

Abstract
A transition‐metal‐free, mild, and highly regioselective synthesis of nitroarenes from arenes has been developed. The products are obtained in a sequential one‐pot reaction by nitration of iodine(III) reagents with two carbon ligands, which are formed in situ from iodine(I). This novel concept has been extended to formation of aryl azides, and constitutes an important step towards catalytic reactions with these hypervalent iodine reagents. An efficient nitration of isolated diaryliodonium salts has also been developed, and the mechanism is proposed to proceed by a [2,2] ligand coupling pathway.
Keywords: arylation, density functional calculations, hypervalent compounds, oxidation, reaction mechanisms
Hypervalent iodine compounds have become important reagents for a wide variety of transformations, as they have a valuable combination of properties including high reactivity, high functional‐group tolerance, and low toxicity.1 The main drawback with hypervalent iodine reagents is the stoichiometric formation of either an iodine(I) or iodine(III) compound as waste, depending on the oxidation state (III or V) of the starting reagent. This aspect lowers the atom efficiency and may lead to product purification problems. The groups of Ochiai and Kita reported the first catalytic uses of hypervalent iodine reagents in 2005 (Scheme 1 a).2 Since then many other systems have appeared, where a catalytic amount of iodoarene is combined with a stoichiometric amount of an oxidant.3
Scheme 1.
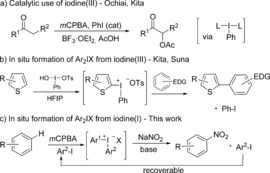
In situ formation of iodine(III) reagents. EDG=electron‐donating group, HFIP=1,1,1,3,3,3‐hexafluoro‐2‐propanol, mCPBA= m‐chloroperbenzoic acid, Ts=4‐toluenesulfonyl.
Iodonium salts and their cyclic counterparts benziodoxol(on)es (e.g. EBX and Togni's reagent) are important iodine(III) reagents for the transfer of carbon ligands (aryl, alkynyl, vinyl, CF3, or CN groups) to a wide range of nucleophiles.4 These reagents are generally synthesized in one or more steps, isolated, and subsequently reacted with a nucleophile. Kita and co‐workers have demonstrated the efficient in situ formation of diaryliodonium salts from other iodine(III) reagents in metal‐free arylations (Scheme 1 b),5 and the group of Suna has utilized the same strategy in metal‐catalyzed arylations.6 However, to the best of our knowledge, catalytic reactions with iodonium salts or benziodoxol(on)es remain unknown.4, 7
In our long‐term quest for catalytic arylation conditions, we reasoned that in situ formation of diaryliodonium salts from arenes and iodoarenes in the presence of an oxidant, followed by arylation of a suitable nucleophile under metal‐free conditions, would constitute an important step towards this difficult goal. Herein we present our results of an efficient arylation of sodium nitrite with diaryliodonium salts, which can be formed in situ from arenes and iodoarenes under oxidative conditions (Scheme 1 c). The methodology compares well to previous routes to nitroarenes, concerning regioselectivity, functional‐group tolerance, and ease of handling.8
Synthetic routes to aromatic nitro compounds are important, as nitroarenes are key intermediates in the preparation of many dyes, plastics, and explosives.9 They can be synthesized from arenes under harsh acidic conditions,9a by treatment with [bis(trifluoroacetoxy)iodo]benzene (PIFA),10 and by rhodium‐catalyzed oxidative C−H activation,11 all of which give regioisomeric mixtures. Regiospecific transformations include palladium‐catalyzed nitration of aryl chlorides,12 and either metal‐mediated or metal‐free conversion of arylboronic acids.13 Nitroarenes have also been obtained from diaryliodonium salts,14 thus making this transformation suitable for our envisioned one‐pot reaction.
The nitration reaction was first investigated using isolated diaryliodonium salts, as a stepwise and general methodology to nitroarenes could be preferable with advanced diaryliodonium salts carrying sensitive functional groups. Various nitrite sources, solvents, and temperatures were screened, and high yields of nitroarenes were obtained with NaNO2 in either DME or EtOAc at 70 °C,8 the latter solvent being preferable from an environmental perspective. The nitration proved insensitive to moisture and air, and can be conveniently carried out without special precautions.
Interestingly, the diaryliodonium counterion had a major impact on the reaction, as shown in Table 1. Diphenyliodonium triflate (X=OTf) and hexafluorophosphate (X=PF6) gave nitrobenzene (2 a) in excellent yields, whereas the corresponding tetrafluoroborate (X=BF4) and tosylate (X=OTs) resulted in poor conversion into 2 a. This result was surprising as tetrafluoroborate salts are usually efficient in both metal‐free and metal‐catalyzed reactions.4e,4f We hypothesized that addition of sodium triflate might improve the outcome, and discovered that 1 equivalent of NaOTf was indeed sufficient to provide 2 a in 94 % yield. The tosylate salt could also be efficiently utilized upon addition of NaOTf, and the trend was verified with other tetrafluoroborate salts.[8] This finding is important, as anion exchange of diaryliodonium salts can be tedious and waste producing. While the exact role of NaOTf remains unknown, the additive effect could be useful in other arylations where the counterion influences the reaction.
Table 1.
Counterion effect under optimized reaction conditions.[a]

| NaOTf | Yield [%][b] | |||||
|---|---|---|---|---|---|---|
| (equiv) | X=OTf | X=PF6 | X=BF4 | X=OTs | ||
| 0 | 94 | 91 | 18 | 18 | ||
| 0.5 | 68 | |||||
| 1.0 | 86 | 94 | 87 | |||
[a] Reaction conditions: NaNO2 (0.16 mmol) was added to 1 (0.14 mmol) and NaOTf (0–1 equiv) in EtOAc (1 mL). The suspension was stirred at 70 °C for 16 hours. [b] Yield determined by 1H NMR analysis using 1,3,5‐trimethoxybenzene as internal standard. Tf=trifluoromethanesulfonyl.
The scope of the arene nitration was subsequently investigated with diaryliodonium salts 1 (Scheme 2).15 Triflate, tetrafluoroborate, and tosylate salts could all be employed, using NaOTf as an additive with the two latter anions. Nitrobenzene (2 a) and the halide‐substituted products 2 b–e were all isolated in high to excellent yields. The reaction showed great tolerance for steric hindrance in the ortho‐position, as exemplified with the halide products 2 d and 2 e, and with the high‐yielding synthesis of the di‐ortho‐alkyl‐substituted nitroarenes 2 f and 2 g. While the electron‐rich products 2 h and 2 i were obtained in moderate yields despite addition of 3 equivalents of NaNO2, the synthesis of 2 h was improved to 79 % yield by changing the solvent. Moderately electron‐donating substituents were well tolerated, and alkyl‐substituted salts were efficiently transformed into 2 f, 2 g, 2 j, and 2 k. The synthesis of 2 k was easily scaled up to 7 mmol, thus displaying the robustness of the reaction. Likewise, products with electron‐withdrawing substituents were obtained in high yields (2 l–o). More elaborate diaryliodonium salts could also be utilized, as demonstrated in the synthesis of the carbonyl‐substituted products 2 p–q, and in the reactions with pyridyl iodonium salts to provide the heterocycles 2 r and 2 s in 78 and 88 % yield, respectively.
Scheme 2.
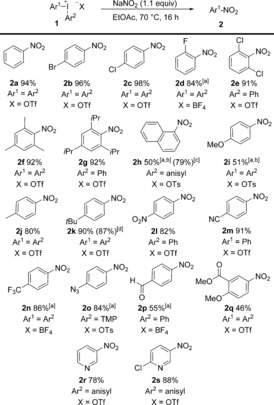
Scope for the nitration with isolated salts 1. Reaction conditions as in Table 1 with 1 (0.5 mmol). Yields are those of isolated products. [a] NaOTf (1 equiv) was added. [b] NaNO2 (3 equiv) was used. [c] In CH2Cl2/H2O 1:1. [d] 7 mmol scale.
Unsymmetric diaryliodonium salts were utilized in several reactions,16 and the phenyl group was an efficient “dummy” group in the nitration of electron‐deficient aryl moieties (2 l, 2 m, 2 p). A strong ortho‐effect4e–4h, 17 was observed in reactions with ortho‐substituted iodonium salts, which proceeded with excellent chemoselectivities (2 e, 2 g, 2 h) despite competing electronic preferences (2 g). p‐Methoxyphenyl (anisyl) or 2,4,6‐trimethoxyphenyl (TMP)17a, 18 dummy groups were efficiently utilized in reactions yielding 2 h, 2 o, 2 r, and 2 s.
The envisioned one‐pot sequence was next investigated. Unfortunately, synthesis of the diaryliodonium salts in either EtOAc or DME was low yielding, hence the one‐pot sequence was further studied in dichloromethane using our established methodology.15 Bromobenzene (3 b) and 4‐bromoiodobenzene (4 b) were treated with mCPBA and TfOH to yield the symmetric 4‐bromophenyl iodonium salt 1 b in situ (Scheme 3 a). Sodium nitrite and NaHCO3 19 were then added, thus providing nitroarene 2 b and unreacted diaryliodonium salt as the major components. Addition of water14d and adjustment of the pH value improved the outcome, and delivered 2 b in 58 % yield with 3 equivalents of NaNO2 within 3 hours.8 Only the p‐substituted product 2 b was observed, and the complete regioselectivity can be attributed to the known, highly p‐selective formation of diaryliodonium salts.4e,4f,4h
Scheme 3.
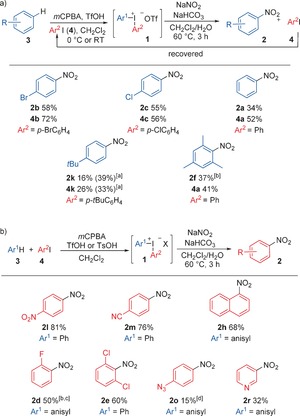
One‐pot nitrations using 4 (0.5 mmol), 3 (1.1 equiv), mCPBA (1.1 equiv), TfOH (2–3 equiv). and NaNO2 (3 equiv). Yields are those of isolated products. [a] Nitration at 80 °C, with <5 % impurity. [b] TsOH⋅H2O (1 equiv) used instead of TfOH. [c] 2 d/2 i>20:1. [d] Anisole (5 equiv) used.
Considering the three steps involved in this sequential one‐pot reaction (oxidation, formation of 1 b, and nitration to 2 b), this metal‐free C−H nitration of bromobenzene is quite efficient (Scheme 3 a). The resulting iodoarene 4 b was isolated in 72 %, thus showing the recyclability of this component. In the same fashion, chlorobenzene was converted into 2 c in 55 % with recovery of the iodoarene 4 c in similar yield, and benzene was transformed into nitrobenzene (2 a). One‐pot C−H nitration of alkyl benzenes proved more difficult, and the three‐step reaction provided 2 k in poor yield, which gratefully was considerably improved at higher temperature.20
The synthesis of unsymmetric diaryliodonium salts (Ar1≠Ar2) is often high yielding,15 hence we investigated the nitration of mesitylene by in situ formation of a mesityl(phenyl)iodonium salt (Scheme 3 a). To our delight, the sterically hindered product 2 f was obtained with excellent chemoselectivity (2 a not observed). In all cases the iodoarenes 4 could be recovered in at least equivalent amounts from the one‐pot reactions, thus allowing reuse in subsequent reactions.21
The scope investigation was continued with a range of unsymmetric diaryliodonium salts, thus delivering functionalized nitroarenes in good yields with complete regioselectivity (Scheme 3 b).22 Nitroarenes with electron‐withdrawing substituents were formed in excellent yields over the three steps (2 l, 2 m). 1‐Nitronaphthalene (2 h), and the ortho‐halogenated products 2 d and 2 e were also obtained with high efficiency, thus illustrating that electron‐donating substituents as well as ortho‐substituents are well tolerated. The p‐azido product 2 o was obtained in an unsatisfactory yield, which is likely due to the moderate‐yielding salt synthesis.8 Heterocyclic products could also be obtained in a one‐pot manner, as exemplified by the synthesis of 3‐nitropyridine (2 r).
The application of this novel one‐pot concept to other nucleophiles is currently underway in our laboratory, and preliminary results with sodium azide23 are promising (Scheme 4).
Scheme 4.
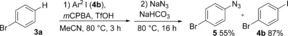
Preliminary investigation with azide as a nucleophile (1H NMR yield).
A radical mechanism is unlikely, as addition of the radical scavengers, 1,1‐diphenylethylene and TEMPO, to the nitration in EtOAc did not hamper the reaction significantly.8 Hence, we suggest that the reaction proceeds by formation of a T‐shaped intermediate containing a Nu−I bond (Scheme 5). The nitrite nucleophile could either form an N−I or O−I intermediate (A and B), followed by ligand coupling to provide the N‐arylated product 2 a or O‐arylated product 6 (not observed). While ligand coupling by the [1,2] pathway is the only possibility with some nucleophiles, recent computational studies have found that four‐ or five‐membered transition states can be favored.17a,17b, 24
Scheme 5.
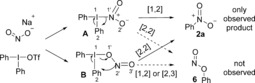
Mechanistic possibilities.
The suggested pathways were studied by DFT computations (Figure 1).8 Ligand exchange by a dissociative pathway leads to a large increase in energy, whereas the associative pathway via four‐coordinated complexes, to give the intermediates B1 and B2, is energetically favorable by 13 and 25 kJ mol−1, respectively. Ligand coupling via the three‐membered TS1 would give N‐arylation, whereas coupling via the three‐membered TS2 or five‐membered TS3 would result in O‐arylation. These pathways are, however, all too high in energy to be plausible. The reaction is suggested to give the N‐arylated product 2 a via the four‐membered TS5 (ΔG ≠ =73 kJ mol−1), which has a lower energy barrier than TS4 (ΔG ≠=87 kJ mol−1). The energy difference corresponds to a 135:1 ratio in favor of 2 a at 70 °C, thus explaining why 6 is not observed.8
Figure 1.
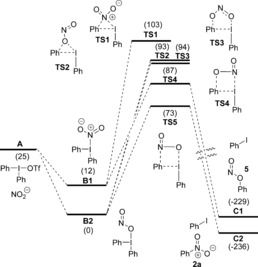
DFT calculated potential energy surfaces for the reaction between 1 a(OTf) and nitrite. Energies are measured in kJ mol−1. −OTf omitted for clarity in B1 and B2, TS1–TS5, and C1 and C2.
To conclude, nitroarenes have been efficiently synthesized by a transition‐metal‐free arylation of sodium nitrite with diaryliodonium salts. An interesting counterion effect was observed, and the unprecedented use of NaOTf as an additive enabled efficient arylations with such salts. The mechanism is suggested to proceed via an O−I intermediate followed by an unusual [2,2] ligand coupling. The reaction has been extended to a sequential one‐pot transformation, where diaryliodonium intermediates are generated from arenes and iodoarenes in a highly regioselective fashion, and subsequently nitrated with recovery of the iodoarene. Azidation also proved possible using the same novel concept, where iodine(III) reagents with two carbon ligands are formed in situ from iodine(I) and subsequently intermolecularly reacted with nucleophiles. This feature constitutes an important step towards catalytic reactions with such hypervalent iodine reagents.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Erik Lindstedt for initial experiments, and Prof. P.‐O. Norrby for mechanistic discussions. This research was conducted using the resources of High Performance Computing Center North (HPC2N). The Swedish Research Council (621‐2011‐3608 and 2015‐04404) and Estonian Research Council (PUTJD114) are kindly acknowledged for financial support.
M. Reitti, P. Villo, B. Olofsson, Angew. Chem. Int. Ed. 2016, 55, 8928.
Contributor Information
Marcus Reitti, http://www.organ.su.se/bo/.
Prof. Berit Olofsson, Email: Berit.Olofsson@su.se.
References
- 1.
- 1a.“Hypervalent Iodine Chemistry” (Ed.: T. Wirth): Top. Curr. Chem 2016, 373; [DOI] [PubMed]
- 1b. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328–3435; [DOI] [PubMed] [Google Scholar]
- 1c. Zhdankin V. V., Hypervalent Iodine Chemistry, Wiley, Chichester, 2013. [Google Scholar]
- 2.
- 2a. Ochiai M., Takeuchi Y., Katayama T., Sueda T., Miyamoto K., J. Am. Chem. Soc. 2005, 127, 12244–12245; [DOI] [PubMed] [Google Scholar]
- 2b. Dohi T., Maruyama A., Yoshimura M., Morimoto K., Tohma H., Kita Y., Angew. Chem. Int. Ed. 2005, 44, 6193–6196; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6349–6352. [Google Scholar]
- 3.For reviews, see:
- 3a. Dohi T., Kita Y., Chem. Commun. 2009, 2073–2085; [DOI] [PubMed] [Google Scholar]
- 3b. Richardson R. D., Wirth T., Angew. Chem. Int. Ed. 2006, 45, 4402–4404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4510–4512; [Google Scholar]
- 3c. Yusubov M. S., Zhdankin V. V., Mendeleev Commun. 2010, 20, 185–191. [Google Scholar]
- 4.
- 4a. Li Y., Hari D. P., Vita M. V., Waser J., Angew. Chem. Int. Ed. 2016, 55, 4436–4454; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4512–4531; [Google Scholar]
- 4b. Waser J., Top. Curr. Chem. 2016, 373, 187–222; [DOI] [PubMed] [Google Scholar]
- 4c. Früh N., Charpentier J., Togni A., Top. Curr. Chem. 2016, 373, 167–186; [DOI] [PubMed] [Google Scholar]
- 4d. Charpentier J., Früh N., Togni A., Chem. Rev. 2015, 115, 650–682; [DOI] [PubMed] [Google Scholar]
- 4e. Olofsson B., Top. Curr. Chem. 2016, 373, 135–166; [Google Scholar]
- 4f. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052–9070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214–9234; [Google Scholar]
- 4g. Aradi K., Tóth B. L., Tolnai G. L., Novák Z., Synlett 2016, 27, 1456–1485; [Google Scholar]
- 4h. Yusubov M. S., Maskaev A. V., Zhdankin V. V., ARKIVOC 2011, 1, 370–409. [Google Scholar]
- 5.
- 5a. Kita Y., Morimoto K., Ito M., Ogawa C., Goto A., Dohi T., J. Am. Chem. Soc. 2009, 131, 1668–1669; [DOI] [PubMed] [Google Scholar]
- 5b. Morimoto K., Yamaoka N., Ogawa C., Nakae T., Fujioka H., Dohi T., Kita Y., Org. Lett. 2010, 12, 3804–3807; see also [DOI] [PubMed] [Google Scholar]
- 5c. Hu B., Miller W. H., Neumann K. D., Linstad E. J., DiMagno S. G., Chem. Eur. J. 2015, 21, 6394–6398. For in situ formation of vinyliodonium salts from iodine(III) see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Dohi T., Kato D., Hyodo R., Yamashita D., Shiro M., Kita Y., Angew. Chem. Int. Ed. 2011, 50, 3784–3787; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3868–3871. [Google Scholar]
- 6.
- 6a. Lubriks D., Sokolovs I., Suna E., J. Am. Chem. Soc. 2012, 134, 15436–15442; [DOI] [PubMed] [Google Scholar]
- 6b. Sokolovs I., Lubriks D., Suna E., J. Am. Chem. Soc. 2014, 136, 6920–6928; [DOI] [PubMed] [Google Scholar]
- 6c. Deprez N. R., Kalyani D., Krause A., Sanford M. S., J. Am. Chem. Soc. 2006, 128, 4972–4973; [DOI] [PubMed] [Google Scholar]
- 6d. Xie F., Zhang Z., Yu X., Tang G., Li X., Angew. Chem. Int. Ed. 2015, 54, 7405–7409; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7513–7517. [Google Scholar]
- 7.The oxidative and often strongly acidic conditions required for synthesis of these reagents from iodine(I) have so far proved incompatible to combine with their use in intermolecular atom transfer reactions. For in situ formation and use of such reagents from iodine(I) in intramolecular reactions, see:
- 7a. Rodríguez A., Moran W. J., Org. Lett. 2011, 13, 2220–2223 (a vinyliodonium salt); [DOI] [PubMed] [Google Scholar]
- 7b. Dohi T., Nakae T., Ishikado Y., Kato D., Kita Y., Org. Biomol. Chem. 2011, 9, 6899–6902 (a vinyliodonium salt); [DOI] [PubMed] [Google Scholar]
- 7c. Hong F., Chen Y., Lu B., Cheng J., Adv. Synth. Catal. 2016, 358, 353–357 (an iodonium ylide). [Google Scholar]
- 8.See the Supporting Information for details.
- 9.
- 9a. Ono N., The Nitro Group in Organic Synthesis Wiley-VCH, New York, 2001; [Google Scholar]
- 9b. Ju K.-S., Parales R. E., Microbiol. Mol. Biol. Rev. 2010, 74, 250–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kloeckner U., Nachtsheim B. J., Chem. Commun. 2014, 50, 10485–10487. [DOI] [PubMed] [Google Scholar]
- 11. Xie F., Qi Z., Li X., Angew. Chem. Int. Ed. 2013, 52, 11862–11866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12078–12082. [Google Scholar]
- 12. Fors B. P., Buchwald S. L., J. Am. Chem. Soc. 2009, 131, 12898–12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Prakash G. K. S., Panja C., Mathew T., Surampudi V., Petasis N. A., Olah G. A., Org. Lett. 2004, 6, 2205–2207; [DOI] [PubMed] [Google Scholar]
- 13b. Wu X.-F., Schranck J., Neumann H., Beller M., Chem. Commun. 2011, 47, 12462–12463; [DOI] [PubMed] [Google Scholar]
- 13c. Chatterjee N., Bhatt D., Goswami A., Org. Biomol. Chem. 2015, 13, 4828–4832. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Beringer F. M., Brierley A., Drexler M., Gindler E. M., Lumpkin C. C., J. Am. Chem. Soc. 1953, 75, 2708–2712; [Google Scholar]
- 14b. Lubinkowski J. J., Gomez M., Calderon J. L., McEwen W. E., J. Org. Chem. 1978, 43, 2432–2435; [Google Scholar]
- 14c. Olah G. A., Sakakibara T., Asensio G., J. Org. Chem. 1978, 43, 463–468; [Google Scholar]
- 14d. Grushin V. V., Kantor M. M., Tolstaya T. P., Shcherbina T. M., Bull. Acad. Sci. USSR Div. Chem. Sci. 1984, 33, 2130–2135. [Google Scholar]
- 15.
- 15a. Bielawski M., Zhu M., Olofsson B., Adv. Synth. Catal. 2007, 349, 2610–2618; [Google Scholar]
- 15b. Bielawski M., Olofsson B., Chem. Commun. 2007, 2521–2523; [DOI] [PubMed] [Google Scholar]
- 15c. Zhu M., Jalalian N., Olofsson B., Synlett 2008, 592–596. [Google Scholar]
- 16.Unsymmetric diaryliodonium salts are often more straightforward to synthesize, and their use avoids the waste of stoichiometric amounts of expensive iodoarenes.
- 17.
- 17a. Malmgren J., Santoro S., Jalalian N., Himo F., Olofsson B., Chem. Eur. J. 2013, 19, 10334–10342; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Gonda Z., Novák Z., Chem. Eur. J. 2015, 21, 16801–16806; [DOI] [PubMed] [Google Scholar]
- 17c. Yamada Y., Okawara M., Bull. Chem. Soc. Jpn. 1972, 45, 2515–2519. [Google Scholar]
- 18. Seidl T. L., Sundalam S. K., McCullough B., Stuart D. R., J. Org. Chem. 2016, 81, 1998–2009. [DOI] [PubMed] [Google Scholar]
- 19. Chan L., McNally A., Toh Q. Y., Mendoza A., Gaunt M. J., Chem. Sci. 2015, 6, 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Details on byproducts, mass balances, and efforts to improve moderate-yielding reactions are given in the Supporting Information.
- 21.This feature could potentially be exploited by attachment of the iodoarene on solid support for large-scale reactions. See:
- 21a. Edwards R., de Vries W., Westwell A. D., Daniels S., Wirth T., Eur. J. Org. Chem. 2015, 6909–6916; [Google Scholar]
- 21b. Chen D.-J., Chen Z.-C., Synlett 2000, 1175–1177. [Google Scholar]
- 22.The synthesis of unsymmetric diaryliodonium salts (Ar1Ar2IX) generally requires combination of the more electron-deficient Ar2I with the more electron-rich Ar1H, while arylation under metal-free conditions is usually chemoselective towards functionalization of the more electron-deficient aryl moiety. The examples in Scheme 3 b hence resulted in a formal substitution of the Ar2I, with concomitant formation of a new iodoarene (Ar1I). We are currently investigating synthetic routes to diaryliodonium salts from electron-rich iodoarenes and electron-deficient arenes to overcome this limitation.
- 23.Azidation of diaryliodonium salts is known. See Ref. [6a, 14b] and
- 23a. Kumar D., Reddy V. B., Synthesis 2010, 1687–1691; [Google Scholar]
- 23b. Wang B., Graskemper J. W., Qin L., DiMagno S. G., Angew. Chem. Int. Ed. 2010, 49, 4079–4083; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4173–4177. The use of azide in chlorinated solvents is unsafe, hence MeCN was used, as reported for synthesis of diaryliodonium salts in Ref. [18]. [Google Scholar]
- 24.
- 24a. Norrby P.-O., Petersen T. B., Bielawski M., Olofsson B., Chem. Eur. J. 2010, 16, 8251–8254; [DOI] [PubMed] [Google Scholar]
- 24b. Ma X.-P., Shi W.-M., Mo X.-L., Li X.-H., Li L.-G., Pan C.-X., Chen B., Su G.-F., Mo D.-L., J. Org. Chem. 2015, 80, 10098–10107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary