

Abstract
The accuracy of human leukocyte antigen (HLA)‐matching algorithms is a prerequisite for the correct and efficient identification of optimal unrelated donors for patients requiring hematopoietic stem cell transplantation. The goal of this World Marrow Donor Association study was to validate established matching algorithms from different international donor registries by challenging them with simulated input data and subsequently comparing the output. This experiment addressed three specific aspects of HLA matching using different data sets for tasks of increasing complexity. The first two tasks targeted the traditional matching approach identifying discrepancies between patient and donor HLA genotypes by counting antigen and allele differences. Contemporary matching procedures predicting the probability for HLA identity using haplotype frequencies were addressed by the third task. In each task, the identified disparities between the results of the participating computer programs were analyzed, classified and quantified. This study led to a deep understanding of the algorithms participating and finally produced virtually identical results. The unresolved discrepancies total to less than 1%, 4% and 2% for the three tasks and are mostly because of individual decisions in the design of the programs. Based on these findings, reference results for the three input data sets were compiled that can be used to validate future matching algorithms and thus improve the quality of the global donor search process.
Keywords: donor registry, hematopoietic stem cell transplantation, human leukocyte antigen, immunogenetics, matching algorithm, patient–donor matching
Introduction
Human leukocyte antigen (HLA) matching between patient and donor is the pivotal factor for the success of an allogeneic hematopoietic stem cell (HSC) transplantation 1, 2, 3. Therefore, the rapid and reliable identification of suitable adult volunteer HSC donor candidates and/or cord blood units for individual patients is of primary importance. This challenging search process is routinely performed in a donor registry or cord blood bank by a computer program in which the HLA‐matching algorithm (HMA) can be regarded as the core element 4. For simplicity, we will use the term ‘donor’ to refer to donors of HSCs from bone marrow or peripheral blood and cord blood units and the term ‘donor registry’ shall include cord blood banks.
Historically, the HMAs were developed independently within each donor registry as a part of a highly specialized individual IT infrastructure 5. Subsequently, HMAs have been developed further to encompass the evolution of the serological and molecular HLA nomenclature and also because of the growing scientific understanding of the clinical matching requirements 6, 7, 8, 9. In particular, the HLA typing resolution required, the number of HLA loci to be considered and the number of donors typed incompletely or at insufficient resolution have promoted a heterogeneous range of matching philosophies. The introduction of probabilistic concepts into the algorithms for predicting high‐resolution HLA allele‐level matching established the current state of the art in patient–donor matching 10, 11, 12, 13.
International cooperation plays an essential role in the HSC donor search process. In 2014, 49% or 8112 of the 16,655 globally shipped bone marrow or peripheral blood stem cells products were imported 14. In this HSC exchange Bone Marrow Donors Worldwide (BMDW) allows for searching of the global repository of currently more than 26 million HSC donors using its own matching procedure 15. In addition, the European Marrow Donor Information System (EMDIS) is of major importance as its 35 member registries provide electronic access by their individual HMAs to 90% of the globally available HSC sources 16.
From the perspective of search coordinators, all individual HMA implementations should present a comparable picture of the donor situation for their patients. However, the long evolution of HMAs has not led to a convergence of their behavior, but instead an increasing number of differences between their algorithms produce diverse results, which can complicate the donor search process.
For this reason, the Matching Validation Subcommittee of the IT working group of the World Marrow Donor Association (WMDA) initiated two projects for the global validation and standardization of HMAs to improve the quality and consistency in global donor searches 17. The first was to define a formal specification of HLA matching using accurate terminology 18. The second effort presented here involves the comparison and cross‐validation of matching algorithms by processing a panel of reference data sets and comparing the results obtained from the different registries' algorithms. This approach had the potential to highlight so far uncharacterized differences between algorithms that cannot be detected based on a high‐level description of their functional components and their underlying assumptions. The relevance of this unique comparative study is underlined by the fact that the largest HLA‐matching providers worldwide have participated (see affiliations).
Material and methods
The Matching Validation Subcommittee defined three matching validation tasks (MVTs). For each task, a new matching validation data set (MVS) comprising the HLA typings of 1000 patients and 10,000 donors was created. The 10 million possible patient–donor pairs of these three MVSs had to be used by the participants as input data for their own HMA. The complexity of the computational tasks with regard to algorithmic requirements and considered data increased from MVT 1 to 3.
All materials and results necessary to run the experiments for newly developed or modified algorithms and to analyze and validate match results are made available for download as part of the Supporting Information. In the following, references to tables and figures in the Supporting Information are denoted with ‘S’.
Simulation of ambiguous HLA genotypes
The genotypes of the patients and the donors for the three tasks of this study were all separately generated by independently drawing two haplotypes from the set of high‐resolution US Caucasoid haplotypes with a probability corresponding to their frequencies given in Maiers et al. 19. For the purpose of having patient and donor genotypes at varying levels of typing resolution, the HLA used in the tasks corresponds to what the known HLA type would have been if those individuals had been typed using representative methods and typing kits. The ambiguities thus introduced are based on IPD‐IMGT/HLA Database 20 releases v2.16.0 for MVS 1 and MVS 2 and v3.4.0 for MVS 3. Patient and donor HLA genotypes are encoded using genotype list (GL) string syntax 21 and multiple allele codes are used to reflect typing ambiguities 22. Identical amino acid sequences in the antigen recognition domain (ARD) were grouped together. The ARD groups have been defined on the basis of the two fields ‘g’‐groups specifically introduced in the Common and Well‐Documented (CWD) alleles catalog 23. The distribution of ambiguously or incompletely typed donors shapes a typical registry profile. Patients are usually typed at high‐resolution upfront. However, in order to challenge the algorithms, a substantial amount of ambiguity has been introduced into the patient HLA assignments.
Matching validation task 1 (mismatch counting)
The goal of the first MVT was to compare the identification of definitive mismatches in ambiguous molecular HLA assignments. MVS 1 was generated based on 3‐locus HLA‐A∼B∼DRB1 haplotypes. Of the donor genotypes, 15% contained only allele assignments or alleles within a single ARD group, 24% had a combination of allele assignments and multiple allele codes and 61% contained first‐field HLA typing results coded as XX‐codes 24. Among the patient genotypes, the corresponding distribution was 49%, 39% and 12%. All donors and patients were typed for all three loci HLA‐A, ‐B and ‐DRB1 (see Tables S1 and S2 for more details).
The task was to report for each patient–donor pair the total number of differences at each HLA locus. No distinction was to be made between antigen and allele mismatches and linkage disequilibrium was not to be considered.
Matching validation task 2 (mismatch grading)
The major refinement of the second task was the requirement to discriminate between mismatches at the antigen (serologic) level and those at the allele level in the counting. The distribution of the HLA assignments was similar to MVS 1 but, as a further complication, in MVS 2 serological assignments instead of XX‐codes were used (see Table S1 for more details). The task was to report for each patient–donor pair at each HLA locus the total number of differences and the number of antigen differences. Again, linkage disequilibrium was not to be considered.
Matching validation task 3 (matching probability)
In addition to the assignment of matching classifications required in tasks 1 and 2, the third task required the calculation of allele‐level match probabilities from ambiguously typed donor and patient HLA genotypes. Implementing such an HMA was a double challenge, first by virtue of the complexity of the task itself and second because of the requirement for computational runtime efficiency as the number of possible high‐resolution haplotype pairs (diplotypes) can be extremely high for incomplete or insufficiently resolved typing data.
MVS 3 was generated based on the above mentioned US Caucasoid 5‐locus HLA‐A∼C∼B∼DRB1∼DQB1 haplotypes using a mixture of HLA typing methods that are typically found in registries with many years of accumulated HLA typings. Here, 6% of the donor genotypes contained only allele or ARD group assignments, 33% had a combination of allele assignments and multiple allele codes and 61% contained only XX‐codes. Among the patient genotypes the corresponding distribution was 71%, 15% and 14%. All patients were typed for all five HLA loci, however, typing for the loci HLA‐C, ‐DRB1 and ‐DQB1 was removed for 80%, 10% and 90% of the donors, respectively, to more closely resemble the practical situation occurring in most donor registries (see Table S1). By design, the genotypes of all individuals were explainable by the haplotypes contained in the frequency table provided.
On the basis of the same 5‐locus haplotype frequencies used for the simulation of the patient and donor population for each patient–donor pair the overall 9/10 and 10/10 matching probability as well as the locus‐specific 2/2 matching predictions each accompanied by a match grade character had to be computed.
General requirements
The resulting file for each MVT had to contain the specific data items for all 107 possible patient–donor pairs in a specific format (see Figure 1). The early stages of the analysis of MVT 1 and 2 showed that very strict and precise specification of the task was necessary to get comparable files. For this purpose, we defined that alleles within the same ARD group had to be considered an allele match. The counting of mismatches for HLA molecular assignments had to be implemented according to the #Max column of Table S3, which was derived from the WMDA HLA matching framework 18.
Figure 1.
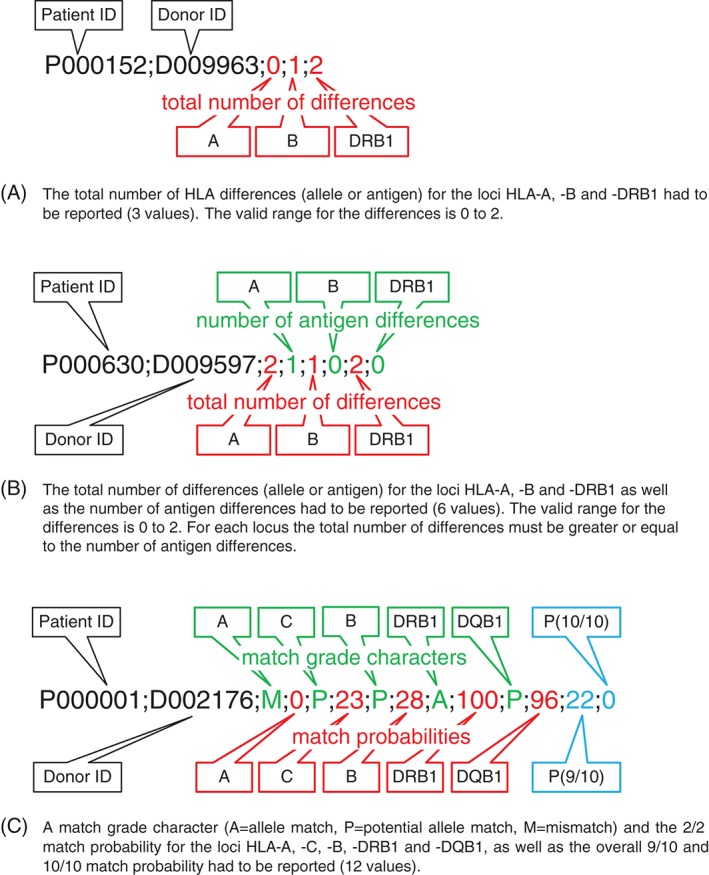
Result file formats for matching validation task (MVT) 1−3 (A to C). A single line had to be reported for each of the 107 possible patient−donor pairs using semicolon as field separator.
For MVT 2 a common mapping of alleles to antigens and vice versa was important for obtaining comparable results. For this purpose, the DNA‐to‐serology mappings defined in the WMDA file rel_dna_ser.txt 25 according to IPD‐IMGT/HLA Database release v3.6.0 have been used. To ensure consistency with World Health Organization (WHO)‐assigned antigen mappings it was decided to disregard the expert‐assigned values of the WMDA mapping file in this experiment.
The differences observed in a first round of analysis for MVT 3 showed the need for an even stricter rule set to achieve comparable results. In summary, the requirements to achieve a ‘baseline’ MVT 3 result are:
-
1
R1 ARD level matching has to be used.
-
2
R2 Mismatches have to be counted according to Table S3.
-
3
R3 WHO HLA nomenclature with WMDA extensions has to be used 24.
-
4
R4 The reported matching probabilities have to utilize simple rounding half up to integers and range between 0 and 100.
-
5
R5 Possible values for the locus‐specific match grade characters are A (allele‐level match), P (potential allele‐level match) and M (mismatch).
-
6
R6 When a locus is not typed, the matching probability and the match grade character have to be calculated and provided.
-
7
R7 The probability for a 9/10 match refers to exactly the probability for a 9/10 match, not for a 9/10 match or better.
-
8
R8 The locus‐specific results have to be calculated on the basis of the possible diplotypes, i.e. linkage disequilibrium has to be considered.
-
9
R9 The locus‐specific results must differentiate between match probabilities that are exact values and those that are the result of rounding. Full allele‐level matches (A) that lack mismatching alternative genotypes are given a result of ‘A;100’. Potential allele‐level matches (P) that do have mismatching genotypes of low likelihood, such that the match probability rounds up to 100, are given a result of ‘P;100’. Likewise, complete mismatches (M) with no possible overlapping genotypes are given a result of ‘M;0’. However, a result of ‘P;0’ is given to cases where overlapping genotypes do exist and the match probability rounds down to 0.
-
10
R10 The calculation of the matching predictions has to consider all theoretically possible haplotypes/diplotypes (i.e. no trimming of the set of possible diplotypes).
-
11
R11 Multiple allele codes that have been discontinued 26 have to be reasonably reinterpreted within the current HLA nomenclature.
Comparison and statistics
In the central analyses for MVT 1 and 2, the overall consistency of the data provided and their compliance with the specification was initially checked. Then, for each patient–donor pair the reported numbers of match differences from the submitted result files were compared separately by means of Perl 27 scripts in which the redundant cases per locus were only considered once. In case of disparity, the values provided by the participants were collated in dedicated spreadsheets in which the cells containing values deviating from the majority (if any) were displayed in color. The discrepancies were manually inspected and categorized. These preliminary findings were discussed and, when necessary, clarified and adjusted in the meetings of the Matching Validation Subcommittee in order to compile a consensus result.
For MVT 3, a two‐step analysis approach was used. First, an overview comparison between the results of every pair of participants was made in which only the number of discrepant results concerning the three areas ‘overall 10/10 and 9/10 matching predictions’, ‘locus‐specific 2/2 matching predictions’ and ‘locus‐specific match grade characters’ were counted. Second, for the detailed analysis Perl scripts were used to collect the specific result items for all discrepant cases in suitably structured spreadsheets including the observed differences and the number of possible diplotypes as indicator for the complexity of the considered patient–donor pair. If necessary, differences concerning probability values were studied in detail by a trace program dissecting the genotypes of a patient–donor pair into their diplotypes and their individual probabilities. This allowed to analyze and classify all occurring differences in detail before visualizing them using the R statistics software package 28.
Characteristics of the HMAs compared
Some characteristics of the matching programs contributed by the participating groups are summarized in Table S4. These descriptions may reflect different snapshots in time due to the improvements during the course of the experiment.
Results
The seven groups participating in this validation experiment were assigned arbitrary numbers from #1 to #7 for the following parts of this publication. Not all groups submitted results for all three MVTs.
Results of MVT 1
All seven participants provided results for the first MVT. The format of the result file is shown in Figure 1A. In order to control the results for consistency and to facilitate the comparison for each locus repeating HLA types within the 1000 patients and the 10,000 donors were identified and combined into unique patient–donor pairs. Identical HLA typing combinations had to have the same outcome variables within each result file. Table 1 shows the number of disparities after reducing the total number of 107 results to unique HLA typing pairs for each locus. The disparities could be ascribed to the following reasons:
Table 1.
Observed disparities between the seven results submitted for MVT 1a
| A tot | B tot | DRB1 tot | |
|---|---|---|---|
| Unique HLA typing pairs | 123,708 | 580,659 | 229,504 |
| Discrepant cases between all result files | 857 | 2450 | 1951 |
| Discrepant cases between all result files (%) | 0.69 | 0.42 | 0.85 |
The number of unique locus‐wise typing pairs, the number of discrepant cases and the percentage of discrepant cases are shown for the total number of differences (tot) for the loci HLA‐A, ‐B and ‐DRB1.
HLA, human leukocyte antigen; MVT, matching validation task.
Missing detection of ARD identical alleles, e.g. B*07:05 vs B*07:06 was reported as mismatch.
Incorrect treatment of multiple allele codes that cross allele groups, e.g. B*56:01 vs B*55:BAXT (=B*55:02/55:12/55:16/56:01) was reported as mismatch.
Algorithmic errors, e.g. in some instances B*14:02 vs B*14:AB (=14:01/14:02) was reported as mismatch.
Virtually all disparities could be attributed to algorithmic issues of implementation #6. The identification and explanation of the remaining disparate cases allowed us to compile a consensus result for this experiment. This result is provided in the Supporting Information.
Results of MVT 2
Participants #1 to #6 provided results for the second MVT. The format of the result file is shown in Figure 1B. MVT 2 was evidently more challenging and showed more disparities between the participating matching algorithms than seen in MVT 1. As a result, several iterations were necessary, including some bug fixes, to narrow down the number of disparities to a manageable volume.
Finally, for 59,236,565 (98.7%) of the 6 × 107 data items an identical number was reported. Table 2 illustrates the number and kind of the remaining disparities after reducing the total number of results to unique pairs of HLA types for each locus.
Table 2.
Observed disparities between the six results submitted for MVT 2a
| A tot | A ag | B tot | B ag | DRB1 tot | DRB1 ag | |
|---|---|---|---|---|---|---|
| Unique HLA typing pairs | 124,950 | 124,950 | 592,800 | 592,800 | 232,311 | 232,311 |
| Discrepant cases between all result files | 661 | 4774 | 2751 | 14,358 | 1018 | 5573 |
| Discrepant cases between all result files (%) | 0.53 | 3.82 | 0.46 | 2.42 | 0.44 | 2.40 |
The number of unique locus‐wise typing pairs, the number of discrepant cases and the percentage of discrepant cases are shown for the total number of differences (tot) and the number of antigen differences (ag) for the loci HLA‐A, ‐B and ‐DRB1.
HLA, human leukocyte antigen; MVT, matching validation task.
Surprisingly, the disparities for the total number of differences were slightly lower than in MVT 1. This can be explained by algorithmic improvements of the implementation discrepant in task 1 that are overcompensating some new discrepancies among all participants in identifying allele mismatches based on serological assignments. The strikingly higher numbers of discrepancies for the number of antigen mismatches was caused by several different approaches in the treatment of serologic assignments. Overall, the differences observed could be attributed to the following reasons:
Usage of different DNA‐to‐serology mapping tables. This is actually a violation of the specification but some participating algorithms could not be adapted to use a configurable mapping table with reasonable effort and time (see also Table S3).
Different decisions on potential allele matches between a serological and a molecular assignment. Those problems occur when broad and split/associated antigens are mixed in the serology‐DNA‐correspondence table entries considered, e.g. donor A25 which is a split antigen of A10 vs patient A*26:ADZV, containing A*26:15 (A10).
Different decisions on potential antigen matches between two molecular assignments. Those problems also occur when broad and split/associated antigens are mixed in the serology‐DNA‐correspondence table entries considered, e.g. donor A*25:01 (A25) vs patient A*26:ADZV, containing A*26:15 (A10).
Different treatment of apparent serological mismatches in the context of alleles bridging serological families, e.g. a donor A24 is an apparent antigen difference with patient A3, however because A*24:18 is showing A24/A3 as corresponding serology, this pair could be classified as a potential allele match. As a consequence, those cases were even classified as potential antigen matches by HMA #5.
Implementation specific features, e.g. no differentiation between B64/65, DR13/14 and DR15/16, i.e. those split antigens are generally mapped back to their broad value. In other words, apparent serologic split differences for such cases were not reported as antigen mismatches.
Although the rate of concordance achieved was quite high, for the above reasons the compilation of a true consensus file was impossible and the result file provided for reference in the Supporting Information reflects the discrepancies observed.
Results of MVT 3
Participants #1 to #5 provided results for the third MVT. The format of the result file is illustrated in Figure 1C. Overview comparisons of the match grades, overall match probabilities and locus‐specific match probabilities of the participating HMAs are presented here. To support the validation of new HMA implementations, we also provide a more detailed comparative analysis with HMA trace output in Appendices S1 and S2, along with a consensus result file in Material S3.
Overview comparison of match grade characters
The comparison of the locus‐specific match grade characters shows virtually identical results for participants #2 and #4 and for participants #1, #3 and #5, respectively (see Table 3). The large discrepancy observed was caused by the latter group not complying with requirement R9, i.e. did not distinguish between rounded and exact values for 0% and 100%. The 52 discrepant cases found within the first group could be tracked down to the different treatment of discontinued multiple allele codes (compliance with R11).
Table 3.
Number and percentage (in brackets) of discrepant patient−donor pairs (1 × 107 data items) for any two participants concerning the locus‐specific match grade characters (A/P/M) for all five HLA loci for MVT 3
| #1 | #2 | #3 | #4 | #5 | |
|---|---|---|---|---|---|
| #1 | × | 9,356,595 (93.6) | 3681 (<0.1) | 9,356,505 (93.6) | 4 (<0.1) |
| #2 | × | 9,355,983 (93.6) | 52 (<0.1) | 9,356,505 (93.6) | |
| #3 | × | 9,355,983 (93.6) | 3677 (<0.1) | ||
| #4 | × | 9,356,505 (93.6) | |||
| #5 | × |
HLA, human leukocyte antigen; MVT, matching validation task.
Overview comparison of match probabilities
The findings of the comparison of the overall and locus‐specific probability values are shown in Tables 4 and 5. The subsequent detailed analysis of the disparities between each pair of participants allowed assigning them to the following reasons corresponding to the superscripts used in these tables:
Discrepant mismatch counting between a homozygous patient and a donor with a null allele or vice versa cases (cf. AA – AN in Table S3).
Discrepant mismatch counting when patient and donor are homozygous (cf. AA – BB in Table S3).
Trimming of the set of possible diplotypes (cf. R10).
Treatment of discontinued multiple allele codes (cf. R11).
Numerical artifacts due to floating point arithmetic in combination with rounding.
Conditional locus‐specific probabilities.
Table 4.
Number of discrepant patient−donor pairs (1 × 107 data items) for any two participants concerning the 9/10 and 10/10 predictions for MVT 3a
| #1 | #2 | #3 | #4 | #5 | |
|---|---|---|---|---|---|
| #1 | × | 454 1,2 | 94 2 | 454 1,2 | 2111 1,2,3 |
| #2 | × | 361 1 | 0 | 1783 3 | |
| #3 | × | 361 1 | 2018 1,3 | ||
| #4 | × | 1783 3 | |||
| #5 | × |
MVT, matching validation task.
All percentages are below 0.1 and not shown. Superscripts indicate reasons for discrepancies as: 1, discrepant mismatch counting between a homozygous patient and a donor with a null allele or vice versa cases; 2, discrepant mismatch counting when patient and donor are homozygous; 3, trimming of the set of possible diplotypes.
Table 5.
Number and percentage (in brackets) of discrepant patient−donor pairs (1 × 107 data items) for any two participants concerning the 2/2 locus‐specific predictions for all five HLA loci for MVT 3a
| #1 | #2 | #3 | #4 | #5 | |
|---|---|---|---|---|---|
| #1 | × |
729 1,2,5
(<0.1) |
n/a 6 |
678 1,2,5
(<0.1) |
186,919 2,3,5
(1.9) |
| #2 | × | n/a 6 |
225 4,5
(<0.1) |
186,453 3,5
(1.9) |
|
| #3 | × | n/a 6 | n/a 6 | ||
| #4 | × |
186,430 3,5
(1.9) |
|||
| #5 | × |
HLA, human leukocyte antigen; MVT, matching validation task; n/a, not applicable.
Superscripts indicate reasons for discrepancies as: 1, discrepant mismatch counting between a homozygous patient and a donor with a null allele or vice versa cases; 2, discrepant mismatch counting when patient and donor are homozygous; 3, trimming of the set of possible diplotypes; 4, treatment of discontinued multiple allele codes; 5, numerical artifacts due to floating point arithmetic in combination with rounding; 6, conditional locus‐specific probabilities.
Overall probabilities
The design of MVT 3 yielded a large number of patient−donor pairs with more than one mismatch implying 9/10 and 10/10 probabilities of zero. For the remaining, almost 14,000 pairs an excellent rate of concordance was achieved (see Table 4). In particular, the results of algorithms #2 and #4 were completely identical and the results of algorithms #1 and #3 were almost identical. The detailed analysis revealed that reason 1 and/or 2 above were the main cause of the higher deviations observed (see Figure 2). The slightly higher rate of disparities for participant #5 could be traced back to reason 3 above. This algorithm had to maintain a trimming threshold to deal with the computational complexity in cases with a high number of possible diplotypes. Actually, the output of algorithm #5 is fully identical to the results of algorithms #2 and #4 when excluding the 1005 donors without HLA‐DRB1 assignments.
Figure 2.
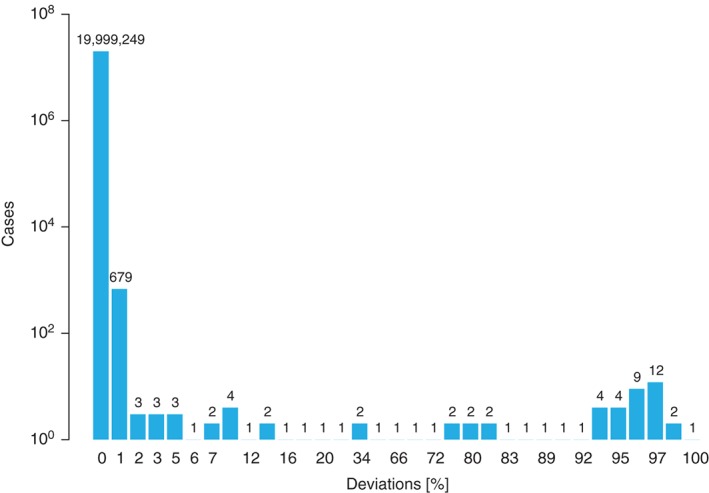
Distribution of the discrepancies for the 9/10 and 10/10 predictions of algorithm #1 compared to algorithm #2 in matching validation task (MVT) 3. The comparison encompasses 2 × 107 data items.
Locus‐specific probabilities
For the probability of locus identity, only a few easily explainable disparities were found (see Table 5). Here, algorithms #2 and #4 altogether showed 225 cases with a deviation of one percentage point in the detailed analysis. Thirteen of these disparities were caused by reason 3 and the rest by reason 5 mentioned above. This is also true for most of the disparities with algorithm #1. However, the discrepant counting addressed in reasons 1 and 2 led to some higher deviations (see Figure 3). The substantial higher rate of disparities observed for participant #5 is again mainly caused by reason 3. When excluding the 1005 donors without HLA‐DRB1 data, the result of algorithm #5 becomes virtually identical to #1, #2 and #4. The locus‐specific probability values provided by participant #3 were not comparable to others because this algorithm returns probabilities for potentially 9/10 matched donors that are conditional on being exactly one mismatch.
Figure 3.
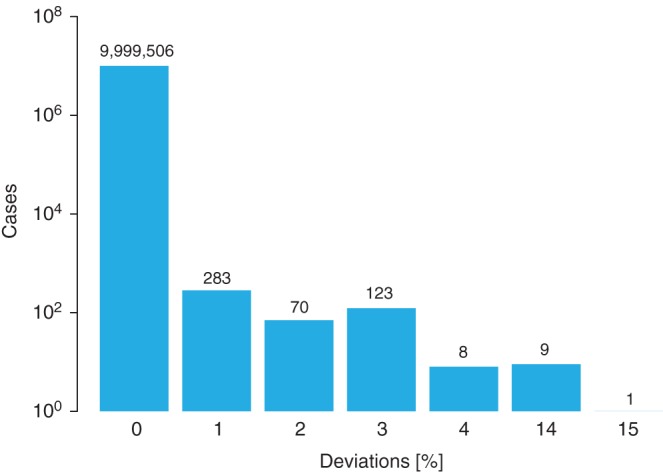
Distribution of the locus‐specific discrepancies for the human leukocyte antigen (HLA)‐C predictions of algorithm #1 compared to algorithm #2 in matching validation task (MVT) 3. The comparison encompasses 1 × 107 data items. For HLA‐C, a substantial number of donors were not typed, complicating the matching computations and therefore leading to higher and more instructive deviations.
Consensus result
In the course of the repeated comparison of gradually refined and/or corrected result files, it finally became apparent that the outputs of all algorithms were converging mainly because the participants started to reduce or altogether avoid the trimming of long diplotype lists. Adapting to the other baseline requirements turned out to be too laborious for some implementations, especially because the advantage for daily use would only be marginal. Since algorithm #2 observed all baseline requirements, it was chosen as reference for the detailed analysis process. This way, all disparities could be explained satisfactorily and have been assessed by the members of the Matching Validation Subcommittee. The result of participant #2 is provided as consensus file for MVT 3 in the Supporting Information. With regard to the accuracy of matching predictions provided in the consensus file, the group agreed that the observed intrinsic deviations of 1% caused by floating point arithmetic 29 in combination with rounding are negligible.
Discussion
The objective of the project was to compare key aspects of the behavior of HMAs when they have to deal with the wide variety of HLA genotype data in today's donor registries and to use the results to identify relevant problems and pitfalls in this complex task. The ultimate goal was to reach a consensus on important underlying principles and the desirable results wherever possible and to otherwise identify the key design decisions where the algorithms or implementations may deliberately deviate from each other. In those cases, the exercise served as a cross‐validation to ensure that the discrepancies between the HMAs are restricted to the intended effects. While HSC donor selection is a complex process that also must consider many factors outside of HLA matching 30, we focused solely on the contribution of HLA match calculations by HMAs to this process.
Other methods of software validation are either impractical (formal methods, code inspection) or less meaningful (examination of specification and documentation) than the functional testing based on a large range of simulated practical input signals carried out in this study. In contrast to HLA typing methodology, there are no established regulatory frameworks in place for HMAs. However, previous validation efforts for the estimation of HLA haplotype frequencies by the WMDA ITWG Registry Diversity working group 31 are not only a building block for this study but are also used in the validation of HLA typing techniques 32.
The major challenges were related to the handling of the complexity of HLA nomenclature in its historical context and to the dealing with complexity of the haplotype‐related calculations for predictive matching. The sequence of the three tasks was necessary to isolate groups of individual problems from each other and to acquire an increasing understanding of the individual design decisions underlying different behaviors.
The first two MVTs addressed the locus‐wise matching decisions between patient and donor without considering frequency information or linkage disequilibrium. In other words, the algorithms were supposed to evaluate potential matches using purely combinatorial methods. Strictly followed, such an exhaustive approach leads to unusable search reports due to extreme unlikely pairs like HLA‐A*02:XX potentially matching HLA‐A*03:XX since both generic groups comprise null alleles (case AN – AN in Table S3 is a match). Apparently, all participating algorithms are intrinsically using heuristics most likely based on allele frequencies or a set of CWD alleles which all lead to identical results for MVT 1.
For MVT 2, however, the differences in the algorithms became apparent since the correspondence between serological and molecular assignments is not unequivocal and well defined for all alleles: some alleles have no official serological correspondence, some have several and some are only attributed to a broad serological specificity. The combinatorial consequences for matching between two serological assignments or between a serological and molecular assignment as well as for assigning a molecular mismatch to the antigen or allele level are quite complex and have been described in details in the result section. Apparently, cases like the examples shown there cannot be decided satisfactorily based on the currently available reference tables 25. As a consequence, MVT 2 did not lead to a proper consensus and the result summary documents a certain degree of variability. MVTs 1 and 2 reflect the dilemma between the historically developed HLA nomenclature that is still the basis of current paradigms in donor selection and the practical requirements in clinical decision‐making.
MVT 3 introduced another level of complexity by requiring the use of haplotype frequencies to distinguish between likely and unlikely potential matches. In the early rounds of analysis, the large number of various discrepancies observed was caused by incomplete or partially disregarded baseline requirements. Later stages showed that adhering to those rules and assumptions makes all relevant discrepancies disappear and lead to a consistent consensus result for MVT 3. This convergence of results required modifying the configuration or implementation details of several participating matching algorithms according to the requirements R1–R11. As a consequence, this exercise does not necessarily reflect their behavior in daily routine where for certain reasons other preferences may be given priority.
In particular, the trimming of the set of possible diplotypes is a usual approach to reduce the computational complexity to save a substantial amount of processing time and give the user an improved response time for display of a new search report. Although in most cases this still gives reasonably precise results, it became apparent that it is impossible to give estimates for the maximum error introduced. This error in the matching probability returned can actually be quite significant and therefore any trimming threshold must be chosen with great care (see Appendix S1). Still, the improvement of the performance of the HMAs will remain a major topic in the refinement of all existing matching programs as long as incomplete and ambiguous donor types have to be dealt with. Their efficiency not only has a major impact on the perceived responsiveness of user interfaces but also for the daily automatic update of thousands of search reports required for highly automated registry services or in the EMDIS communication system 33, 34. Deployment of high‐performance computing clusters and development of HMAs that use parallel computing paradigms may allow registries to meet these big data challenges.
The participants have benefitted in multiple ways from this series of experiments. First, several errors became apparent and were fixed, but moreover many other more or less intended properties underwent a critical review. So eventually the quality of all HMAs was confirmed in certain aspects and improved in others. Second, future updates of all HMAs can refer back to the consensus results to ensure that no regressions or unintended features have been introduced. Similarly, the developers of new HMAs can validate their implementation using the Supporting Information accompanying this article. Lastly, the baseline result of MVT 3 allows for the measurement of the impact of any performance tweaks used in the real‐life version of an HMA on the speed and quality of the results to make a sound cost−benefit judgment. Eventually, it will be up to the registry community cooperating within the WMDA to decide to which extent the MVTs can become a part of their global assurance efforts 35. Moreover, the experience gained in this study could be incorporated into the specifications for the HMA of the global search system of BMDW.
This study provides the first major contribution to the practical validation of HMAs, but it still leaves certain aspects uncovered and will have to be refined when HMAs further evolve. The most relevant limitation of this study is the fact that all patient and donor genotypes are simulated from the same set of haplotypes that are later used with a positive frequency. However in practice, a substantial fraction of the patients and more often the donors cannot be explained by the set(s) of haplotypes with known positive frequency. All probabilistic HMAs need fallback strategies for such situations that were not addressed in this experiment.
More challenges will arise when haplotype frequency tables specific to a population or a population subgroup become available and individual patients or donors cannot be clearly assigned. Such an approach would model the reality of matching in a global donor pool more realistically 36, but the ultimate benchmark for all haplotype frequency estimation efforts is the validation of real‐matching predictions with typing outcome data. Nevertheless, the findings of this study are independent from the population haplotype frequencies used since the causes of the differences observed are either algorithmic or intrinsic to the HLA nomenclature.
Although such comparative analyses do not prove the correctness of any program, they do provide a strong indication of correctness due to the consensus results of independent implementations. The authors are aware that ‘even when the experts all agree, they may well be mistaken’ and that ‘when the experts are agreed, the opposite opinion cannot be held to be certain’ 37.
Conflicts of interest
The authors have declared no conflicting interests.
Supporting information
Table S1. Characteristics of the matching validation tasks and associated data sets.
Table S2. Types of HLA assignments used and their resolution.
Table S3. Counting mismatches for 2 by 2 comparisons of patient and donor molecular HLA assignments.
Table S4. Software and hardware environments of the HMA implementations.
Appendix S1. PDF file with the detailed analysis of MVT 3 (including Figures S1–S3).
Appendix S2. Tar GZip file with an example trace for MVT 3 as Excel file.
Material S1. Tar GZip file containing README, MVS 1 and consensus result.
Material S2. Tar GZip file containing README (including the coding of disparities), MVS 2, DNA‐to‐serology mapping table and reference result.
Material S3. Tar GZip file containing README, MVS 3, list of ARD groups, haplotype frequency table and consensus result.
Acknowledgments
LG, JS and MM have been funded by Office of Naval Research Grant N00014‐13‐1‐0039.
References
- 1. Fürst D, Müller CR, Vucinic V et al. High‐resolution HLA matching in hematopoietic stem cell transplantation: a retrospective collaborative analysis. Blood 2013: 122: 3220–9. [DOI] [PubMed] [Google Scholar]
- 2. Lee SJ, Klein J, Haagenson M et al. High‐resolution donor‐recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 2007: 110: 4576–83. [DOI] [PubMed] [Google Scholar]
- 3. Woolfrey AE, Lee SJ, Gooley TA et al. HLA‐allele matched unrelated donors compared to HLA‐matched sibling donors: role of cell source and disease risk category. Biol Blood Marrow Transplant 2010: 16: 1382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steiner D. Computer algorithms in the search for unrelated stem cell donors. Bone Marrow Res 2012: 2012: 175419–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hurley CK, Maiers M, Marsh SGE, Oudshoorn M. Overview of registries, HLA typing and diversity, and search algorithms. Tissue Antigens 2007: 69 (Suppl 1): 3–5. [DOI] [PubMed] [Google Scholar]
- 6. Ottinger HD, Müller CR, Goldmann SF et al. Second German consensus on immunogenetic donor search for allotransplantation of hematopoietic stem cells. Ann Hematol 2001: 80: 706–14. [DOI] [PubMed] [Google Scholar]
- 7. Bray RA, Hurley CK, Kamani NR et al. National marrow donor program HLA matching guidelines for unrelated adult donor hematopoietic cell transplants. Biol Blood Marrow Transplant 2008: 14: 45–53. [DOI] [PubMed] [Google Scholar]
- 8. Spellman SR, Eapen M, Logan BR et al. A perspective on the selection of unrelated donors and cord blood units for transplantation. Blood 2012: 120: 259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Müller CR, Mytilineos J, Ottinger HD et al. Deutscher Konsensus 2013 zur immungenetischen Spenderauswahl für die allogene Stammzelltransplantation. Transfusionsmedizin 2014: 4: 190–6. [Google Scholar]
- 10. Bochtler W, Beth M, Eberhard H‐P, Mueller CR. OptiMatch – a universally configurable HLA matching framework. Tissue Antigens 2008: 71: 321. [Google Scholar]
- 11. Tiercy JM. Unrelated hematopoietic stem cell donor matching probability and search algorithm. Bone Marrow Res 2012: 2012: 695018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt AH, Sauter J, Pingel J, Ehninger G. Toward an optimal global stem cell donor recruitment strategy. PLoS One 2014: 9: e86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gragert L, Eapen M, Williams E et al. HLA match likelihoods for hematopoietic stem‐cell grafts in the U.S. Registry. N Engl J Med 2014: 371: 339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WMDA annual report for stem cell donor registries 2014. https://www.wmda.info/about‐us/publications.
- 15.BMDW. www.bmdw.org.
- 16.EMDIS. www.emdis.net.
- 17. Maiers M, Bakker JNA, Bochtler W et al. Information technology and the role of WMDA in promoting standards for international exchange of hematopoietic stem cell donors and products. Bone Marrow Transplant 2010: 45: 839–42. [DOI] [PubMed] [Google Scholar]
- 18. Bochtler W, Maiers M, Bakker JNA et al. World Marrow Donor Association framework for the implementation of HLA matching programs in hematopoietic stem cell donor registries and cord blood banks. Bone Marrow Transplant 2010: 44: 1–6. [DOI] [PubMed] [Google Scholar]
- 19. Maiers M, Gragert L, Klitz W. High‐resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007: 68: 779–88. [DOI] [PubMed] [Google Scholar]
- 20. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 2015: 43: D423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milius RP, Mack SJ, Hollenbach JA et al. Genotype List String: a grammar for describing HLA and KIR genotyping results in a text string. Tissue Antigens 2013: 82: 106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allele code lists. https://bioinformatics.bethematchclinical.org/HLA‐Resources/Allele‐Codes/Allele‐Code‐Lists/.
- 23. Cano P, Klitz W, Mack SJ et al. Common and well‐documented HLA alleles: report of the Ad‐Hoc committee of the american society for histocompatibility and immunogenetics. Hum Immunol 2007: 68: 392–417. [DOI] [PubMed] [Google Scholar]
- 24. Bochtler W, Maiers M, Oudshoorn M et al. World Marrow Donor Association guidelines for use of HLA nomenclature and its validation in the data exchange among hematopoietic stem cell donor registries and cord blood banks. Bone Marrow Transplant 2007: 39: 737–41. [DOI] [PubMed] [Google Scholar]
- 25.HLA nomenclature in WMDA file format. http://hla.alleles.org/wmda/index.html.
- 26. Bochtler W, Maiers M, Bakker JNA et al. An update to the HLA Nomenclature Guidelines of the World Marrow Donor Association, 2012. Bone Marrow Transplant 2013: 48: 1387–8. [DOI] [PubMed] [Google Scholar]
- 27.The perl programming language. www.perl.org.
- 28.The R project for statistical computing. www.r‐project.org.
- 29. Buontempo F. Floating point fun and frolics. Overload 2009: 17: 4–8. [Google Scholar]
- 30. Kollman C, Spellman SR, Zhang M‐J et al. The effect of donor characteristics on survival after unrelated donor transplantation for hematologic malignancy. Blood 2016: 127: 260–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eberhard H‐P, Madbouly AS, Gourraud PA et al. Comparative validation of computer programs for haplotype frequency estimation from donor registry data. Tissue Antigens 2013: 82: 93–105. [DOI] [PubMed] [Google Scholar]
- 32. Osoegawa K, Mack SJ, Udell J et al. HLA haplotype validator for quality assessments of HLA typing. Hum Immunol 2016: 77: 273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Müller CR. Computer applications in the search for unrelated stem cell donors. Transpl Immunol 2002: 10: 227–40. [DOI] [PubMed] [Google Scholar]
- 34. Steiner D. European marrow donor information system: concept and praxis. Transplant Proc 2010: 42: 3255–7. [DOI] [PubMed] [Google Scholar]
- 35. Hurley CK, Foeken L, Horowitz M et al. Standards, regulations and accreditation for registries involved in the worldwide exchange of hematopoietic stem cell donors and products. Bone Marrow Transplant 2010: 45: 819–24. [DOI] [PubMed] [Google Scholar]
- 36. Maiers M, Gragert L, Madbouly AS et al. 16(th) IHIW: global analysis of registry HLA haplotypes from 20 million individuals: report from the IHIW Registry Diversity Group. Int J Immunogenet 2013: 40: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Russell BAW. Introduction In: Sceptical Essays. London: George Allen & Unwin, 1928. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the matching validation tasks and associated data sets.
Table S2. Types of HLA assignments used and their resolution.
Table S3. Counting mismatches for 2 by 2 comparisons of patient and donor molecular HLA assignments.
Table S4. Software and hardware environments of the HMA implementations.
Appendix S1. PDF file with the detailed analysis of MVT 3 (including Figures S1–S3).
Appendix S2. Tar GZip file with an example trace for MVT 3 as Excel file.
Material S1. Tar GZip file containing README, MVS 1 and consensus result.
Material S2. Tar GZip file containing README (including the coding of disparities), MVS 2, DNA‐to‐serology mapping table and reference result.
Material S3. Tar GZip file containing README, MVS 3, list of ARD groups, haplotype frequency table and consensus result.