

Abstract
Aims
Brentuximab vedotin, an antibody–drug conjugate (ADC), selectively delivers the microtubule‐disrupting agent monomethyl auristatin E (MMAE) into CD30‐expressing cells. The pharmacokinetics of brentuximab vedotin have been characterized in patients with CD30‐positive haematologic malignancies. The primary objective of this phase 1 open label evaluation was to assess the pharmacokinetics of brentuximab vedotin in patients with hepatic or renal impairment.
Methods
Systemic exposures were evaluated following intravenous administration of 1.2 mg kg–1 brentuximab vedotin in patients with CD30‐positive haematologic malignancies and hepatic (n = 7) or renal (n = 10) impairment and compared with those of unimpaired patients (n = 8) who received 1.2 mg kg–1 brentuximab vedotin in another arm of the study.
Results
For any hepatic impairment, the ratios of geometric means (90% confidence interval) for AUC(0,∞) were 0.67 (0.48, 0.93) for ADC and 2.29 (1.27, 4.12) for MMAE. Mild or moderate renal impairment caused no apparent change in ADC or MMAE exposures. Severe renal impairment (creatinine clearance <30 ml min–1; n = 3) decreased ADC exposures (0.71 [0.54, 0.94]) and increased MMAE exposures (1.90 [0.85, 4.21]). No consistent pattern of specific adverse events was evident, but analysis of the safety data was confounded by the patients' poor baseline conditions. Five patients died due to adverse events considered unrelated to brentuximab vedotin. All had substantial comorbidities and most had poor baseline performance status.
Conclusions
Hepatic impairment and severe renal impairment may cause decreases in brentuximab vedotin ADC exposures and increases in MMAE exposures.
Keywords: antibody‐drug conjugate, brentuximab vedotin, CD30 antigen, hepatic impairment, lymphoma, renal impairment
What is Already Known about this Subject
Brentuximab vedotin was initially approved in the United States in 2011 for the treatment of relapsed Hodgkin's lymphoma and systemic anaplastic large cell lymphoma.
Its anticancer activity is due primarily to the release of MMAE within CD30‐expressing cells.
MMAE is excreted primarily via faeces. The remainder is recovered in urine.
What this Study Adds
MMAE exposures were increased 2.3 and 1.9 fold in patients with hepatic or severe renal impairment, respectively. ADC exposures decreased in the same patients.
Poor individual adverse event outcomes observed in some patients may be attributable, at least in part, to comorbidities and poor performance status at baseline.
Introduction
Brentuximab vedotin (ADCETRIS®) is an antibody–drug conjugate (ADC), consisting of a CD30‐directed antibody, cAC10, conjugated by a protease‐cleavable linker to a microtubule‐disrupting agent, monomethyl auristatin E (MMAE). The anticancer activity of brentuximab vedotin is due primarily to the binding of the ADC to CD30‐expressing cells, followed by internalization of the ADC‐CD30 complex and the release of MMAE via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within the cell, subsequently inducing cell cycle arrest and apoptotic death of the cell 1, 2.
Brentuximab vedotin was initially approved in the United States in 2011 for the treatment of relapsed Hodgkin's lymphoma and systemic anaplastic large cell lymphoma and is currently approved in more than 50 countries, including Canada, Japan and members of the European Union. In clinical trials, including two pivotal phase 2 studies in patients with relapsed or refractory Hodgkin's lymphoma and systemic anaplastic large cell lymphoma, brentuximab vedotin showed substantial efficacy and an acceptable safety profile when administered at 1.8 mg kg–1 every 3 weeks 3, 4.
The pharmacokinetics (PK) of brentuximab vedotin have been characterized by monitoring serum concentrations of total antibody (TAb, the sum of ADC and cAC10) and ADC and plasma concentrations of released MMAE 5, 6, 7. The exposures of all three analytes were approximately dose proportional in the therapeutic dose range. Peak concentrations typically occurred at the end of infusion for ADC and TAb and approximately 2 to 3 days post‐dose for MMAE. The TAb PK profile was similar to that of the ADC and its exposures were generally higher than ADC. Steady‐state was achieved for both ADC and MMAE by approximately 21 days, consistent with the half‐life estimates of 4 to 6 days and 3 to 4 days, respectively. On a mass basis, MMAE exposures were approximately 2000‐fold lower than those of the ADC 7. Minimal to no accumulation was observed for ADC after the second or third dose of brentuximab vedotin, whereas the MMAE exposures decreased to approximately 50% to 80% relative to the first dose 8.
Similar to other antibody‐based therapeutics, the elimination of brentuximab vedotin ADC and unconjugated cAC10 are presumably through proteolytic degradation into amino acids and recycling into other proteins 9. The elimination of the unconjugated small molecule MMAE, on the other hand, occurs primarily through the liver and kidneys. A clinical excretion study 10 of brentuximab vedotin demonstrated that the primary excretion route of MMAE was via faeces, which accounted for a median of 72% (range 59 to 77%) of the recovered MMAE over a 1 week period. The rest of the MMAE was recovered in urine. Intact MMAE was the primary species excreted in both faeces and urine. Approximately 23.5% of the intact MMAE was recovered after administration of the ADC. All other species were below the limit of quantitation in faecal and urine extracts, although 10‐fold concentration of the extracts allowed the detection of eight human metabolites.
Patients enrolled in early clinical studies of brentuximab vedotin typically had normal hepatic and renal function and thus did not provide adequate information on the pharmacokinetic differences that might arise with hepatic or renal impairment. The main objectives of this phase 1 open label evaluation were to assess the PK and safety of brentuximab vedotin in patients with hepatic or renal impairment.
Methods
Patient eligibility
In addition to the requisite hepatic and/or renal impairment, patients were adults (≥18 years) with histologically‐confirmed CD30‐positive haematologic malignancy and relapsed, refractory or progressive disease following more than one prior systemic therapy. Treatment‐naive patients with hepatic impairment also were eligible if they were unable to tolerate intensive regimens. Recovery from treatment‐related toxicities due to prior therapies and a baseline absolute neutrophil count ≥1000 μl–1 and platelet count ≥50 000 μl–1 were required. Effective inducers and strong inhibitors of CYP3A4 were prohibited beginning 2 weeks prior to first dose of brentuximab vedotin and during the study, as inhibition of CYP3A4 using ketoconazole has been shown to increase MMAE concentrations 10. Patients with congestive heart failure (New York Heart Association Class III or IV), primary cutaneous anaplastic large cell lymphoma, acute or chronic graft vs. host disease or active systemic infection requiring antibiotics were excluded from enrolment.
Additional entry criteria for patients with hepatic impairment included serum bilirubin >2 mg dl–1, stable hepatic function prior to study entry as assessed by the investigator at the screening visit, Child‐Pugh assessment group A or B and Eastern Cooperative Oncology Group (ECOG) performance status ≤3. Patients with a history of cirrhosis could be eligible.
Additional entry criteria for patients with renal impairment included moderate or severe renal impairment (i.e. estimated creatinine clearance [CLcr] ≤50 ml min–1), stable hepatic and renal function prior to study entry as assessed by the investigator at the screening visit and ECOG performance status ≤2.
Study design and treatment
This phase 1 evaluation was conducted at six clinical sites in the United States. The protocol was reviewed and approved by the local Institutional Review Boards at Healthcare Corporation of America (HCA)‐HealthONE (Glendale, CO, USA), New York University Medical Center (New York, NY, USA), and Wayne State University (Detroit, MI, USA), and by the Western Institutional Review Board (Olympia, WA, USA). Written informed consent was obtained from all patients prior to any study‐specific procedures, in accordance with the Declaration of Helsinki. Data from patients with hepatic and renal impairment were collected at six clinical sites from January 2010 to May 2012.
The primary study objective was to assess the PK of brentuximab vedotin in patients with hepatic or renal impairment. Secondary objectives were to evaluate the safety, tolerability and immunogenicity of brentuximab vedotin in these patients. In addition, other treatment arms in this clinical pharmacology study (ClinicalTrials.gov NCT01026415) evaluated potential CYP3A‐mediated drug–drug interactions and determined the primary route of excretion for MMAE 10.
Patients received a dose of 1.2 mg kg–1 brentuximab vedotin rather than the typical starting dose (1.8 mg kg–1) to minimize potential adverse effects related to increased exposures in the event that hepatic or renal impairment affected PK. Patients were scheduled to receive two cycles of brentuximab vedotin 1.2 mg kg–1 intravenously (i.v.). Doses were given on day 1 of each 21 day cycle and infused over 30 min in an outpatient setting. Disease characteristics were assessed by the investigator at baseline and patients with clinical benefit who were free from unacceptable toxicity could receive extended treatment with brentuximab vedotin in a separate trial (ClinicalTrials.gov NCT00947856).
Baseline hepatic impairment was categorized using the Child–Pugh classification scale, originally developed for surgical evaluation of patients with alcoholic cirrhosis 11, 12. Patients were assessed as good operative risk (group A, mild), moderate operative risk (group B, moderate) or poor operative risk (group C, severe).
Baseline renal impairment was based on estimated CLcr as determined using the Cockroft–Gault formula 13 and categorized as mild (CLcr >50 to 80 ml min–1), moderate (CLcr 30 to 50 ml min–1) or severe (CLcr <30 ml min–1).
Study assessments
Brentuximab vedotin PK was evaluated over the first 21 days of the study only (i.e. cycle 1) and PK parameters were estimated using non‐compartmental methods. Systemic exposures were compared with those of unimpaired patients (n = 8) who received 1.2 mg kg–1 brentuximab vedotin i.v. in another arm of the study and had no hepatic or renal impairment (‘unimpaired group’). Ratios of geometric means (hepatic or renal impairment/patients without impairment) were calculated.
Performance status was assessed at baseline and at the end of treatment using the ECOG performance status scale, which ranges from 0 (normal activity level) to 5 (dead) 14. In this study, baseline ECOG status for patients with renal impairment could be 0, 1 (ambulatory but with symptoms) or 2 (up and about >50% of waking hours). Baseline ECOG status for patients with hepatic impairment could range from 0 to 3 (capable of limited self care and in bed >50% of the time).
Safety parameters (adverse events [AEs], routine laboratory tests and antitherapeutic antibody assessments) were evaluated over both treatment cycles. Adverse events were summarized using the Medical Dictionary for Regulatory Activities (MedDRA), Version 13.0. The last available data point prior to first dose (e.g. pre‐dose on day 1) was considered to be the baseline value for laboratory tests. Adverse events and laboratory results were graded using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 3.0.
No efficacy data were collected in this study.
Sample collection and bioanalytical methods
ADC, TAb and antitherapeutic antibodies were measured in serum and MMAE was measured in plasma, using sensitive and validated assays. The assay methods for all analytes have been described previously 10.
Briefly, serum samples for ADC and TAb analyses were collected predose, at the end of infusion, at 2, 4, and 36 h and at 3, 7, 14, 17 and 21 days post‐infusion. Concentrations were measured with enzyme‐linked immunosorbent assays in a sandwich format. The limits of quantitation were 12.5 to 400 ng ml–1 for both ADC and TAb.
Plasma samples for MMAE analysis were collected predose, at the end of infusion, at 2, 4, 8, 12, 24 and 36 h and at 2, 3, 4, 7, 10, 14, 17 and 21 days post‐infusion. Concentrations were measured by liquid chromatography and tandem mass spectrometry following solid‐phase extraction. The limits of quantitation for MMAE were 0.025 to 1 ng ml–1.
Serum samples for antitherapeutic antibody analysis were collected on day 1 of both treatment cycles prior to brentuximab vedotin administration and at the end‐of‐treatment visit. Antitherapeutic antibodies were detected with an electrochemiluminescence assay with a sensitivity of 4 ng ml–1 anti‐brentuximab vedotin monoclonal antibody and drug tolerance of 3125 ng ml–1 brentuximab vedotin. For screen positive samples, antitherapeutic antibody titre and specificity were evaluated and then the presence was confirmed in a competitive inhibition assay in which samples were pre‐exposed to saturating concentrations of brentuximab vedotin before assay.
Statistical analysis
There were no formal statistical hypotheses. All analyses were descriptive.
Brentuximab vedotin PK evaluations included ADC and MMAE. TAb also was assessed but is not reported here due to the similarity to ADC PK profiles. PK parameters were estimated by non‐compartmental analysis using WinNonlin 6.3 (Pharsight, Mountain View, CA, USA). The area under the concentration vs. time curve (AUC(0,∞)) was estimated as the sum of AUC(0,t last) (AUC to the last time point) and the quotient of the last measured concentration and λ (the terminal rate coefficient, estimated by regression of the terminal log‐linear portion of the concentration vs. time profile). The maximum observed concentration (C max) was determined by inspection of the concentration data. The effect of hepatic or renal impairment was evaluated by comparing AUC and C max during cycle 1 for evaluable patients with those of the unimpaired group. The ratios of the geometric means of AUC and C max were summarized for impaired hepatic or renal function/unimpaired function, with 90% confidence intervals (CIs).
Evaluable patients with hepatic or renal impairment were those who received brentuximab vedotin at the intended dose level at cycle 1 day 1, did not receive any effective inducers or strong inhibitors of CYP3A during the PK evaluation period and had adequate PK samples for estimation of the appropriate PK parameters.
For patients with hepatic impairment who had insufficient data to estimate AUC(0,∞) for both ADC and MMAE, single imputation was performed based on data from patients with sufficient data (t last ≥7.0 days) to estimate AUC using linear regression. The dependent variable was AUC and important covariates were selected from the following variables using stepwise selection: AUC(0,2.5 days), C max, baseline age, gender and baseline antitherapeutic antibody status. The stepwise selection was done in SAS 9.2 using procedure GLMSELECT with default model selection criterion (i.e.Schwarz Bayesian Information criterion) and additional options (i.e. intercept suppressed and AUC(0,2.5 days) forced to be included in the model). The fitted values with the final selected model were used as the imputed values for missing AUC values.
Results
Patients
A total of 17 patients with hepatic (n = 7) or renal (n = 10) impairment were treated with brentuximab vedotin. All 17 patients were evaluable for both safety and PK.
Table 1 presents baseline characteristics for the patients with hepatic and renal impairment, as well as for the unimpaired group of patients used as a comparator for PK evaluations. The majority of patients had Hodgkin's lymphoma. Two patients, one with hepatic and one with renal impairment, had systemic anaplastic large cell lymphoma. The median age was 55 years in patients with hepatic impairment, 56.5 years in patients with renal impairment and 33.5 years for the unimpaired group.
Table 1.
Patient baseline characteristics
| Hepatic impairment (n = 7) | Renal impairment (n = 10) | Unimpaired group (n = 8) | |
|---|---|---|---|
| Age, median (range), years | 55.0 (22–72) | 56.5 (29–78) | 33.5 (24–56) |
| Gender, n (%) | |||
| Male | 4 (57) | 5 (50) | 4 (50) |
| Female | 3 (43) | 5 (50) | 4 (50) |
| Weight, median (range), kg | 74.30 (43.3–101.7) | 84.30 (47.2–134.0) | 72.85 (46.7, 103.1) |
| ECOG status, n (%) * | |||
| 0 | 0 | 3 (30) | 5 (63) |
| 1 | 2 (29) | 5 (50) | 3 (38) |
| 2 | 1 (14) | 2 (20) | 0 |
| 3 | 4 (57) | 0 | 0 |
| Disease diagnosis, n (%) | |||
| Hodgkin lymphoma | 6 (86) | 9 (90) | 8 (100) |
| Anaplastic large cell lymphoma | 1 (14)† | 1 (10)‡ | 0 |
| Duration of disease, median (range), months § | 35.0 (4–70) | 55.0 (1–98) | 38.0 (15–146) |
| Prior therapies | |||
| Systemic chemotherapy, n (%) | 7 (100) | 9 (90) | 8 (100) |
| Median (range) number of regimens ¶ | 2.0 (1–5) | 5.0 (0–7) | 5.0 (2–13) |
| Radiotherapy, n (%) | 2 (29) | 4 (40) | 5 (63) |
| Prior haematopoietic stem cell transplant, n (%) | 2 (29) | 5 (50) | 8 (100) |
| Autologous only, n (%) ** | 1 (50) | 4 (80) | 7 (88) |
| Allogeneic only, n (%) ** | 0 | 0 | 1 (13) |
| Both autologous and allogeneic, n (%) ** | 1 (50) | 1 (20) | 0 |
| Child–Pugh score, n (%) †† | |||
| A (mild) | 1 (14) | – | – |
| B (moderate) | 5 (71) | – | – |
| C (severe) | 1 (14)‡‡ | – | – |
| Creatinine clearance, median (range), ml min–1 §§ | 108.5 (40–216) | 34.6 (20–59) | 123.9 (90–163) |
| Renal impairment category, n (%) | |||
| Unimpaired (CLcr >80 ml min–1) | 6 (86) | 0 | 8 (100) |
| Mild (CLcr >50 to ≤80 ml min–1) | 0 | 4 (40)¶¶ | 0 |
| Moderate (CLcr ≥30 to ≤50 ml min–1) | 1 (14) | 3 (30) | 0 |
| Severe(CLcr <30 ml min–1) | 0 | 3 (30) | 0 |
CLcr creatinine clearance; ECOG Eastern Cooperative Oncology Group.
The maximum ECOG status allowed was 3 for patients with hepatic impairment, 2 for patients with renal impairment and 1 for unimpaired patients.
Anaplastic lymphoma kinase (ALK) positive.
ALK negative.
Time from initial diagnosis to first dose of brentuximab vedotin.
Excludes stem cell mobilization therapy.
Number of patients (percent of patients with any prior stem cell transplant).
Calculated using bilirubin, albumin, and prothrombin time data from the clinical database (if available) and site assessments for encephalopathy, ascites and any unavailable labs.
Although patients with Child–Pugh assessment group C were to be excluded from study enrolment, an exception was granted to one patient.
Calculated from baseline serum creatinine, weight, age and gender using the Cockroft–Gault formula.
Four patients with renal impairment met the entry criterion of moderate or severe impairment at screening, but had estimated CLcr corresponding to mild impairment on day 1, prior to first dose of brentuximab vedotin.
All but one patient had previously received at least one regimen of systemic chemotherapy. The median number of prior systemic regimens was 2 (range 1–5) for patients with hepatic impairment, 5 (range 0–7) for patients with renal impairment and 5 (range 2–13) for the unimpaired group.
The eligibility criteria for the protocol required a baseline ECOG status ≤2 for patients with renal impairment, but allowed an ECOG status of up to 3 for patients with hepatic impairment. Four of seven patients with hepatic impairment had an ECOG status of 3 at baseline. In contrast, eight of 10 patients with renal impairment and all eight patients in the unimpaired group were generally ambulatory and able to perform normal activities without assistance, as indicated by a baseline ECOG performance status of 0 or 1.
At baseline, five of the seven patients in the hepatic impairment group had moderate impairment, one had mild impairment and one had severe impairment (an exception was granted for enrolment). For five of these seven patients, the underlying lymphoma was noted to have contributed to hepatic dysfunction.
Among the 10 patients with renal impairment, three had moderate impairment and three had severe impairment. The other four patients had mild renal impairment at baseline, defined as the closest measurement prior to first dose of brentuximab vedotin. Although these four patients met the entry criterion of moderate or severe renal impairment at the screening visit, all four had an estimated CLcr corresponding to mild impairment on day 1, prior to the first dose of brentuximab vedotin.
Pharmacokinetics
In the unimpaired group, the geometric means (%CV) for the ADC were 51.39 (19) μg ml–1 day and 23.39 (28) μg ml–1 for AUC(0,∞) and C max, respectively. For MMAE, the geometric means (%CV) were 27.23 (80) ng ml–1 day and 4.05 (75) ng ml–1 for AUC(0,∞) and C max, respectively.
Effect of hepatic impairment
All seven patients with hepatic impairment were evaluable for PK (Table 2). Imputation was performed for two of these patients who had insufficient data to estimate AUC(0,∞). Based on the stepwise model selection, the final linear regression for imputation of MMAE AUC(0,∞) included AUC(0,2.5 days) as the only covariate. For ADC, AUC(0,2.5 days), C max, age, gender and baseline antitherapeutic antibody status were included as covariates for the final linear regression model. Exploratory analyses were performed to determine whether exclusion of patients with partial or imputed PK parameters from analysis would affect the ratios of geometric means for brentuximab vedotin ADC and MMAE AUC. The results of these analyses conducted on patients with full PK profiles only were consistent with those conducted on the PK analysis set.
Table 2.
Ratio of geometric means by degree of hepatic impairment
| Degree of hepatic impairment | |||||
|---|---|---|---|---|---|
| Unimpaired (n = 8) | Mild (n = 1) | Moderate (n = 5) | Severe (n = 1) | Any (n = 7) | |
| ADC | |||||
| AUC(0,∞) (μg ml–1 day) | |||||
| GM (%CV) | 51.39 (19) | 29.26 (‐) | 33.47 (61) | 46.62 (‐) | 34.43 (51) |
| Ratio of GMs * (90% CI) | – | 0.57 (0.39, 0.84) | 0.65 (0.45, 0.95) | 0.91 (0.62, 1.33) | 0.67 (0.48, 0.93) |
| Cmax (μg ml–1) | |||||
| GM (%CV) | 23.39 (28) | 20.70 (‐) | 18.93 (19) | 18.70 (‐) | 19.14 (16) |
| Ratio of GMs * (90% CI) | – | 0.88 (0.51, 1.55) | 0.81 (0.63, 1.05) | 0.80 (0.46, 1.40) | 0.82 (0.66, 1.01) |
| MMAE | |||||
| AUC(0,∞) (ng ml–1 day) | |||||
| GM (%CV) | 27.23 (80) | 95.49 (‐) | 60.30 (71) | 48.08 (‐) | 62.34 (61) |
| Ratio of GMs * (90% CI) | – | 3.51 (0.86, 14.35) | 2.21 (1.11, 4.44) | 1.77 (0.43, 7.22) | 2.29 (1.27, 4.12) |
| Cmax (ng ml–1) | |||||
| GM (%CV) | 4.05 (75) | 11.30 (‐) | 6.59 (54) | 4.92 (‐) | 6.83 (51) |
| Ratio of GMs * (90% CI) | – | 2.79 (0.73, 10.69) | 1.63 (0.87, 3.05) | 1.21 (0.32, 4.66) | 1.68 (0.98, 2.89) |
ADC antibody–drug conjugate; CI confidence interval; %CV coefficient of variation as a percentage of the GM or ratio of GMs; GM geometric mean; MMAE monomethyl auristatin E.
Ratio of GMs for impaired hepatic function/unimpaired hepatic function.
When ADC exposures were assessed for all seven evaluable patients combined, the ratios of geometric means (90% CI) were 0.67 (0.48, 0.93) for AUC(0,∞) and 0.82 (0.66, 1.01) for C max. Similarly, when ADC exposures were evaluated only for patients with moderate hepatic impairment (n = 5 patients), the ratios of geometric means (90% CI) were 0.65 (0.45, 0.95) for AUC(0,∞) and 0.81 (0.63, 1.05) for C max. Thus, a decrease in ADC exposures was observed in the patients with hepatic impairment (Figure 1A).
Figure 1.
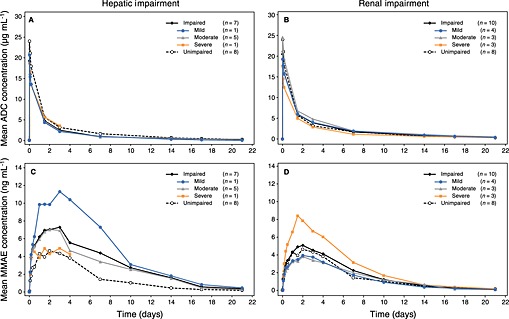
Mean serum ADC concentration–time profiles by degree of (A) hepatic impairment and (B) renal impairment. Mean plasma MMAE concentration–time profiles by degree of (C) hepatic impairment and (D) renal impairment. ◆ impaired patients;  mild impairment;
mild impairment;  moderate impairment;
moderate impairment;  severe impairment; ○ unimpaired patients
severe impairment; ○ unimpaired patients
When MMAE exposures were evaluated in all patients with hepatic impairment, the ratios of geometric means (90% CI) were 2.29 (1.27, 4.12) for AUC(0,∞) and 1.68 (0.98, 2.89) for C max. When MMAE exposures were evaluated only for the patients with moderate impairment (n = 5) the ratios of geometric means (90% CI) were 2.21 (1.11, 4.44) for AUC(0,∞) and 1.63 (0.87, 3.05) for C max. Thus, MMAE exposures increased approximately 2.3 fold (Figure 1C).
Effect of renal impairment
All 10 patients with renal impairment had full PK profiles and were evaluable for PK (Table 3).
Table 3.
Ratio of geometric means by degree of renal impairment
| Degree of renal impairment | |||||
|---|---|---|---|---|---|
| Unimpaired (n = 8) | Mild (n = 4)* | Moderate (n = 3) | Severe (n = 3) | Any (n = 10)* | |
| ADC | |||||
| AUC(0,∞) (μg ml–1day) | |||||
| GM (%CV) | 51.39 (19) | 44.66 (45) | 62.55 (21) | 36.57 (31) | 46.75 (38) |
| Ratio of GMs † (90% CI) | – | 0.87 (0.63, 1.21) | 1.22 (0.96, 1.55) | 0.71 (0.54, 0.94) | 0.91 (0.70, 1.17) |
| Cmax (μg ml–1) | |||||
| GM (%CV) | 23.39 (28) | 18.78 (28) | 24.44 (12) | 17.37 (20) | 19.98 (24) |
| Ratio of GMs † (90% CI) | – | 0.80 (0.57, 1.13) | 1.04 (0.76, 1.43) | 0.74 (0.54, 1.03) | 0.85 (0.69, 1.06) |
| MMAE | |||||
| AUC(0,∞) (ng ml–1 day) | |||||
| GM (%CV) | 27.23 (80) | 23.10 (79) | 29.61 (41) | 51.65 (39) | 31.68 (65) |
| Ratio of GMs † (90% CI) | – | 0.85 (0.39, 1.84) | 1.09 (0.49, 2.42) | 1.90 (0.85, 4.21) | 1.16 (0.68, 1.98) |
| Cmax (ng ml–1) | |||||
| GM (%CV) | 4.05 (75) | 3.15 (95) | 3.73 (28) | 8.38 (17) | 4.44 (74) |
| Ratio of GMs † (90% CI) | – | 0.78 (0.35, 1.71) | 0.92 (0.44, 1.95) | 2.07 (0.99, 4.33) | 1.10 (0.63, 1.90) |
ADC antibody–drug conjugate; CI confidence interval; %CV coefficient of variation as a percentage of the GM or ratio of GMs; GM geometric mean; MMAE monomethyl auristatin E.
One patient with mild impairment was not evaluable for ADC exposure. Thus, for ADC, n = 3 for mild impairment and n = 9 for any impairment.
Ratio of GMs for impaired renal function/unimpaired renal function.
When ADC exposures were assessed for all 10 evaluable patients combined, the ratios of geometric means (90% CI) were 0.91 (0.70, 1.17) for AUC(0,∞) and 0.85 (0.69, 1.06) for C max. For patients with mild or moderate impairment (n = 6), the ratios of geometric means for ADC ranged from 0.80 to 1.22 for both AUC(0,∞) and C max. The 90% CIs are provided in Table 3. For the patients with severe renal impairment (n = 3), the ratios of geometric means (90% CI) for ADC were 0.71 (0.54, 0.94) for AUC(0,∞) and 0.74 (0.54, 1.03) for C max. Thus, mild or moderate renal impairment caused no apparent change in ADC exposure and a decrease in ADC exposures was observed only in patients with severe renal impairment (Figure 1B).
When MMAE exposures were assessed for all 10 evaluable patients combined, the ratios of geometric means (90% CI) were 1.16 (0.68, 1.98) for AUC(0,∞) and 1.10 (0.63, 1.90) for C max. For patients with mild or moderate impairment (n = 7), the ratios of geometric means for MMAE ranged from 0.78 to 1.09 for both AUC(0,∞) and C max. The 90% CIs are provided in Table 3. For the three patients with severe renal impairment, the ratios of geometric means (90% CI) for MMAE were 1.90 (0.85, 4.21) for AUC(0,∞) and 2.07 (0.99, 4.33) for C max. Thus, MMAE exposures were not altered by mild or moderate renal impairment, but increased approximately 1.9 fold in patients with severe renal impairment (CLcr <30 ml min–1; Figure 1D).
Safety
This study was not designed to evaluate differences between treatment arms, and differences in baseline factors, such as age and ECOG performance status, confounded some of the comparisons. However, some apparent differences in safety observations were noted. An overview of adverse events is provided in Table 4. Relative to the AE profile of the unimpaired group, individual AE outcomes appeared generally less favourable in the patients with hepatic or renal impairment, although no consistent pattern of specific AEs was evident. Additionally, patients in this study who had hepatic impairment tended to have worse ECOG performance status at baseline and worse individual AE outcomes than those with renal impairment. These AE observations may be due, at least in part, to differences in the maximum baseline ECOG status allowed per protocol (ECOG 3 for patients with hepatic impairment, ECOG 2 for patients with renal impairment and ECOG 1 for unimpaired patients).
Table 4.
Overview of treatment‐emergent adverse events
| Hepatic impairment (n = 7) | Renal impairment (n = 10) | Unimpaired group (n = 8) | |
|---|---|---|---|
| Patients with any AE, n (%) | 7 (100) | 9 (90) | 8 (100) |
| Brentuximab vedotin‐related AE, n (%) * | 4 (57) | 7 (70) | 7 (88) |
| Maximum severity of AE, n (%) | |||
| Grade 1 | 0 | 3 (30) | 4 (50) |
| Grade 2 | 1 (14) | 0 | 1 (13) |
| Grade 3 | 0 | 2 (20) | 3 (38) |
| Grade 4 | 3 (43)† | 3 (30)‡ | 0 |
| Grade 5 | 3 (43)§ | 1 (10)¶ | 0 |
| ≥ Grade 3 | 6 (86) | 6 (60) | 3 (38) |
| Patients with serious AE (SAE), n (%) ** | 6 (86) | 4 (40) | 1 (13) |
| Brentuximab vedotin‐related SAE, n (%) ** | 2 (29) | 1 (10) | 1 (13) |
| Discontinued treatment due to AE, n (%) | 4 (57)†† | 0 | 0 |
| Median number of unique preferred terms per patient ‡‡ | 14 | 3 | 3.5 |
AE adverse event; SAE serious adverse event.
Related to brentuximab vedotin at any time during the safety reporting period, as assessed by the investigator.
Unrelated hyperbilirubinaemia and related neutropenia, unrelated thrombocytopenia, unrelated hypokalaemia and unrelated tumour pain [SAE].
Unrelated aspiration [SAE] and unrelated hypoxia, unrelated hyperuricaemia, related hypokalaemia.
Three patients died on study after receiving one dose of brentuximab vedotin (unrelated brain herniation, unrelated fungal lower respiratory tract infection, unrelated cryptococcal fungaemia that was later determined to have been present at baseline). A fourth patient with a maximum grade 4 AE while on study died of progressive Hodgkin's lymphoma after withdrawing consent but within 3 weeks of the first dose of brentuximab vedotin.
One patient with a history of coronary artery disease, class II congestive heart failure and an abnormal ejection fraction died of unrelated myocardial infarction.
All events, from time of informed consent to the end of the safety reporting period.
Three patients had grade 5 events resulting in death and one patient discontinued treatment due to a grade 3 SAE of steatohepatitis.
All patients in the safety population were included (patients without AEs were considered to have 0 events).
Sixteen of the 17 patients with hepatic or renal impairment experienced at least one treatment‐emergent AE (Table 5). The most commonly reported AEs for these 17 patients were alopecia and diarrhoea (five patients overall, 29% each), and the most common event ≥ grade 3 was thrombocytopenia (four patients overall, 24%). A single treatment‐emergent peripheral neuropathy event (unrelated grade 1 hypoaesthesia) occurred during this two cycle study. No safety analyses of specific AEs were conducted for the unimpaired group, as their demographic characteristics were not well matched with those of the patients with hepatic or renal impairment.
Table 5.
Treatment‐emergent adverse events occurring in ≥20% of patients in either treatment arm
| Hepatic impairment (n = 7) n (%) | Renal impairment (n = 10) n (%) | |
|---|---|---|
| Any event | 7 (100) | 9 (90) |
| Preferred term | ||
| Alopecia | 1 (14) | 4 (40) |
| Diarrhoea | 3 (43) | 2 (20) |
| Dyspnoea | 4 (57) | 0 |
| Fatigue | 3 (43) | 1 (10) |
| Hypokalaemia | 2 (29) | 2 (20) |
| Thrombocytopenia | 3 (43) | 1 (10) |
| Anaemia | 2 (29) | 1 (10) |
| Constipation | 2 (29) | 1 (10) |
| Cough | 3 (43) | 0 |
| Oral candidiasis | 3 (43) | 0 |
| Acidosis | 2 (29) | 0 |
| Arthralgia | 0 | 2 (20) |
| Flank pain | 0 | 2 (20) |
| Headache | 0 | 2 (20) |
| Herpes zoster | 0 | 2 (20) |
| Oropharyngeal pain | 2 (29) | 0 |
| Pleural effusion | 2 (29) | 0 |
| Pyrexia | 2 (29) | 0 |
| Tachycardia | 2 (29) | 0 |
| Weight decreased | 2 (29) | 0 |
| Weight increased | 0 | 2 (20) |
Haematology and blood chemistry values ≥ grade 3 were common, both prior to the first dose and during the study (Table 6). The titres of all confirmed positive antitherapeutic antibody test results in this study were ≤125. Due to the short duration of the study, the transience or persistence of the antitherapeutic antibodies could not be assessed.
Table 6.
Haematology and blood chemistry test results ≥ grade 3
| Hepatic impairment (n = 7) n (%) | Renal impairment (n = 10) n (%) | |
|---|---|---|
| Haematology | ||
| Prior to first dose | 5 (71) | 2 (20) |
| Treatment emergent | 6 (86) | 4 (40) |
| Blood chemistry | ||
| Prior to first dose | 7 (100) | 3 (30) |
| Treatment emergent | 4 (57) | 2 (20) |
An extensive medical review of by‐patient listings was conducted in an attempt to identify potential contributing factors to the poor individual AE outcomes observed in this study.
Among the seven patients with hepatic impairment, four patients died after receiving one dose of brentuximab vedotin (Table 4). All four of these patients had a Child–Pugh score of B or C (indicating moderate or severe disease), a baseline ECOG status of 3 and substantial comorbidities at baseline. No associations with other factors were evident. In contrast, two other patients with hepatic impairment, a baseline ECOG status of 1 or 2 and no significant baseline comorbidities tolerated brentuximab vedotin therapy and completed the study. One subsequently enrolled in a separate trial to receive additional treatment cycles.
Among the 10 patients with renal impairment, one patient died and three others had grade 4 treatment‐emergent AEs (Table 4). No clear contributing factors were identified, but all four of these patients had grade 2 or grade 3 renal failure and substantial comorbidities at baseline. All four had a baseline ECOG status of 1 or 2 and no associations with other factors were evident. Nine of the 10 patients with renal impairment completed the study and subsequently enrolled in a separate trial for further brentuximab vedotin therapy.
Among all 17 patients, the poor individual AE outcomes did not appear to be associated with patient age or gender. None of the patients with poor outcomes had a prior allogeneic stem cell transplant. For patients with hepatic impairment, no conclusions could be drawn regarding poor outcomes and baseline hepatic impairment severity because five of seven patients had a Child–Pugh score of B. For patients with renal impairment, the poor outcomes did not appear to be associated with the degree of baseline renal impairment. No conclusions could be drawn regarding diagnosis (all but two patients had Hodgkin's lymphoma). Although the study was not powered to detect any relationship between brentuximab vedotin exposures and individual AE outcomes, no apparent trends in ADC or MMAE exposures were noted among the subset of patients with poor AE outcomes.
Discussion
This study assessed the PK of brentuximab vedotin in patients with both advanced haematologic malignancies as well as hepatic or renal impairment. A total of 17 patients, seven with hepatic impairment and 10 with renal impairment, were enrolled from the relatively small pool of potential study participants. To minimize potential increased exposures in the event that hepatic or renal impairment affected PK, patients received a dose of 1.2 mg kg–1 brentuximab vedotin rather than the typical starting dose (1.8 mg kg–1) every 3 weeks. Exposures were compared with those of unimpaired patients (n = 8) who received 1.2 mg kg–1 brentuximab vedotin i.v. in another arm of the study.
For patients with hepatic impairment, a decrease in ADC exposures was observed and MMAE exposures increased approximately 2.3 fold. Hepatic impairment is associated with low serum concentrations of albumin, a component of the Child–Pugh score. Clearance of other biologics, such as trastuzumab, ado‐trastuzumab emtansine and infliximab, has been shown to be inversely correlated with albumin concentrations using population pharmacokinetics 15, 16, 17. For the patients with hepatic impairment enrolled in this study, baseline albumin concentrations were lower than in the unimpaired group (mean of 2.6 mg dl–1 vs. 3.8 mg dl–1). Thus, hypoalbuminaemia may be associated with increased clearance of brentuximab vedotin although the potential mechanism(s) is not clear. For MMAE, the observed increase in exposure may be related to increased ADC catabolism and drug release, decreased hepatic excretion or a combination of these factors. The liver is the primary organ for MMAE metabolism and excretion 10.
Mild or moderate renal impairment caused no apparent change in ADC and MMAE exposure. However, for patients with severe renal impairment (CLcr <30 ml min–1), a decrease in ADC exposures was observed and MMAE exposures increased approximately 1.9 fold. It is possible that ADC was cleared more rapidly in patients with severe renal impairment due to proteinuria. This cannot be determined as urine protein concentrations were not measured in this study. Of note, the three patients with severe renal impairment did not have significant hypoalbuminaemia (2.7, 3.6 and 3.9 g dl–1). The observed increase in MMAE exposure in patients with severe renal impairment was greater than anticipated based on clinical excretion evaluations and preclinical data. However, severe renal impairment has been associated with changes in hepatic drug metabolism, plasma protein binding, transport and tissue distribution with other compounds 18, factors that may have contributed to decreased MMAE clearance.
This study was not designed to evaluate differences between treatment arms. However, individual AE outcomes appeared generally less favourable in the patients with hepatic or renal impairment, relative to those of the unimpaired group, although no consistent pattern of specific AEs was evident. This observation may be attributable, at least in part, to baseline comorbidities in the patients with hepatic or renal impairment evaluated in this study. Additionally, the patients with a baseline ECOG status of 3, all of whom had hepatic impairment, tended to have worse individual AE outcomes than those with baseline ECOG status ≤2 regardless of the degree of organ impairment. With the exception of baseline comorbidities in the patients with hepatic or renal impairment and ECOG status in the patients with hepatic impairment, the poor individual AE outcomes observed in this study did not appear to be associated with the other parameters evaluated, such as age, gender and degree of impairment.
Analysis of the safety data was confounded by several factors, including the inherent difficulty of assessing safety in small numbers of patients, the short dosing and observation periods and the patients' poor baseline condition. Five patients, four of whom had a baseline ECOG status of 3, died due to adverse events considered unrelated to brentuximab vedotin. Among patients with baseline ECOG status ≤2, however, two of three patients with hepatic impairment and nine of 10 patients with renal impairment in this study tolerated brentuximab vedotin therapy.
Determination of whether the ADC or unconjugated MMAE may be responsible for AEs is challenging because ADC and MMAE exposures are correlated. However, no apparent trends in ADC or MMAE exposures were noted among individual patients with poor AE outcomes, suggesting that the poor outcomes may be more dependent on baseline factors or comorbidities than on differences in exposure.
After the i.v. administration of 1.2 mg kg–1 of brentuximab vedotin, MMAE exposures were not altered by mild or moderate renal impairment but were increased 2.3 fold in patients with hepatic impairment and 1.9 fold in patients with severe renal impairment (CLcr <30 ml min–1). Based on the results of this study, dose modifications may be considered for some patients with hepatic or renal impairment. Recommendations may be found in the approved local product labels 19, 20, 21.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare direct funding for this research was issued by Seattle Genetics, Inc. through the joint financial support of Seattle Genetics, Inc. and Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceuticals Limited. Seattle Genetics, Inc. provided research funding to the institutions of RC, OAO, AKG, RR, AG and JVM. RC and RR have been on the speakers' bureau for Seattle Genetics, Inc. AKG has been a consultant to Gilead and Pfizer; additional research funding has been provided by Biomarin, Bristol‐Myers Squibb, Gilead, Janssen, Merck, Pfizer, Spectrum, and Teva. AG has been on the speakers' bureau and has been an advisory board member for Celgene, Pharmacyclics/JNJ and Takeda Pharmaceuticals International Co.; Celgene and Pharmacyclics/JNJ also provided research funding to the institution. AAF is employed by and has equity ownership in Takeda Pharmaceuticals International Co. TJM and BZ are employed by and have equity ownership in Seattle Genetics, Inc. THH was employed by Seattle Genetics, Inc. at the time the work was performed and has equity ownership in Seattle Genetics, Inc.
Contributors
RC was the principal investigator responsible for review of the final clinical study report. RC, OAO, AKG, RR, AG and JVM were principal investigators at the clinical study sites and participated in the conduct of the clinical trial, including patient care and data collection. TJM was the sponsor's medical monitor, responsible for the medical and safety aspects of the clinical study report. BZ, AAF and THH were the clinical pharmacologists responsible for the study design, PK data analysis and interpretation, and writing the study report and other associated documents. All authors have reviewed and approved the manuscript. Laurie Grove contributed to study design and conduct, Yinghui Wang provided statistical guidance and Roberta Connelly provided medical writing assistance, all under the sponsorship of Seattle Genetics, Inc.
Zhao, B. , Chen, R. , O'Connor, O. A. , Gopal, A. K. , Ramchandren, R. , Goy, A. , Matous, J. V. , Fasanmade, A. A. , Manley, T. J. , and Han, T. H. (2016) Brentuximab vedotin, an antibody–drug conjugate, in patients with CD30‐positive haematologic malignancies and hepatic or renal impairment. Br J Clin Pharmacol, 82: 696–705. doi: 10.1111/bcp.12988.
References
- 1. Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003; 102: 1458–65. [DOI] [PubMed] [Google Scholar]
- 2. Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, et al. Intracellular activation of SGN‐35, a potent anti‐CD30 antibody‐drug conjugate. Clin Cancer Res 2010; 16: 888–97. [DOI] [PubMed] [Google Scholar]
- 3. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Brentuximab vedotin (SGN‐35) in patients with relapsed or refractory systemic anaplastic large cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30: 2190–6. [DOI] [PubMed] [Google Scholar]
- 4. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol 2012; 30: 2183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, et al. Brentuximab vedotin (SGN‐35) for relapsed CD30‐positive lymphomas. N Engl J Med 2010; 363: 1812–21. [DOI] [PubMed] [Google Scholar]
- 6. Fanale MA, Forero‐Torres A, Rosenblatt JD, Advani RH, Franklin AR, Kennedy DA, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30‐positive hematologic malignancies. Clin Cancer Res 2012; 18: 248–55. [DOI] [PubMed] [Google Scholar]
- 7. Han TH, Kennedy D, Hayes S, Lynch CM. The pharmacokinetics of brentuximab vedotin (SGN‐35), an antibody‐drug conjugate (ADC) [abstract]. Clin Pharmacol Ther 2012; 91 (suppl 1): PII–1. [Google Scholar]
- 8. Han TH, Kennedy DA, Hayes S, Lynch CM. The pharmacokinetics of brentuximab vedotin (SGN‐35), an antibody‐drug conjugate (ADC). Poster session presented at: 113th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics; 2012. March 14‐17; National Harbor, MD, USA.
- 9. Han TH, Zhao B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody‐drug conjugates. Drug Metab Dispos 2014; 42: 1914–20. [DOI] [PubMed] [Google Scholar]
- 10. Han TH, Gopal AK, Ramchandren R, Goy A, Chen R, Matous JV, et al. CYP3A‐mediated drug‐drug interaction potential and excretion of brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive hematologic malignancies. J Clin Pharmacol 2013; 53: 866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973; 60: 646–9. [DOI] [PubMed] [Google Scholar]
- 12. FDA Guidance for Industry . Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling; 2003.
- 13. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31–41. [DOI] [PubMed] [Google Scholar]
- 14. Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982; 5: 649–55. [PubMed] [Google Scholar]
- 15. Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol 2009; 65: 1211–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta M, Lorusso PM, Wang B, Yi JH, Burris HA 3rd, Beeram M, et al. Clinical implications of pathophysiological and demographic covariates on the population pharmacokinetics of trastuzumab emtansine, a HER2‐targeted antibody‐drug conjugate, in patients with HER2‐positive metastatic breast cancer. J Clin Pharmacol 2012; 52: 691–703. [DOI] [PubMed] [Google Scholar]
- 17. Cosson VF, Ng VW, Lehle M, Lum BL. Population pharmacokinetics and exposure‐response analyses of trastuzumab in patients with advanced gastric or gastroesophageal junction cancer. Cancer Chemother Pharmacol 2014; 73: 737–47. [DOI] [PubMed] [Google Scholar]
- 18. FDA Guidance for Industry . Pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling; 2010.
- 19. ADCETRIS . Canadian product monograph. Bothell, WA, US: Seattle Genetics, Inc., 2016. [Google Scholar]
- 20. ADCETRIS . EU summary of product characteristics. Taastrup, Denmark: Takeda Pharma A/S, 2015. [Google Scholar]
- 21. ADCETRIS . US package insert. Bothell, WA, US: Seattle Genetics, Inc., 2016. [Google Scholar]