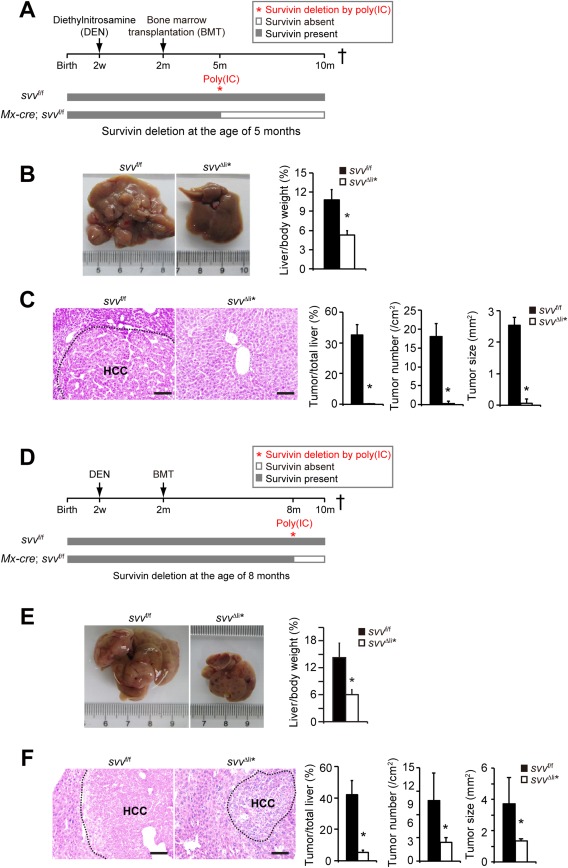
Survivin is essential in HCC development. (A) Experimental design for the DEN‐induced HCC model. Mx‐cre; svv f/f and svv f/f control mice were treated with DEN at 2 weeks of age and γ‐irradiated, followed by transplantation with bone marrows from svv f/f mice at the age of 2 months. No HCCs were detectable at the time of bone marrow transplantation. Five‐month‐old Mx‐cre; svv f/f and control mice were injected with poly(IC) intraperitoneally to induce liver‐specific Survivin gene depletion. Mx‐cre; svv f/f mice were referred to as svv Δli* after poly(IC)‐induced depletion of Survivin. All mice were sacrificed for HCC analysis at the age of 10 months. (B,C) DEN‐induced HCCs were analyzed in svv Δli* and svv f/f mice 5 months after poly(IC) treatment. (B) Liver/body weight ratios were measured. n = 5 for svv f/f mice; n = 6 for svv Δli* mice. (C) Hematoxylin and eosin (H&E) staining of liver sections. HCC was encircled with a dashed line. HCCs were quantified. n = 5 for svv f/f mice; n = 5 for svv Δli* mice. (D) Similar treatment as described in (A) was applied to Mx‐cre; svv f/f and svv f/f mice. However, Survivin was depleted with poly(IC) in Mx‐cre; svv f/f mice at the age of 8 months. (E,F) DEN‐induced HCC was analyzed in svv Δli* mice at the age of 10 months. (E) Liver/body weight ratios were measured. n = 7 for svv f/f mice; n = 5 for svv Δli* mice. (F) H&E staining of liver sections. HCCs were encircled with dashed lines, and HCCs were quantified. n = 5 for svv f/f mice; n = 5 for svv Δli* mice. Scale bars, 100 μm. * P < 0.05, t test.
To validate this phenotype of HCC inhibition in Survivin‐deficient livers, HCCs were induced in another liver‐specific Survivin knockout mouse line, Alb‐cre, svv f/f. We previously showed that Survivin was deleted in >90% of hepatocytes within 2 weeks after birth in these mice.24 Notably, DEN induced very few HCCs in Alb‐cre, svv f/f mice compared to control svv f/f mice (Supporting Fig. S2A,B), and Alb‐cre, svv f/f mice showed improved liver function and survival (Supporting Fig. S2C,D). The robustness of our findings was demonstrated with a c‐Met and ΔN90‐β‐Catenin‐induced HCC model (Supporting Fig. S10), in which we also observed reduced HCC formation in svv Δli* mice (Supporting Fig. S2E). The results from different mouse models indicate that lack of Survivin results in marked inhibition of HCC and provide compelling genetic evidence that targeting Survivin might be an effective therapeutic strategy.
SURVIVIN DEPLETION TRIGGERS MITOSIS DEFECT AND SENESCENCE IN HCC CELLS
Although a potentially attractive therapeutic oncotarget, it is yet difficult to develop inhibitors against Survivin.22, 23 YM155 is the most clinically advanced Survivin inhibitor.25 However, YM155 failed to show clear efficacy in phase II clinical trials for primary melanomas and non‐small‐cell lung cancers.26, 27 Moreover, remarkable toxicity of YM155 was reported in clinical trials. In line with these findings, we found that YM155, at the dose of 5 mg/kg body weight per day, showed significant toxicity on HCC‐bearing mice, which lost over 30% of their body weight after 1 month of treatment (Supporting Fig. S3A) and imposed poorly repressive effects on primary HCCs (Supporting Fig. S3B). We therefore explored the mechanisms underlying Survivin deletion‐induced inhibition of HCC growth, with a view to utilizing those mechanisms for the development of novel anti‐HCC strategies. HCCs were analyzed in svv Δli* mice with Survivin depletion at the age of 8 months, which was relevant to treatment of late‐stage HCCs. We first asked whether Survivin controls the function of CPC complex and mitotic transition in HCCs. We found that the number of p‐H3S10‐positive HCC cells was decreased in svv Δli* HCCs 1 week after Survivin depletion (Fig. 2A). Moreover, immunofluorescent staining showed that Aurora‐B kinase, the enzymatic member of the CPC complex, diffused in chromosome arms and failed to locate to kinetochores in svv Δli* HCC cells (Supporting Fig. S4A). These data suggest that Survivin deficiency causes impaired mitosis in HCC cells.
Figure 2.
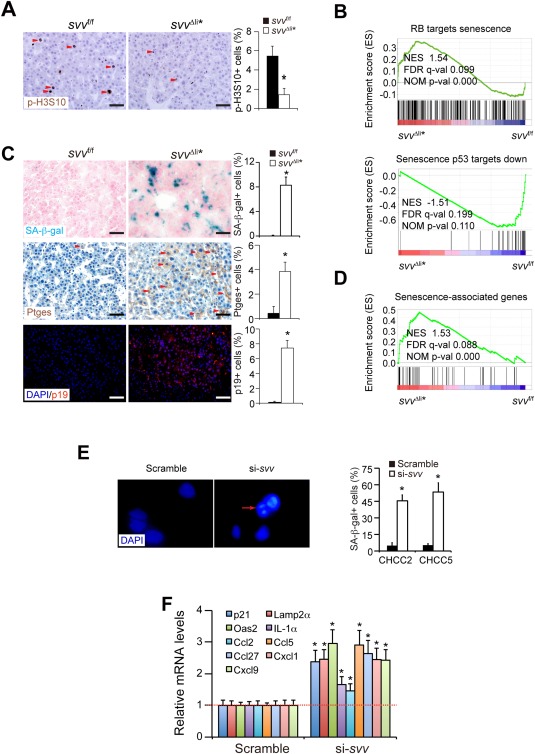
Survivin deficiency induces a mitosis defect and cellular senescence. (A) Mitotic HCC cells were analyzed by immunohistochemical staining for phosphorylated H3S10 (p‐H3S10). HCCs were harvested 1 week after poly(IC)‐induced Survivin depletion. (B) GSEA analysis showed that the expression profile of RB‐related senescence and p53‐related senescence were enriched in svv Δli* liver cancers. (C) SA‐β‐gal staining (blue) on cryosections of HCCs from svv Δli* mice 1 week after polyIC treatment. SA‐β‐gal‐positive HCC cells were quantified. Cancer cells expressing senescence marker genes Ptges and p19Arf were determined by immune staining on paraffin sections. Arrowheads indicate Ptges‐positive cancer cells. Quantification of Ptges‐ and p19Arf‐positive cancer cells are shown. n = 4 for each group for (A) and (C). (D) GSEA of expression profiles of senescence‐associated genes in DEN‐induced HCCs from svv f/f and svv Δli* mice 1 week after poly(IC) treatment. Senescence‐associated genes were taken from the study of Pribluda et al.28 (E) Two primary human HCC cells were transfected with Survivin‐specific siRNAs to silence Survivin. SAHFs were characterized by DAPI‐DNA staining. SAHF are indicated by arrows. SA‐β‐gal‐positive cells were quantified 72 hours after siRNA transfection. All results represent three independent experiments. * P < 0.05, t test. (F) qPCR analysis of senescence‐associated genes in Survivin‐silenced human HCC cells. Results represent three independent experiments. Scale bars, 50 μm. * P < 0.05, t test. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; FDR, false discovery rate; NES, normalized enrichment score; NOM, nominal.
Whole‐genome expression profiling of svv Δli* HCCs 1 week after Survivin depletion was performed by microarray assay. Gene set enrichment analysis (GSEA) showed that genes involved in mitosis and cell‐cycle transition were significantly down‐regulated in svv Δli* HCCs (Supporting Fig. S4B). Moreover, expression of genes involved in RB‐related senescence and p53‐related senescence were enriched in svv Δli* HCCs (Fig. 2B). Because mitotic arrest has been previously shown as a cause of cellular senescence,11 we characterized senescence in Survivin‐deficient HCCs. Senescence‐associated β‐galactosidase activity (SA‐β‐gal) was markedly increased in svv Δli* HCCs (Fig. 2C). Senescence marker genes, including prostaglandin E synthase (Ptges) and p19Arf, were similarly up‐regulated in svv Δli* HCCs28 (Fig. 2C). Furthermore, a panel of senescence‐associated genes28 were up‐regulated in svv Δli* HCCs, as shown by GSEA analysis (Fig. 2D) and confirmed by quantitative PCR (qPCR; Supporting Fig. S4C). Overall, these data indicate that Survivin depletion induces senescence in HCC cells.
To assess the clinical relevance of our findings, primary human HCC cells were transfected with small interfering RNAs (siRNAs) targeting Survivin. Efficient knockdown of Survivin was confirmed by qPCR (Supporting Fig. S4D). Survivin knockdown resulted in impaired mitosis in primary human HCC cells at 24 to 48 hours (Supporting Fig. S4E). After impaired mitosis, prominent senescence was validated with senescence‐associated heterochromatin foci (SAHF) staining and SA‐β‐gal staining (Fig. 2E) and increased expression of senescence‐associated genes in Survivin‐deficient human HCC cells by qPCR (Fig. 2F). Together, our data indicate that Survivin deficiency causes mitosis defect‐associated senescence in HCC cells.
SURVIVIN PROTECTS HCC CELLS FROM TNFα‐INDUCED CELL DEATH
Previous studies showed that senescent cancer cells have a role in increase local inflammatory responses in tumors.7, 8 Accordingly, increased numbers of macrophages and CD3+ T lymphocytes were found in svv Δli* HCCs (Fig. 3A). Moreover, several inflammation response‐related pathways were remarkably enriched in svv Δli* HCCs by GSEA analysis (Fig. 3B). Importantly, we found that genes downstream of the TNFα pathway were strikingly increased (Fig. 3B). Given that TNFα is a prominent cell death inducer, we focused our study on this cytokine. Increased messenger RNA (mRNA) and protein levels of TNFα were validated in svv Δli* HCCs (Fig. 3C). To characterize whether senescent cells have a role in inducing TNFα in inflammatory cells, we generated senescent cells by silencing Survivin and applied supernatant from the culture of senescent cells to macrophages. It was notable that macrophages produced higher TNFα upon addition of the supernatant. These data suggest a causative role of senescence in TNFα induction (Fig. 3D).
Figure 3.
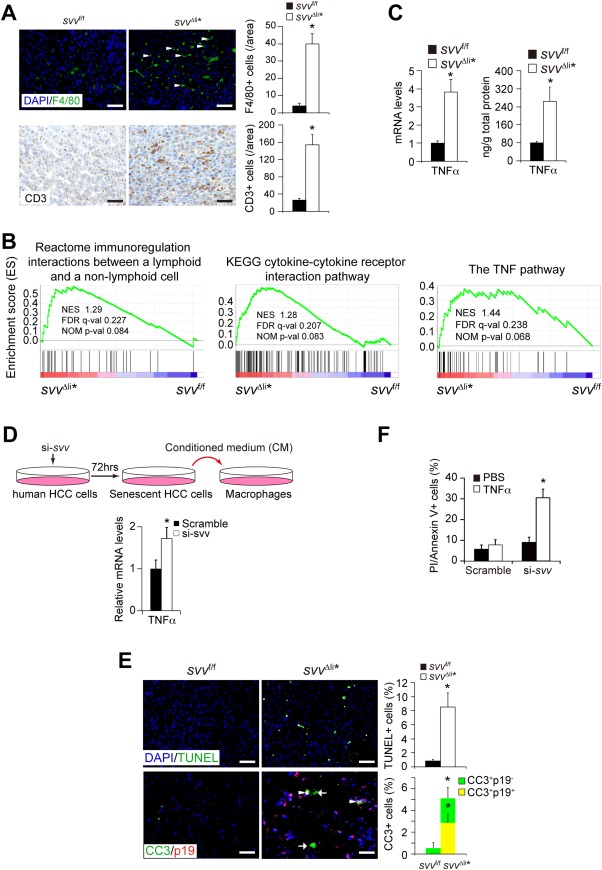
Survivin deficiency induces senescence‐associated inflammation and TNFα expression. (A) Immunofluorescent staining of F4/80 and CD3 showed increased infiltration of macrophages and T cells, respectively, in svv Δli* liver cancers. F4/80‐positive and CD3‐positive cells were quantified. n = 4 for each group. (B) GSEA analysis showed enrichment of inflammation pathway and the TNFα pathway genes in svv Δli* liver cancers. (C) TNFα mRNA and protein levels in svv Δli* HCCs were determined by qPCR and enzyme‐linked immunosorbent assay. n = 4 for each group. (D) Conditioned medium of senescent human HCC cells stimulates THP1‐derived macrophages to secret TNFα. Human HCC cells were induced to be senescent by siRNA against Survivin. Conditioned medium from senescent culture were collected at 72 hours. THP‐1‐derived macrophages were cultured with conditioned medium of senescent HCC cells for another 24 hours. mRNA levels of TNFα were analyzed by qPCR. Results represent two independent experiments. * P < 0.05, t test. (E) Cell death was detected by TUNEL staining in HCCs 1 week after poly(IC) treatment. TUNEL‐positive cancer cells were quantified. n = 4 for each group. Costaining of cleaved caspase 3 (CC3; for dead cells) and p19Arf (p19; for senescent cells) on HCCs 1 week after poly(IC) treatment. CC3‐positive p19Arf‐negative cells are indicated by arrows. CC3‐positive p19Arf‐positive cancer cells are indicated by arrowheads. Quantification of CC3‐positive and p19Arf‐positive cells showed both senescent and neighboring nonsenescent cells undergoing cell death. n = 3 for each group. (F) Two primary human HCC cells were transfected with siRNAs against Survivin and then treated with 10ng/mL of human TNFα (hTNFα) to induce cell death. Dead cells were stained by propidium iodide and Annexin V and then analyzed by fluorescence‐activated cell sorting. Results represent three independent experiments. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; FDR, false discovery rate; KEGG, Kyoto Encyclopedia of Genes and Genomes; NES, normalized enrichment score; NOM, nominal; PBS, phosphate‐buffered saline.
Because Survivin protects cells from death ligand‐triggered cell death, we considered whether senescence‐associated TNFα might cause enhanced cell death in Survivin‐deficient HCCs. Both terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and immunostaining for cleaved caspase 3 revealed extensive cell death in svv Δli* HCCs (Fig. 3E and Supporting Fig. S5A). In addition, gene expression profiling showed that a panel of cell death and apoptosis genes were increased in svv Δli* HCCs (Supporting Fig. S5B). It is also noteworthy that both senescent and neighboring nonsenescent HCC cells underwent cell death in svv Δli* tumors (Fig. 3E), suggesting that a paracrine effect to induce cell death in Survivin‐deficient HCC cells.
We additionally analyzed these findings using cultured primary human HCC cells. TNFα treatment alone induced a low level of cell death in primary HCC cells, which were consistent with previous findings that cancer cells are resistant to TNFα‐induced cell death.29 In contrast, siRNA‐mediated Survivin silencing notably augmented TNFα‐induced cell death (Fig. 3F). These results indicated that Survivin‐deficient cells are hypersensitive to TNFα‐induced cell death, which may explain the dramatic HCC repression in svv Δli* mice.
These findings together suggested a role of TNFα in mediating mitosis defect‐induced senescence and sensitized cell death in Survivin‐deficient HCC cells. To validate this hypothesis, we analyzed the anticancer effect of TNFα in vivo by blocking TNFα signaling with the TNFα antagonist, etanercept.30 (Fig. 4A). We found that expression of TNFα was already reduced in svv Δli* HCCs after 2 weeks of etanercept treatment (Supporting Fig. S5C), confirming an inhibitory effect of etanercept on TNFα in vivo.31 Moreover, decreased number of SA‐β‐gal‐positive cells further validated the blockage of TNFα (Supporting Fig. S5D), because TNFα has a positive‐feedback effect on senescence.32 Importantly, cell death in svv Δli* HCCs was significantly attenuated after 2 weeks of etanercept treatment (Fig. 4B). As a consequence of reduced cell death, HCC regression in svv Δli* mice was largely reverted after etanercept treatment for 2 months (Fig. 4F). In contrast, etanercept had no effect on liver tumorigenesis in svv f/f mice (Fig. 4C), supporting a key role for Survivin in antagonizing cell death induced by senescence‐related TNFα. Based on these findings, we proposed that Survivin targeting causes mitosis arrest‐induced senescence and increased TNFα levels in HCCs. Moreover, Survivin depletion additionally sensitizes cancer cells to TNFα‐induced cell death (Fig. 4D).
Figure 4.
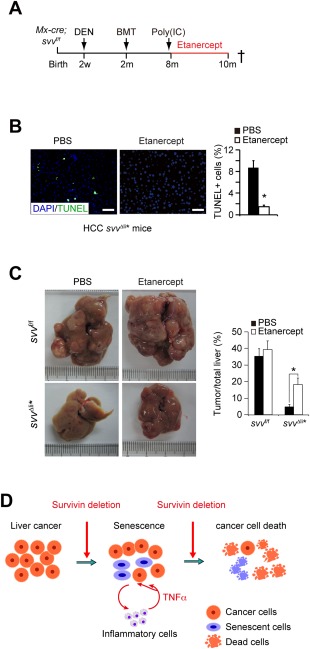
Survivin protects HCC cells from inflammatory TNFα‐mediated cell death. (A) Experimental scheme of etanercept treatment. Survivin was deleted in HCC‐bearing Mx‐cre; svv f/f at the age of 8 months. These mice were then treated with etanercept at 10 mg/kg body weight intraperitoneally twice a week. (B) Cell death in svv Δli* HCCs was dramatically reduced after etanercept treatment for 1 week as determined by TUNEL staining. (C) HCC development was analyzed in svv Δli* mice after etanercept treatment. HCCs were quantified. n = 3 for each group. * P < 0.05, t test. (D) Schematic of TNFα‐mediated synergistic lethal effect of senescence and apoptosis sensitization; Survivin depletion induced impaired mitosis and senescence in cancer cells, whereas infiltration of inflammatory cells and senescence‐associated TNFα triggered cell death in senescent and neighboring nonsenescent cancer cells lacking Survivin. Abbreviations: BMT, bone marrow transplantation; DAPI, 4′,6‐diamidino‐2‐phenylindole; PBS, phosphate‐buffered saline.
COMBINATION OF MITOSIS INHIBITOR AND SMAC MIMETIC ELIMINATES MURINE HCCs
We asked whether we could take advantage of the anticancer mechanisms by mimicking mitosis arrest‐caused senescence and sensitization to TNFα‐induced cell death. We hypothesized that combination of a drug triggering mitosis arrest and senescence with a drug enhancing TNFα‐induced cell death may phenocopy the Survivin deficiency effect on HCC. To that end, we used the PLK inhibitor, BI2536, to induce mitosis arrest and senescence and the SMAC mimetic, LCL161, to sensitize HCC cells to TNFα‐induced cell death.
BI2536 is a highly selective PLK1 inhibitor,33 which targets PLK1, a critical activator of the CPC complex and causes mitosis defect.34 Exposure of primary human HCC cells to BI2536 caused mitosis defect (Supporting Fig. S6A) and senescence, as shown by SA‐β‐gal staining (Supporting Fig. S6B), SAHF (Supporting Fig. S6C) and augmented expression of senescence‐associated genes (Supporting Fig. S6D). We analyzed the in vivo effect of BI2536 on wild‐type mice bearing DEN‐induced HCCs. A decrease in p‐H3S10‐positive cell numbers was found in HCCs after BI2536 treatment for 2 weeks at a dose of 25 mg per kilogram body weight (mg/kg) every other day (Fig. 5A). BI2536‐treated HCCs showed senescence, evidenced by SA‐β‐gal staining and increased expression of senescence‐associated genes (Fig. 5B and Supporting Fig. S6E). Notably, HCCs from BI2536‐treated mice were infiltrated with inflammatory cells (Supporting Fig. S6F), and TNFα levels were elevated in BI2536‐treated HCCs (Fig. 5C). However, BI2536 treatment induced a much lower level of cell death than in svv Δli* HCCs (Fig. 5D), suggesting that the HCC cells may be protected by anticell death mechanisms. These data were in line with recent reports that BI2536 used alone did not appear to be an effective agent for cancer therapy.14, 35
Figure 5.
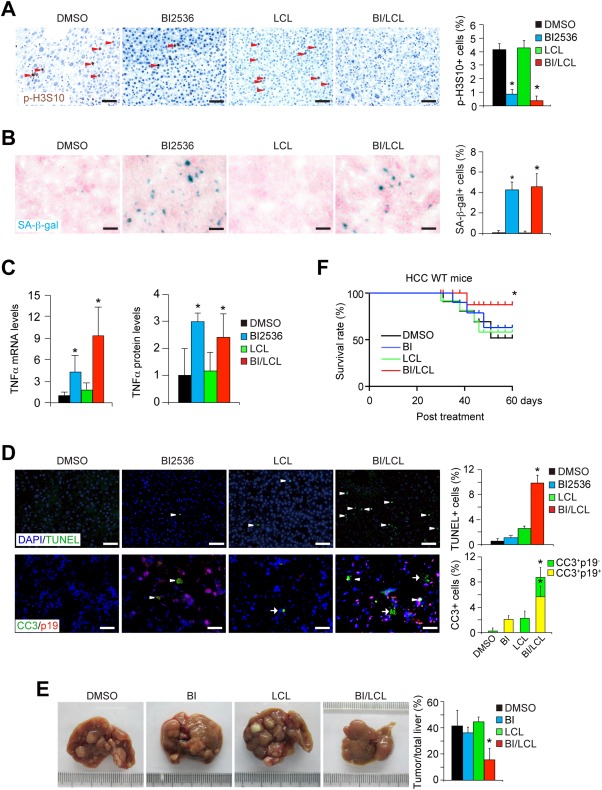
Regression of HCCs by combined treatment with mitotic inhibitor and SMAC mimetic. (A,B) HCCs were induced in wild‐type mice at the age of 2 weeks:. 25 mg/kg body weight BI2536 (BI), LCL161 (LCL) alone, or combination with BI2536 and LCL161 (BI/LCL) were injected into cancer‐bearing mice every other day at the age of 8 months. Mitosis and senescence in HCCs were characterized 2 weeks after BI2536 treatment. Mitosis of HCC cells was analyzed by p‐H3S10 immunohistochemical staining. p‐H3S10‐positive cancer cells were quantified (A). n = 4 for each group. SA‐β‐gal staining showed increased senescence in BI2536‐treated or BI/LCL combined‐treated HCCs. SA‐β‐gal‐positive HCC cells were quantified (B). (C) TNFα mRNA and protein levels in HCCs from mice treated by BI2536, LCL161 alone, or BI/LCL combination for 2 weeks were determined by qPCR and enzyme‐linked immunosorbent assay. n = 4 for each group. (D) Cell death was determined by TUNEL staining of HCCs 2 weeks after treatment. Positively stained cancer cells were quantified. Cleaved caspase 3 (CC3) and p19Arf were costained on HCC sections. CC3‐positive p19Arf‐negative cells are indicated by arrows. CC3‐positive p19Arf‐positive cancer cells are indicated by arrowheads. CC3‐positive cells and p19Arf‐positive cells were quantified. n = 3 for each group. (E) Therapeutic effects were monitored and HCCs were quantified after 2 months after BI2536, LCL161 alone, or BI/LCL combination treatment. n = 5 for DMSO group; n = 4 for BI group; n = 4 for LCL group; n = 5 for BI/LCL group. (F) Kaplan‐Meier survival analysis showed that combined treatment of BI2536 and LCL161 significantly improved the survival of HCC‐bearing mice (log‐rank test, P < 0.01). n = 11 for DMSO group; n = 10 for BI2536 group; n = 7 for LCL group; n = 12 for BI/LCL group. * P < 0.05, t test. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide.
To boost the sensitivity to TNFα‐induced cell death, we chose a clinically advanced SMAC mimetic, LCL161.36, 37 We first confirmed that LCL161 increased sensitivity to TNFα‐induced cell death in primary human HCC cells (Supporting Fig. S6G). However, when LCL161 alone was applied to mice bearing DEN‐induced HCC, it had a negligible effect on mitosis and senescence in HCC cells (Fig. 5A,B), and LCL161 only induced a mildly increased cell death in HCCs (Fig. 5D). As a consequence, LCL161 alone did not repress the progress of DEN‐induced primary HCCs at a dose of 25 mg/kg every other day for 2 months (Fig. 5E).
We then analyzed the therapeutic effect of combined use of BI2536 and LCL161 in vivo. mRNA levels of Survivin were not significantly changed in HCCs by BI2536 treatment or combined treatment of BI2536 and LCL161 (Supporting Fig. S6H). Combination treatment with BI2536 and LCL161, each administered at a dose of 25 mg/kg every other day for 2 weeks, triggered mitotic arrest (Fig. 5A). Moreover, cellular senescence was enhanced in HCC cells, detected by SA‐β‐gal staining (Fig. 5B) and senescence‐associated gene expression (Supporting Fig. S6I). Increased levels of TNFα were also found in combined treatment with BI2536 and LCL161 (Fig. 5C). Importantly, the combination treatment induced substantially more cell death in both senescent and neighboring nonsenescent HCC cells, when compared to that observed with either BI2536 or LCL161 alone (Fig. 5D).
HCC‐bearing mice were sacrificed after continuous treatment of both BI2536 and LCL161 for 2 months. Although HCCs were detectable in these mice, HCC progression was markedly arrested after combined treatment with BI2536 and LCL161, a finding that largely phenocopied Survivin‐deficiency in HCCs (Fig. 5E). Moreover, combination therapy extended the life span of cancer‐bearing mice (Fig. 5F). Overall, the combination of BI2536‐induced cellular senescence and LCL161‐sensitized cell death mimics the therapeutic effects of Survivin depletion in HCCs.
BI2536 AND LCL161 SYNERGISTICALLY INHIBIT HUMAN HCCs
To further assess the clinical utility of our findings, we tested whether the combination of BI2536 and LCL161 could restrain progression of human HCCs in vivo in the human PDX mouse model, in which primary human HCC tissues are transplanted into athymic nude mice (Fig. 6A). When HCC volume reached ∼800 mm3, HCC‐bearing mice were treated with BI2536 and LCL161 each at 25 mg/kg every other day for 1 month. Tumor size was monitored every 5 days (Fig. 6A). Intriguingly, single treatment with either BI2536 or LCL161 slowed the growth of PDX HCC, suggesting that these inhibitors alone have better therapeutic effects on xenografted HCCs than de novo HCCs. We speculated that this might be because the PDX HCC is a selective model for cancer cells with a higher mitotic index compared to de novo HCCs (compare Fig. 6C with Figs. 2A and 5A). Mice bearing the PDX HCCs were sacrificed after continuous treatment of combined BI2536 and LCL161 for 1 month. Importantly, combination treatment with BI2536 and LCL161 caused PDX HCCs to stop growing within 2 weeks and regress to ∼75% of their original size by the end of the experiment (Fig. 6A). The findings suggest that these two agents act synergistically to achieve a beneficial response when combined.
Figure 6.
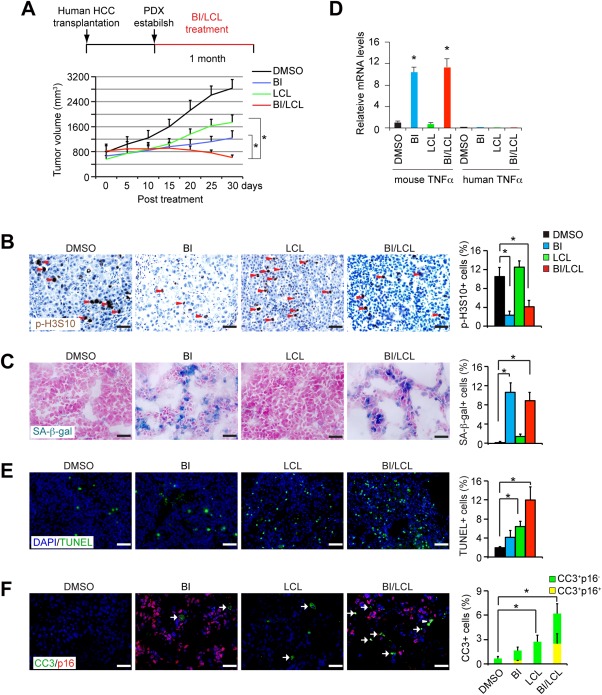
Mitotic inhibitor and SMAC mimetic synergistically eliminate human PDX HCCs. (A) PDX HCCs developed in athymic nude mice and treated with BI2536 and LCL161 alone or combined for 1 month. For each experimental group, we transplanted 5 human HCCs with 2 PDX tumors for each HCC. Growth of the PDX HCC was monitored during combination treatment with BI2536 (25 mg/kg) and LCL161 (25 mg/kg) for 1 month. n = 10 for each group. (B,C) Cell mitosis and senescence in PDX cancers were determined by p‐H3S10 (B) and SA‐β‐gal (C) staining, respectively. Positively stained cancer cells were quantified. n = 3 for each group. (D) PDX liver cancers were treated with BI2536, LCL161, or combination of BI2536 and LCL161. Species‐specific qPCR was applied to analyzed mouse and human TNFα mRNA levels in PDX cancers. n = 3 for each group. (E) Cell death was determined by TUNEL staining. Positively stained HCC cells were quantified. n = 3 for each group. (F) Cleaved caspase 3 (CC3) and p16 were costained on HCC tissues. CC3‐positive p16‐negative cells are indicated by arrows. CC3‐positive p16‐positive cancer cells are indicated by arrowheads. CC3‐positive cells and p16‐positive cells were quantified. n = 3 for each group. Scale bars, 50 μm. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide.
The mechanisms of the responses were further evaluated. BI2536 treatment alone or in combination with LCL161 resulted in a profound cell cycle arrest and caused prominent cellular senescence in PDX cancers (Fig. 6B,C) without affecting expression of Survivin (Supporting Fig. S7A). High expression of human senescence‐associated genes (Supporting Fig. S7B) and increased infiltration of inflammatory macrophages (Supporting Fig. S7C) were detected in PDX HCCs receiving BI2536 alone or the combination treatment. The expression level of mouse TNFα, rather than human TNFα, increased in the BI2536 treatment alone and in the combination treatment (Fig. 6D), indicating that mouse inflammatory cells, but not the human HCC cells, represent the sources of TNFα. In PDX HCCs treated with LCL161 alone, impairments in mitosis and senescence‐associated features were not detectable (Fig. 6B,C). However, LCL161 treatment alone did increase the level of cell death in human PDX HCCs (Fig. 6E,F). Notably, combination treatment with BI2536 and LCL161 induced the highest level of cell death in both senescent and neighboring nonsenescent HCC cells (Fig. 6E,F), reinforcing the concept that synergistically boosting senescence and TNFα‐triggered cell death is of value in treating HCC.
Discussion
In this study, genetic deletion of Survivin induced mitotic defect and senescence in HCCs, which then caused inflammation and TNFα induction. Moreover, Survivin inactivation sensitized TNFα‐induced cell death in both senescent and nonsenescent HCC cells, which led to a marked regression of HCCs (Fig. 4D). Based on these findings, we proposed a new therapy to mimic the multiple anti‐HCC mechanisms of Survivin deletion by triggering mitosis defect and senescence and enhancing TNFα‐induced cell death. To exploit these findings, we used the PLK1 inhibitor, BI2536, to induce mitosis defect and senescence and the SMAC mimetic, LCL161, to sensitize HCC cells to TNFα‐induced cell death (Fig. 7). Importantly, combined treatment with the mitotic inhibitor and the SMAC mimetic phenocopied Survivin deletion by inducing senescence and sensitizing TNFα‐dependent cell death therefore supporting the proposed mechanistic model. Also, it provided a strategy bypassing direct targeting Survivin. Several important points are worth noting. First, senescence is an essential trigger of HCC regression by inducing inflammatory cytokine TNFα. Second, TNFα is critical for cell death in both cytostatic and proliferating HCC cells. Finally, depletion of Survivin or blocking the antiapoptotic pathway caused a dramatic increase in cell death in response to TNFα. Our results therefore demonstrate a paradigm to harness the synergistic power of mitosis arrest‐induced senescence and boosting TNFα‐induced cell death.
Figure 7.
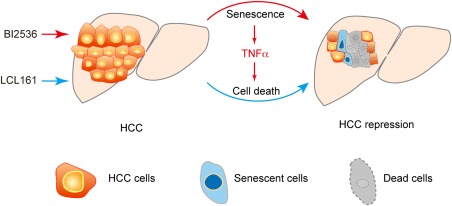
Proposed therapeutic strategy. Schematic summarization of the proposed HCC therapeutic strategy, which shows that PLK1 inhibitor BI2536 induced mitosis defect and senescence, and the SMAC mimetic, LCL161, sensitized HCC cells to TNFα‐induced cell death. Combined treatment with these two drugs markedly repressed HCC.
Both Survivin depletion and PLK1 inhibition caused mitotic defects and senescence in HCC cells, consistent with an earlier report showing that mitosis defect is efficient in induction of senescence.38 Moreover, genetic depletion or chemical inhibition of Aurora kinase caused aberrant mitosis, which mediated senescence in cancer cells.11, 39 Notably, both p53‐ and Rb‐associated senescence pathways were activated in Survivin‐deficient HCCs. The underlying mechanisms of mitosis defect‐associated senescence could be even more complicated, because senescence induced by Aurora kinase inhibition were independent of the function of p53 and Rb.11, 39 Although it remains unclear by which mechanisms mitosis arrest induces senescence, these findings indicate that mitotic inhibition is a plausible approach to induce senescence in vivo.
Our findings showed that cytostatic and proliferating HCC cells could be eliminated by sensitizing to TNFα‐induced cell death. TNFα expression was strongly associated with senescence and infiltration of inflammatory cells. It was notable that Survivin‐deficient HCC cells had heightened sensitivity to TNFα‐induced cell death. TNFα has been reported to play multiple roles in liver tumorigenesis.40 In the initiation of HCCs, and in the setting of nuclear factor kappa B inhibition, inflammation and TNFα cause cell death and compensatory proliferation; the latter contributes to tumorigenesis.21, 41 Although secondary tumorigenesis may be a concern post‐TNFα induction, its major effect in our study appears to be to eliminate malignant HCC cells. After etanercept treatment, HCC cell death was markedly blocked, which led to partially restored liver tumorigenesis. However, etanercept treatment did not fully restore the inhibited HCC phenotype in svv Δli* mice, likely attributed to the fact that the Survivin‐deficient HCC cells did not proliferate normally.
Roles for Survivin in mitosis and cell death have been recognized in tumorigenesis for many years; however, it is unclear whether and how these two seemingly separate functions are intertwined.42 Using a genetic model, our results showed, for the first time, that Survivin promotes tumorigenesis in both a cell autonomous and noncell autonomous manner. This has not previously been fully appreciated using xenograft models,43 but is important because development of Survivin‐specific inhibitors has so far been unsuccessful.22, 23 By uncovering the unique mechanisms regulated by Survivin, it is possible to bypass direct targeting of Survivin. Indeed, we proposed and tested a new therapeutic approach that mimics the anticancer mechanisms of Survivin‐deletion by triggering mitosis defect and senescence and enhancing TNFα‐induced cell death.
In this study, we tested the combination of the PLK1 inhibitor, BI2536, and the SMAC mimetic, LCL161, for HCC therapy. It is worth noting that we only attempted to use these compounds to mimic mitosis defect and senescence and cell death sensitization. It is not necessarily relevant whether PLK1 and IAPs are direct targets of Survivin. Interestingly, both compounds have shown limited therapeutic responses in clinical trials when used as a monotherapy.14, 35, 36 Others have proposed that BI2536 or LCL161 might be used in combination with other cytotoxic compounds.15, 36, 44 Our data strongly support the use of these two newly developed compounds in combination for HCC treatment. Furthermore, no adverse effects on liver functions or body weight were found with longer‐term combined treatment in HCC‐bearing mice (Supporting Fig. S8A,B) and in mice with 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC)‐induced liver injury (Supporting Fig. S8C,D). Pharmacokinetic studies reveal that clearance of BI2536, when combined with LCL161, was slightly increased, and thus total exposure dose was decreased compared to single use (Supporting Fig. S9A). On the other hand, the clearance and total exposure dose of LCL161 were unchanged (Supporting Fig. S9A). Moreover, there were no drug‐drug interactions between BI2536 and LCL161 in mice with DDC‐induced liver injury (Supporting Fig. S9B). The pharmacological effects of BI2536 and LCL161 remained in combination treatment (Supporting Fig. S9C,D). Therefore, the synergistic anti‐HCC effect of combination treatment is not likely attributed to drug‐drug interactions between BI2536 and LCL161.
Based on our findings, we expect that additional experiments will reveal whether combinations of other mitotic inhibitors or proapoptotic compounds would exhibit similar therapeutic benefits (Fig. 7). We recognize that such an approach requires careful monitoring and dosing, so as to avoid adversely affecting healthy cells by inducing unwanted senescence and cell death. Overall, we speculate that the combination of a mitotic inhibitor and a SMAC mimetic will not only provide new ammunition against HCC, but also prove useful for the treatment of a range of other inflammation‐related cancers.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28637/suppinfo.
Supporting Information
Acknowledgments
We thank Zhengye Yang (USTC, China) for comments on mitosis and senescence, Liming Sun (SIBCB, China) and Zheng‐Gang Liu (NIH, USA) for comments on cell death, and Dangsheng Li (Cell Research) for thorough discussion of the manuscript.
Potential conflict of interest: Nothing to report.
This study is funded by the Ministry of Science and Technology (MOST) of China (2014CB910601, 2012CB945001, and 2012ZX10002), the National Science Foundation of China (NSFC) (31225016, 31501111, 81221061, 81471948, and 81471948), Shanghai Science and Technology Committee (15JC1400200, 14XD1404200). Dr. Dan Li is supported by the Postdoctor Research Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2014KIP302), and the China Postdoctoral Science Foundation (2015M580361).
[The copyright line for this article was changed on July 26, 2016, after original online publication.]
Contributor Information
Yuan Ji, Email: ljhui@sibcb.ac.cn.
Hong‐Yang Wang, Email: hywangk@vip.sina.com.
Lijian Hui, Email: ji.yuan@zs-hospital.sh.cn.
REFERENCES
- 1. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 3. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656‐665. [DOI] [PubMed] [Google Scholar]
- 4. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012;379:1245‐1255. [DOI] [PubMed] [Google Scholar]
- 5. Kuilman T, Peeper DS. Senescence‐messaging secretome: SMS‐ing cellular stress. Nat Rev Cancer 2009;9:81‐94. [DOI] [PubMed] [Google Scholar]
- 6. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature 2011;479:547‐551. [DOI] [PubMed] [Google Scholar]
- 7. Acosta JC, Gil J. Senescence: a new weapon for cancer therapy. Trends Cell Biol 2012;22:211‐219. [DOI] [PubMed] [Google Scholar]
- 8. Perez‐Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer 2014;14:547‐558. [DOI] [PubMed] [Google Scholar]
- 9. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell 2008;134:657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y, Hawkins OE, Su Y, Vilgelm AE, Sobolik T, Thu YM, et al. Targeting aurora kinases limits tumour growth through DNA damage‐mediated senescence and blockade of NF‐kappaB impairs this drug‐induced senescence. EMBO Mol Med 2013;5:149‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012;13:789‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lens SM, Voest EE, Medema RH. Shared and separate functions of polo‐like kinases and aurora kinases in cancer. Nat Rev Cancer 2010;10:825‐841. [DOI] [PubMed] [Google Scholar]
- 14. Mross K, Dittrich C, Aulitzky WE, Strumberg D, Schutte J, Schmid RM, et al. A randomised phase II trial of the Polo‐like kinase inhibitor BI 2536 in chemo‐naive patients with unresectable exocrine adenocarcinoma of the pancreas ‐ a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer 2012;107:280‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ellis PM, Chu QS, Leighl N, Laurie SA, Fritsch H, Gaschler‐Markefski B, et al. A phase I open‐label dose‐escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non‐small‐cell lung cancer. Clin Lung Cancer 2013;14:19‐27. [DOI] [PubMed] [Google Scholar]
- 16. Schoffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo‐like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer 2010;46:2206‐2215. [DOI] [PubMed] [Google Scholar]
- 17. Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto‐oncogene and inhibition of aurora‐B kinase. Proc Natl Acad Sci U S A 2010;107:13836‐13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tunquist BJ, Woessner RD, Walker DH. Mcl‐1 stability determines mitotic cell fate of human multiple myeloma tumor cells treated with the kinesin spindle protein inhibitor ARRY‐520. Mol Cancer Ther 2010;9:2046‐2056. [DOI] [PubMed] [Google Scholar]
- 19. Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin‐5. Cancer Res 2008;68:3269‐3276. [DOI] [PubMed] [Google Scholar]
- 20. Walczak H. Death receptor‐ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol 2013;5:a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation is controlled by AP‐1 through SIRT6‐dependent inhibition of survivin. Nat Cell Biol 2012;14:1203‐1211. [DOI] [PubMed] [Google Scholar]
- 22. Altieri DC. Targeting survivin in cancer. Cancer Lett 2013;332:225‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holmes D. Cancer drug's survivin suppression called into question. Nat Med 2012;18:842‐843. [DOI] [PubMed] [Google Scholar]
- 24. Li D, Cen J, Chen X, Conway EM, Ji Y, Hui L. Hepatic loss of survivin impairs postnatal liver development and promotes expansion of hepatic progenitor cells in mice. Hepatology 2013;58:2109‐2121. [DOI] [PubMed] [Google Scholar]
- 25. Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. YM155, a novel small‐molecule survivin suppressant, induces regression of established human hormone‐refractory prostate tumor xenografts. Cancer Res 2007;67:8014‐8021. [DOI] [PubMed] [Google Scholar]
- 26. Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, et al. A multi‐center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs 2011;29:161‐166. [DOI] [PubMed] [Google Scholar]
- 27. Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small‐molecule suppressor of survivin, in patients with advanced, refractory, non‐small‐cell lung cancer. J Clin Oncol 2009;27:4481‐4486. [DOI] [PubMed] [Google Scholar]
- 28. Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, et al. A senescence‐inflammatory switch from cancer‐inhibitory to cancer‐promoting mechanism. Cancer Cell 2013;24:242‐256. [DOI] [PubMed] [Google Scholar]
- 29. Wang L, Du F, Wang X. TNF‐alpha induces two distinct caspase‐8 activation pathways. Cell 2008;133:693‐703. [DOI] [PubMed] [Google Scholar]
- 30. Goldenberg MM. Etanercept, a novel drug for the treatment of patients with severe, active rheumatoid arthritis. Clin Ther 1999;21:75‐87; discussion, 71‐72. [DOI] [PubMed] [Google Scholar]
- 31. Zalevsky J, Secher T, Ezhevsky SA, Janot L, Steed PM, O'Brien C, et al. Dominant‐negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol 2007;179:1872‐1883. [DOI] [PubMed] [Google Scholar]
- 32. Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T‐helper‐1‐cell cytokines drive cancer into senescence. Nature 2013;494:361‐365. [DOI] [PubMed] [Google Scholar]
- 33. Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, et al. BI 2536, a potent and selective inhibitor of polo‐like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007;17:316‐322. [DOI] [PubMed] [Google Scholar]
- 34. Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt‐Dias M. Polo‐like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 2014;15:433‐452. [DOI] [PubMed] [Google Scholar]
- 35. Sebastian M, Reck M, Waller CF, Kortsik C, Frickhofen N, Schuler M, et al. The efficacy and safety of BI 2536, a novel Plk‐1 inhibitor, in patients with stage IIIB/IV non‐small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open‐label, randomized phase II clinical trial. J Thorac Oncol 2010;5:1060‐1067. [DOI] [PubMed] [Google Scholar]
- 36. Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB. Phase I dose‐escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 2014;32:3103‐3110. [DOI] [PubMed] [Google Scholar]
- 37. Houghton PJ, Kang MH, Reynolds CP, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2012;58:636‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johmura Y, Shimada M, Misaki T, Naiki‐Ito A, Miyoshi H, Motoyama N, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell 2014;55:73‐84. [DOI] [PubMed] [Google Scholar]
- 39. Perez de Castro I, Aguirre‐Portoles C, Fernandez‐Miranda G, Canamero M, Cowley DO, Van Dyke T, Malumbres M. Requirements for Aurora‐A in tissue regeneration and tumor development in adult mammals. Cancer Res 2013;73:6804‐6815. [DOI] [PubMed] [Google Scholar]
- 40. Karin M, Greten FR. NF‐kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749‐759. [DOI] [PubMed] [Google Scholar]
- 41. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005;121:977‐990. [DOI] [PubMed] [Google Scholar]
- 42. Altieri DC. Survivin and IAP proteins in cell‐death mechanisms. Biochem J 2010;430:199‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mesri M, Wall NR, Li J, Kim RW, Altieri DC. Cancer gene therapy using a survivin mutant adenovirus. J Clin Invest 2001;108:981‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fulda S. Molecular pathways: targeting inhibitor of apoptosis proteins in cancer—from molecular mechanism to therapeutic application. Clin Cancer Res 2014;20:289‐295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28637/suppinfo.
Supporting Information