

Abstract
Apart from efficacy and toxicity, many drug development failures are imputable to poor pharmacokinetics and bioavailability. Gastrointestinal absorption and brain access are two pharmacokinetic behaviors crucial to estimate at various stages of the drug discovery processes. To this end, the Brain Or IntestinaL EstimateD permeation method (BOILED‐Egg) is proposed as an accurate predictive model that works by computing the lipophilicity and polarity of small molecules. Concomitant predictions for both brain and intestinal permeation are obtained from the same two physicochemical descriptors and straightforwardly translated into molecular design, owing to the speed, accuracy, conceptual simplicity and clear graphical output of the model. The BOILED‐Egg can be applied in a variety of settings, from the filtering of chemical libraries at the early steps of drug discovery, to the evaluation of drug candidates for development.
Keywords: blood–brain barrier, chemoinformatics, drug absorption, medicinal chemistry, physicochemical properties
Any input to support the critical daily choice of which compound to synthesize, test, and promote is of utmost importance to identify those compounds with the highest probability of overcoming all obstacles in drug discovery and development, and to ultimately become a marketed medicine for the patient′s benefit. Apart from efficacy and toxicity, many failures during drug development are related to pharmacokinetics, i.e., the fate of the compound in the organism.1 Nowadays, by monitoring physicochemical profiles of lead compounds it is possible to increase the quality of clinical candidates.2 The individual consideration of absorption, distribution, metabolism and excretion (ADME) behaviors at the early stages of drug discovery has decreased the fraction of global pharmacokinetics‐related failures in later phases of development. As a consequence, today, drug candidates reach the market more efficiently.3
Although there are different routes of drug administration, oral dosing is highly preferred for the patient′s comfort and compliance. Early estimation of oral bioavailability, i.e., the fraction of the dose that reaches the bloodstream after oral administration, is a key decision‐making criterion at various steps of the discovery process. Bioavailability is highly multifactorial, but is primarily driven by gastrointestinal absorption.4
The large number of molecules and the small physical sample amount at initial stage of medicinal chemistry projects, together with the need to limit animal testing, prevent systematic recourse to experiments. This has fostered computational models that are able to predict pharmacokinetic parameters, especially bioavailability.5 The eminent rule‐of‐five by Lipinski and co‐workers provides physicochemical margins outside of which the probability for a molecule to become an oral drug is low.6 Despite criticism, often due to over‐interpretation, the rule‐of‐five shed light on the relationship between bioavailability and physicochemical properties, settling the concept of drug‐likeness, and inspired many simple rule‐based models. Later, more sophisticated and precise models based on machine‐learning methods were built. However, these latter share the severe drawback of being “black boxes” difficult to interpret and to translate into molecular design.7
An elegant compromise between these two types of models was proposed by Egan et al.,8 who developed a descriptive representation to discriminate between well‐absorbed and poorly absorbed molecules based on their lipophilicity and polarity, described by the n‐octanol/water partition coefficient (log P) and the polar surface area (PSA). The delineation exists in a region of favorable properties for gastrointestinal absorption on a plot of two computed descriptors: ALOGP989 versus PSA.10 Because the region most populated by well‐absorbed molecules is elliptical, it was called the Egan egg. The advantages of this representation are related to its simple concept, straightforward interpretation, and direct translation into molecular design (unlike machine‐learning methods). In contrast to rule‐based models and thanks to its 2D graphical nature, it not only provides thresholds, but also a clear picture of how far a molecular structure is from the ideal physicochemical region for good absorption. As lipophilicity and polarity are often inversely correlated properties, the sometimes‐tricky chemical modifications simultaneously impacting log P and PSA are efficiently supported by the model, which is rapid enough to allow trial‐and‐error iterations. These practical benefits make the Egan egg widely used in industrial and academic contexts, as indicated by its implementation in commercial packages (e.g., Discovery Studio, Dassault Systèmes BIOVIA, San Diego, CA, USA) and numerous citations of the seminal articles.8, 11 Successful applications include, for example, the discovery and development of the groundbreaking drug against hepatitis C, telaprevir,12 and a detailed pharmacokinetic analysis leading to anti‐tuberculosis agents.13
However, Egan′s method comes with some concerns. Although routinely applied as a prediction tool, it was developed as a delineation, merely descriptive and without evaluation of predictive power. Indeed, the computation of the ellipse took into account well‐absorbed molecules but neglected poorly absorbed molecules. The resulting confidence region merely depicts the dispersion of properties related to good absorption and lacks an assessment of accuracy. Additionally, several points hinder the reproducibility of the published methodology: the dataset was not fully disclosed; the values of ALOGP989 were obtained through a closed‐source commercial implementation; and the details of PSA calculations relying on tridimensional geometries were not described.
Given the undeniable practicality of Egan′s egg and its effectiveness for drug discovery projects, we sought to amend these methodological aspects, to assess the predictive power of the model for gastrointestinal passive absorption, and to complement it with the prediction for brain access by passive diffusion to finally lay the BOILED‐Egg (Brain Or IntestinaL EstimateD permeation predictive model).
We curated recent human intestinal absorption (HIA) data4 by literature, patent, and database cross‐checks (refer to Methods S1 in the Supporting Information) to gather 660 small molecules (567 well‐ and 93 poorly absorbed) with cleansed structures and reliable measurements of the fraction absorbed by human (FA), excluding actively transported compounds. This HIA dataset is given in Table S1 in the Supporting Information.
All 660 molecular structures were subject to log P and PSA computation (Figure 1; see Methods S3 in the Supporting Information for details). The log P method developed by Wildman and Crippen (WLOGP) was chosen, because it is closely related to ALOGP98, but with exhaustive chemical description, which makes its implementation straightforward.14 Likewise, we calculated the topological polar surface area (tPSA), a well‐described technique to estimate PSA based on a 2D fragmental system.10
Figure 1.
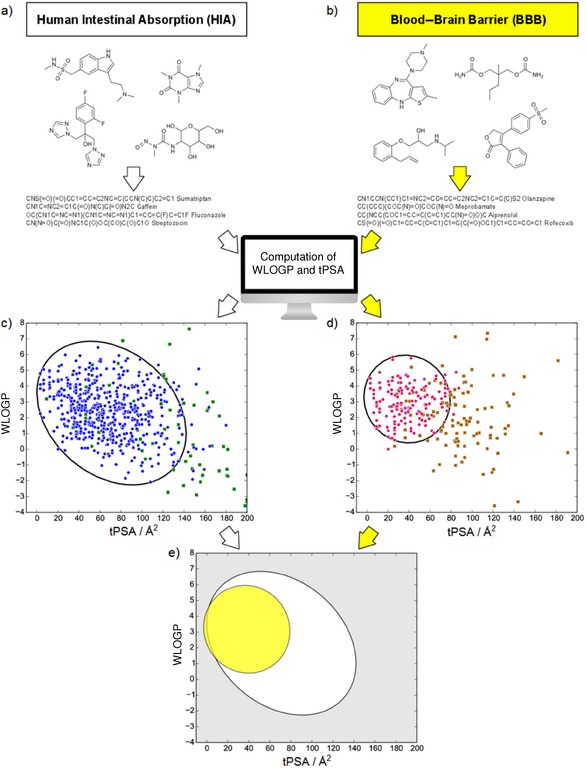
Overview of the BOILED‐Egg construction. a) Gastrointestinal absorption and b) brain penetration datasets (HIA and BBB in Tables S1 and S2, respectively) cleansed, neutralized, standardized, converted into SMILES notation were subject to lipophilicity (WLOGP) and polarity (tPSA) computation. Best classification ellipse for well‐ and poorly absorbed molecules (blue points and green squares, respectively, in (c) and Figure S1) as well as for brain penetrant and non‐penetrant molecules (pink points and brown squares, respectively, in (d) and Figure S2). e) Combining both best ellipses yields the BOILED‐Egg predictive model. The white region is the physicochemical space of molecules with highest probability of being absorbed by the gastrointestinal tract, and the yellow region (yolk) is the physicochemical space of molecules with highest probability to permeate to the brain. Yolk and white areas are not mutually exclusive.
The ellipse that best classifies the 660 molecules of the HIA dataset was computed by including as many well‐absorbed and as less poorly absorbed compounds as possible on the WLOGP versus tPSA plot (details in Methods S4, S5, and S6 in the Supporting Information). Five parameters defining the ellipse—the Cartesian coordinates of the foci (x 1, x 2); (y 1, y 2) and the major axis (or largest diameter, d)—were submitted to Monte‐Carlo (MC) optimization, evaluated by the Matthews correlation coefficient (MCC, ranging from −1 to 1 for perfect classification, see Methods S7). After about 100 000 independent MC runs of 100 000 cycles each, with starting parameters spanning the desired physicochemical space, the optimal ellipse was obtained with an excellent MCC=0.70 (Figure 1 a and 1 c, and Data S1 and Figure S1 in the Supporting Information). Reasons for misclassification can be attributed to either technical issues, i.e., WLOGP or tPSA do not accurately describe the lipophilicity and polarity of particular compounds, or to conceptual issues, i.e., other unrelated properties impact absorption. These latter properties, if linked to the molecular structure (e.g., its charge), could eventually be considered by additional orthogonal axes. Physicochemical description issues could explain part of the 26 false positives (structures depicted in Figure S5), as many of these bear positive charges. In other cases, the neglected properties are most probably physiological. Even a high‐quality dataset is influenced by the state of knowledge at the time of curation. This can explain part of the 20 false negatives, as a given molecule could be considered as absorbed passively just because its active transporter remains to be discovered15 (structures depicted in Figure S6).
Our passive absorption model, with an internal accuracy of 93 %, was further assessed by 10‐fold cross‐validation. The high cross‐validated MCC, MCCCV=0.65, and cross‐validated accuracy of 92 % (see Methods S8 and Table S3 in the Supporting Information) together with the fact that the ten ellipses show a large overlap (Figure S3) ascertains the robustness of classification. Finally, our model confirms and refines the guidelines for good absorption,7 the ellipse being encompassed in the commonly accepted rectangular limits of PSA lower than 142 Å2 and log P between −2.3 and +6.8.
These results encouraged us to extend the approach to blood–brain barrier (BBB) permeation, which is fundamental for the distribution of central‐acting molecules, or reversely for limited unwanted effects of peripheral drugs. Similarly to bioavailability and given the substantial effort to measure BBB permeation, several computational methods were developed.16 Again, they can be divided in “Lipinski‐like” rule‐based and in machine‐learning models, but so far no “Egan‐like” approach has been published.
The BBB can be considered as a shield protecting the brain by a “physical” barrier (e.g., tight junctions in endothelial cells preventing paracellular penetration) and a “biochemical” barrier consisting of enzymatic activities and active efflux (e.g., P‐glycoprotein pumping out substrates from central nervous system (CNS) tissues). Although active transport is important, passive diffusion is the major route for drugs to access the brain from the bloodstream.17 Substantial curation of a recent dataset18 supported by specialized databases was required to build our passive BBB‐permeation model (refer to Methods S2 in the Supporting Information). We collected 260 molecules (156 permeant and 104 non‐permeant) with cleansed structures and reliable measurements of blood–brain partition (log BB). This BBB dataset is provided in Table S2. The same methodology as for the absorption classification was applied. Massively parallelized MC runs yielded the best classifying ellipse on the WLOGP versus tPSA graph for BBB permeation (MCC=0.79, Figure 1 b and 1 d, and Data S2 and Figure S2 in the Supporting Information). Our model is in accordance with and refines the established simple guidelines giving PSA thresholds for BBB permeation.16, 19 Indeed, we show that moderately polar (PSA<79 Å2) and relatively lipophilic (log P from +0.4 to +6.0) molecules have a high probability to access the CNS. Similarly to the gastrointestinal model, some of the 22 false positives can be attributed to the limitations of the WLOGP and tPSA descriptors (Figure S7 in the Supporting Information). The imperfect current state of knowledge about active transport for discovery or early development compounds could explain the five false negatives (Figure S8). However, with an internal classification accuracy of 90 %, our BBB permeation model shows a brilliant descriptive ability. A 10‐fold cross‐validation returned a MCCCV=0.75 and a cross‐validated accuracy of 88 % (refer to Table S4 in the Supporting Information). The robustness of our BBB classification model is further confirmed by the large overlap of the ten ellipses (Figure S4).
As an illustration, the 46 non‐prodrug new chemical entities (NCEs) with clear oral bioavailability accepted by the FDA between January 2014 and September 2015 (FDA dataset in Table S5) were mapped onto the BOILED‐Egg (Figure 2 a). The vast majority of gastrointestinal absorption predictions are sensible, as indicated by a classification accuracy reaching 83 %. The same appears true for BBB permeation, as most NCEs with evidence for brain penetration lie inside the yellow ellipse.
Figure 2.
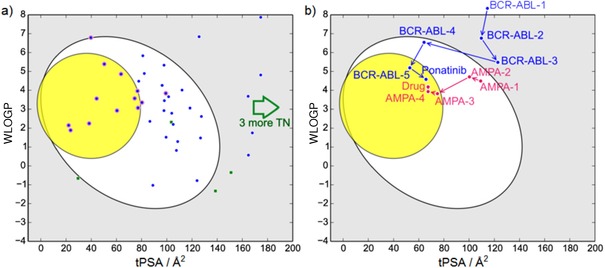
Test and illustrative uses of the BOILED‐Egg. a) Plot of 46 compounds accepted as NCEs by the FDA in 2014 and 2015 (FDA dataset in Table S5). Well‐ and poorly absorbed molecules (blue points and green squares, respectively) are predicted with an accuracy of 83 % (three true negatives (TN) are outside the range). Drugs with good evidence of brain access are circled in red. b) Optimization path of BCR‐ABL inhibitors leading to the oral anticancer drug ponatinib (in blue) and optimization path of AMPA receptor modulators leading to a brain penetrant investigational drug under clinical evaluation (in pink). Chemical structures and a more detailed description are provided in Figures S9 and S10.
In our practice, the BOILED‐Egg is of great support for lead optimization. The following two cases illustrate how it can steer property‐based lead optimization to improve pharmacokinetics. The first example is the optimization of third‐generation BCR‐ABL kinase inhibitors, starting from the lead BCR‐ABL‐1 with poor pharmacokinetics, distant from the egg, to finally obtain the oral anticancer drug ponatinib.20 Ponatinib correctly lies inside the white ellipse, but inside the BOILED‐Egg′s yolk, too (blue path in Figures 2 b and S9). This agrees with experimental data suggesting that ponatinib crosses the BBB.21 The second example is the optimization of AMPA receptor modulators to enhance synaptic activity. The optimization started from an orally bioavailable, but BBB non‐permeant lead AMPA‐1. The physicochemical modifications to finally obtain a brain‐penetrant investigational drug22 correctly located in the yolk can be followed (pink path in Figures 2 b and S10).
The BOILED‐Egg model delivers a rapid, intuitive, easily reproducible yet statistically unprecedented robust method to predict the passive gastrointestinal absorption and brain access of small molecules useful for drug discovery and development. The BOILED‐Egg is depicted in Figure 1 c, and the coordinates of respective ellipses are given in Figures S1 and S2 in the Supporting Information. Finally, an Excel file is provided as Data S3 (described in the Supporting Information), including the Cartesian coordinates of both ellipses’ trace. The user has the possibility to add the WLOGP and tPSA for up to 100 molecules, and the corresponding points are mapped onto the BOILED‐Egg (detailed protocol in Methods S9).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
The authors are thankful to the SIB Swiss Institute of Bioinformatics (www.sib.swiss) for funding and to its high‐performance computing center (Vital‐IT, www.vital‐it.ch) for providing computational resources. This work was also supported by the Solidar‐Immun Foundation. Our profound gratitude is expressed to Prof. Olivier Michielin for helpful and supportive discussions, as well as to Dr. Ute Röhrig for a thorough review of the manuscript. We acknowledge ChemAxon Ltd. (www.chemaxon.com) for the academic license agreement. Graphical plots were generated with the matplotlib python library (matplotlib.org).
A. Daina, V. Zoete, ChemMedChem 2016, 11, 1117.
References
- 1. Kubinyi H., Nat. Rev. Drug Discovery 2003, 2, 665–668. [DOI] [PubMed] [Google Scholar]
- 2. Waring M. J., Arrowsmith J., Leach A. R., Leeson P. D., Mandrell S., Owen R. M., Pairaudeau G., Pennie W. D., Pickett S. D., Wang J., Wallace O., Weir A., Nat. Rev. Drug Discov. 2015, 14, 475–486. [DOI] [PubMed] [Google Scholar]
- 3. Jorgensen W. L., Science 2004, 303, 1813–1818. [DOI] [PubMed] [Google Scholar]
- 4. Newby D., Freitas A. A., Ghafourian T., Eur. J. Med. Chem. 2015, 90, 751–765. [DOI] [PubMed] [Google Scholar]
- 5. Tian S., Wang J., Li Y., Li D., Xu L., Hou T., Adv. Drug Delivery Rev. 2015, 86, 2–10. [DOI] [PubMed] [Google Scholar]
- 6. Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J., Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- 7. Ursu O., Rayan A., Goldblum A., Oprea T. I., WIREs Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar]
- 8. Egan W. J., Merz K. M., Baldwin J. J., J. Med. Chem. 2000, 43, 3867–3877. [DOI] [PubMed] [Google Scholar]
- 9. Ghose A. K., Viswanadhan V. N., Wendoloski J. J., J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar]
- 10. Ertl P., Rohde B., Selzer P., J. Med. Chem. 2000, 43, 3714–3717. [DOI] [PubMed] [Google Scholar]
- 11. Egan W. J., Lauri G., Adv. Drug Delivery Rev. 2002, 54, 273–289. [DOI] [PubMed] [Google Scholar]
- 12. Kwong A. D., Kauffman R. S., Hurter P., Mueller P., Nat. Biotechnol. 2011, 29, 993–1003. [DOI] [PubMed] [Google Scholar]
- 13. Lakshminarayana S. B., Huat T. B., Ho P. C., Manjunatha U. H., Dartois V., Dick T., Rao S. P. S., J. Antimicrob. Chemother. 2015, 70, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wildman S. A., Crippen G. M., J. Chem. Inf. Model. 1999, 39, 868–873. [Google Scholar]
- 15. Sugano K., Kansy M., Artursson P., Avdeef A., Bendels S., Di L., Ecker G. F., Faller B., Fischer H., Gerebtzoff G., Lennernaes H., Senner F., Nat. Rev. Drug Discov. 2010, 9, 597–614. [DOI] [PubMed] [Google Scholar]
- 16. Rankovic Z., J. Med. Chem. 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- 17. Di L., Artursson P., Avdeef A., Ecker G. F., Faller B., Fischer H., Houston J. B., Kansy M., Kerns E. H., Krämer S. D., Lennernäs H., Sugano K., Drug Discov. Today 2012, 17, 905–912. [DOI] [PubMed] [Google Scholar]
- 18. Brito-Sánchez Y., Marrero-Ponce Y., Barigye S. J., Yaber-Goenaga I., Morell Pérez C., Le-Thi-Thu H., Cherkasov A., Mol. Inf. 2015, 34, 308–330. [DOI] [PubMed] [Google Scholar]
- 19. Ghose A. K., Herbertz T., Hudkins R. L., Dorsey B. D., Mallamo J. P., ACS Chem. Neurosci. 2012, 3, 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang W.-S., Metcalf C. A., Sundaramoorthi R., Wang Y., Zou D., Thomas R. M., Zhu X., Cai L., Wen D., Liu S. et al., J. Med. Chem. 2010, 53, 4701–4719. [DOI] [PubMed] [Google Scholar]
- 21. Gaur S., Torabi A.-R., Corral J., In Vivo 2014, 28, 1149–1153. [PubMed] [Google Scholar]
- 22. Ward S. E., Harries M., Aldegheri L., Andreotti D., Ballantine S., Bax B. D., Harris A. J., Harker A. J., Lund J., Melarange R., Mingardi A., Mookherjee C., Mosley J., Neve M., Oliosi B., Profeta R., Smith K. J., Smith P. W., Spada S., Thewlis K. M., Yusaf S. P., J. Med. Chem. 2010, 53, 5801–5812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary