

Abstract
β‐Amyloid (Aβ) oligomers are neurotoxic and implicated in Alzheimer's disease. Neuronal plasma membranes may mediate formation of Aβ oligomers in vivo. Membrane components sphingomyelin and GM1 have been shown to promote aggregation of Aβ; however, these studies were performed under extreme, non‐physiological conditions. We demonstrate that physiological levels of GM1, organized in nanodomains do not seed oligomerization of Aβ40 monomers. We show that sphingomyelin triggers oligomerization of Aβ40 and that GM1 is counteractive thus preventing oligomerization. We propose a molecular explanation that is supported by all‐atom molecular dynamics simulations. The preventive role of GM1 in the oligomerization of Aβ40 suggests that decreasing levels of GM1 in the brain, for example, due to aging, could reduce protection against Aβ oligomerization and contribute to the onset of Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid beta-peptides, diffusion coefficients, fluorescence spectroscopy, neuroprotectives
Oligomers of the β‐amyloid (Aβ) peptide are thought to spark neuronal dysfunction, cell death, and Alzheimer's disease (AD).1 Oligomerization of Aβ occurs spontaneously at high concentration in solution.2 However, it is likely that plasma membranes mediate the oligomerization of nanomolar (nm) concentrations of Aβ in the brain.
Sphingomyelin (Sph) was shown to promote membrane‐mediated aggregation of micromolar concentrations of Aβ. The in vitro studies used rigid bilayers of Sph and Sph/cholesterol (Chol) (gel and liquid ordered phases)3 as well as phase‐separated ternary mixtures.4a Aggregation of Aβ was also shown to occur preferentially in microscopic liquid ordered phases.4 Therefore, it is not clear whether Sph has a specific effect or if aggregation is promoted by the physical state of the membrane. Another component of neuronal membranes proposed to enhance aggregation of Aβ is the monosialoganglioside GM1. It was suggested that GM1 clusters seed formation of amyloid fibrils and are involved in development of AD.5 However, the in vitro studies used concentrations of GM1 above 20 mol %, while total ganglioside expression in neuronal cells is below 10 mol % of total membrane lipids.6 Reported levels of GM1, the most abundant ganglioside in neurons and white matter, are 2–4 mol %.7 Importantly, ganglioside concentration in the brain decreases with AD development, and GM1 is known to have neuroprotective and neurorestorative effects.8 Recent reports state that GM1 can reduce toxicity induced by Aβ peptides in vivo.8a, 9
The membranes of high rigidity used in all cited in vitro studies are commonly justified as models for cellular membrane “rafts”. However, the level of order of such in vitro systems is yet to be found in living cells. It is nowadays more widely accepted that cellular “rafts” must not be viewed as rigid, highly ordered domains but as dynamic nanoscopic entities.10
In this work, we address how Sph and GM1 influence in‐membrane oligomerization of Aβ40 at the molecular level. Employing well‐controlled model systems, we emulate more physiological conditions by using: a) nm concentrations of fluorescently labeled Aβ40 monomers (similar to conditions in the brain); b) GM1 levels at maximum of 4 mol % (levels of GM1 in neurons);7 c) membranes of increasing complexity that contain transient nanoheterogeneities rather than large‐scale segregation.10 Single‐molecule fluorescence techniques reveal the triggering of oligomerization of Aβ40 by Sph and its inhibition by GM1. We propose a model for the underlying mechanisms of triggering and inhibition of oligomerization with insights obtained from molecular dynamics simulations.
The Aβ solutions used were 12 nm monomeric dispersions [of Aβ40‐HiLyteFluor488 (g‐Aβ) and/or Aβ40‐HiLyteFluor647 (r‐Aβ)] as concluded from analytical ultracentrifugation and supported by three‐dimensional diffusion coefficients of Aβ in solution (see the Supporting Information (SI)). Oligomerization of Aβ was detected by monitoring changes in its lateral diffusion coefficient (D 2D) using Z‐scan fluorescence correlation spectroscopy (Z‐FCS), and by cross‐correlation FCS (FCCS). Z‐FCS provides precise and absolute diffusion coefficients, and overcomes positioning and calibration problems associated with FCS measurements in planar systems. Z‐FCS also resolves simultaneous two‐ and three‐dimensional diffusion.11 FCS data for Aβ were fitted to a model containing both two‐ and three‐dimensional diffusion to account for the fraction of peptides that remained in solution, unbound to the lipid membrane (SI Note 1). FCCS experiments, achieved by mixing (1:1) g‐ and r‐Aβ monomer solutions, have the ability to detect codiffusion of differently labeled peptides, that is, an oligomer. For proof that monomeric Aβ at nm concentrations binds to neutral lipid bilayers, two‐color Z‐FCS experiments were performed on giant unilamellar vesicles (GUVs) of DOPC (1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine), POPC (1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine), OSPC (1‐oleoyl‐2‐stearoyl‐sn‐glycero‐3‐phosphocholine), and DOPC/Chol mixtures. The g‐Aβ was found to bind and diffuse freely in the plane of all membranes. Moreover, its lateral diffusion was sensitive to membrane viscosity, which was simultaneously gauged via the fluorescent lipid tracer DiD (Figure S3). The lateral diffusion of Aβ did not vary, meaning that no oligomerization of the peptide occurs on simple model bilayers (Figure S3). The results also demonstrate that the labels do not induce oligomerization of Aβ by themselves, in agreement with literature.2b
Striving to emulate physiological conditions, we used lipid bilayers composed of relevant lipids of the neuronal plasma membrane (DOPC, Chol, Sph, and GM1). Special care was taken to avoid the liquid ordered (Lo)/liquid disordered (Ld) phase separation since the Lo phase of model membranes does not seem physiologically relevant.10 The ternary lipid bilayers [DOPC, 25 mol % Chol and (5, 8, 10) mol % of Sph] are below the phase separation point according to the phase diagram (SI Note 2). Nonetheless, we applied a fluorescence lifetime Förster resonance energy transfer (FLIM‐FRET) approach that allows determination of lipid domain sizes at the nanometer scale12 in order to assess the existence of phase separation below the optical resolution limit. In this method, the donor fluorescence decay is obtained from FLIM data and analyzed using Monte Carlo simulations (SI Note 3).13 Measurements of donor–acceptor pair FL‐ and 564/570‐ bodipy‐head‐labeled GM1 molecules (g‐ and r‐GM1) were performed and analyzed as reported previously (SI Note 3).12 The g‐/r‐GM1 do not cluster on their own (Table 1).
Table 1.
Results from Monte Carlo simulations of FLIM‐FRET data using the donor–acceptor pair FL and 564/570‐bodipy‐head‐labeled GM1. Lipid components indicated as mol %. All compositions contained 2 % biotinylated lipid for immobilization of GUVs. The term “domain” is used in reference to simulation terminology.
| DOPC [%] | Sph [%] | Chol [%] | GM1 label [%] | Extra GM1 [%] unlabeled | Domain radius [nm] | Domain area [%] |
|---|---|---|---|---|---|---|
| 100 | 0 | 0 | 1 | 0 | homogeneous | |
| 75 | 0 | 25 | 1 | 0 | homogeneous | |
| 95, 92 | 5, 8 | 0 | 1 | 0 | homogeneous | |
| 90 | 10 | 0 | 1 | 0 | 8±1[a] 12±3[a] | 37±10[a] 55±5[a] |
| 70, 67, 65 | 5, 8, 10 | 25 | 1 | 0 | 9±1 | 45±5 |
| 100 | 0 | 0 | 1 | 1, 2, 4 | 6±1 | 40±10 |
| 75 | 0 | 25 | 1 | 1, 2, 4 | 6±1 | 40±10 |
| 70, 67, 65 | 5, 8, 10 | 25 | 1 | 1, 2, 4 | 26±2 | 30±5 |
[a] Two global minima obtained.
FLIM‐FRET data revealed heterogeneities of 9 nm average radius in the ternary mixtures of DOPC/Chol/Sph (Figure S5a). Their size does not vary with increase of Sph content (Table 1). FLIM‐FRET experiments of the donor–acceptor pair g‐GM1 and DiD reported no segregation of the “Ld‐marker” DiD from the nanoheterogeneities (Figure S6 a). This indicates there is no significant Lo/Ld phase separation driving apart DiD and g‐GM1 (which have different affinities to Lo phase). Moreover, Z‐FCS measurements show that diffusion of DiD senses the increase of Sph content and mobility of both DiD and g‐GM1 is significantly higher than mobility in the Lo phase11 (Figure 1 a). In conclusion, even though the DOPC/Chol/Sph bilayers contain nanoheterogeneities, the g‐GM1 and DiD FLIM‐FRET and FCS results show no evidence of a nanoscopic Lo phase.
Figure 1.

a) Lateral diffusion coefficients, D 2D, of membrane‐bound Aβ (g‐Aβ) and lipid tracers (DiD and g‐GM1) in DOPC/Chol/Sph GUVs containing 0 or 4 % added GM1. A 2D component model describes well the diffusion of Aβ and lipid tracers. D 2D of Aβ decreases with time (green arrow symbols; see next panel) in DOPC/Chol/Sph bilayers, indicating oligomerization of Aβ. In the quaternary compositions [(DOPC/Chol/Sph)+4 %GM1)] no changes of Aβ diffusion are observed. b) Time evolution of D 2D of membrane‐bound Aβ (t“; 0 h, addition of Aβ monomers). DOPC/Chol/Sph compositions: circles (70:25.5); triangles (67:25:8); squares (65:25:10). Each point is the weighted average of D 2D results obtained from at least five independent two‐color Z‐FCS measurements (each composed of 15–20 scans). Error bars are the standard deviation within the sample of D 2D results obtained for each composition. Where D 2D of Aβ varies with time, error bars for Aβ values are the standard deviation obtained from the fitting procedure of 15 scans obtained via Z‐FCS.
Aβ oligomerized spontaneously on DOPC/Chol/Sph GUVs. A time‐dependent change in the diffusion of bound Aβ is found only in, and for all, Sph‐containing bilayers (Figure 1). The variation of diffusion with time (Figure 1 b) resembles a typical profile of aggregation phenomena.14 FCCS experiments were performed to corroborate oligomerization as the cause of the observed decrease of Aβ’s diffusion coefficient with time. After addition of the monomeric g‐/r‐Aβ mixture to DOPC/Chol/Sph membranes, the cross‐correlation function (Gx) amplitude increased with time (Figure 2). Such data imply formation of hybrid oligomers of g‐/r‐Aβ and show that the probability of finding oligomers increases with time. The cross‐correlation functions are not all parallel to each other (Figure 2 b), suggesting a heterogeneous population of oligomers. Likewise, FCCS experiments were performed on GUVs composed of DOPC and Sph (5, 8, 10 mol %) also below the phase‐separation point (SI Note 2). In the binary systems, FLIM‐FRET did not resolve heterogeneities below 10 mol % of Sph (Table 1). Cross‐correlation results show that Aβ spontaneously oligomerizes in DOPC/Sph bilayers as opposed to pure DOPC, and DOPC/Chol membranes (Figure 2, Figures S3 and S4). As a parallel to the DOPC/Sph binary system, we used bilayers of DOPC/DSPC (1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine) under the phase‐separation point (SI Note 2). DSPC is a glycerol lipid analogue of Sph (i.e. has the same headgroup but the similar fatty acid chains are bridged by a glycerol moiety). Cross‐correlation experiments with DSPC‐containing bilayers revealed no oligomerization of Aβ (Figure 2).
Figure 2.
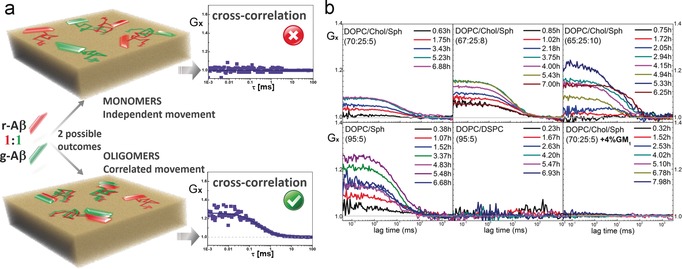
a) Illustration of cross‐correlation experiments using a 1:1 mixture of r‐ and g‐Aβ monomers. Top: Membrane‐bound r‐ and g‐Aβ monomers diffuse independently; thus red and green signal fluctuations are not correlated, resulting in null cross‐correlation function (Gx), that is, Gx=1. Bottom: Formation of Aβ oligomers implies co‐diffusion of peptides. If oligomers are formed of both r‐ and g‐Aβ, red and green signal fluctuations become correlated, resulting in positive cross‐correlation, that is, Gx>1. b) Summary of results. Top: DOPC/Chol/Sph membranes show positive cross‐correlation. Bottom left: DOPC/Sph bilayers show positive cross‐correlation, example DOPC/Sph (95:5). Bottom middle: DOPC/DSPC membranes show no cross‐correlation, example DOPC/DSPC (95:5). Bottom right: DOPC/Chol/Sph+GM1 bilayers show no cross‐correlation, example [DOPC/Chol/Sph (70:25:5)+4 %GM1].
Our findings show a specific role of Sph in the regulation of Aβ’s oligomerization. Devanathan et al.4a had suggested that aggregation of membrane‐bound Aβ may be promoted by Sph in gel phase, gel/Lo, or Lo/Ld phase separated supported lipid bilayers (using μm concentrations of Aβ, likely in oligomeric states). Here, for the first time, Sph is shown to induce oligomerization of membrane bound Aβ monomers (at physiological relevant amounts) in Ld phase membranes containing transient nanoheterogeneities. The model systems preclude putative impact of Lo phase and support a direct impact of Sph on oligomerization.
It was suggested that clusters of GM1 are a membrane binding site for Aβ and that the binding of Aβ to GM1 could seed formation of amyloid fibrils.5 Kakio and co‐workers observed increased seeding and fast formation of Aβ fibrils on model membranes containing GM1.15 The used bilayers contained over 20 mol % of GM1, meaning the bilayer surface was covered by the sugar headgroups of the ganglioside.16 Nevertheless, at physiological GM1 concentrations (≤4 mol %) a bilayer is one fluid phase with “island‐like” structures enriched in GM1.16 We have shown previously12 that GM1 (1–4 mol %) forms clusters of 5–7 nm radius in DOPC and DOPC/Chol (70:30) free‐standing bilayers. Such GM1 clusters contain high amounts of DOPC and Chol, and do not exhibit Lo phase characteristics. Thus, use of physiological levels of GM1 in in vitro studies is important as high levels of GM1 may mask the membrane surface.
Progressing in bilayer composition, an additional 4 mol % of GM1 was added to the DOPC/Chol/Sph systems described previously. The addition of GM1 led to an increase in radius of the heterogeneities present in the bilayer from 9 to 26 nm (Table 1, Figure S5 b). FLIM‐FRET experiments with g‐GM1 and DiD reported no segregation of DiD from the GM1‐containing heterogeneities (Figure S6 b). The addition of GM1 slowed diffusion of g‐GM1 and DiD (Figure 1), consistent with an increase in order of the nanoheterogeneities, which is related to the decrease in their calculated area from 45 % to 30 % (Table 1). As in DOPC/Chol/Sph bilayers, the g‐GM1 and DiD FLIM‐FRET and FCS results show no evidence of nanoscopic Lo phase.
After incubation of Aβ with the quaternary lipid membranes notably there wasno evidence of oligomerization. In the DOPC/Chol/Sph membranes containing extra 4 mol % of GM1, the diffusion coefficient of Aβ is stable over the course of the experiments (9 h) (Figure 1). Moreover, FCCS results confirm no oligomerization of Aβ in ganglioside‐containing bilayers (Figure 2). The behavior of Aβ was also monitored in GUVs of DOPC and DOPC/Chol (75:25) containing 2 and 4 mol % of GM1 in the form of small clusters (radius≈6 nm, Table 1).12 Such fluid nanoscale GM1 clusters have no effect on the lateral diffusion of Aβ and no oligomerization occurs (Figure S4). Contrary to reports in which high concentrations of GM1 are used,5, 15 low and closer to physiological amounts of the ganglioside do not seed oligomerization of Aβ. Moreover, the presence of GM1 de facto prevents the spontaneous oligomerization of Aβ observed in DOPC/Chol/Sph membranes.
In order to understand the underlying mechanism of the triggering of oligomerization of Aβ by Sph at the molecular level, we performed all‐atom molecular dynamics simulations of the peptide in lipid bilayers of DOPC and DOPC/Sph (90:10) (see Figures S7–S19). Figure S8 depicts the final configurations of Aβ after 1.5 μs in eight independent simulations. The C‐terminal of Aβ seems to have a higher tendency to form a β‐sheet in the presence of Sph (Figure S14). The β‐sheet conformations of Aβ (from the membrane with Sph) fully or partially unfolded within 1 μs when placed in a pure DOPC membrane (Figure S15). Such instability of the β‐sheet conformation indicates the role of Sph in inducing the conformational change.
Our simulations of Aβ in DOPC bilayers containing GM1 demonstrate a strong interaction between Aβ and the ganglioside, in agreement with previous studies17 (Figure S20). Aβ bound specifically to the sugar moiety of GM1 with the hydrogen‐bonded histidine residue playing an important role.17b The strong binding of Aβ to GM1 and the involvement of the β‐sheet residues in the binding might explain why Aβ is unable to oligomerize in DOPC/GM1 (and DOPC/Chol/GM1) membranes. The sequestering of the peptide by GM1 would also explain why addition of low concentrations of GM1 to DOPC/Chol/Sph membranes effectively inhibits the oligomerization of Aβ. In contrast, high surface densities of GM1 (>20 mol %) can accelerate the rate of aggregation.5 At such ganglioside concentrations the bilayer is covered by the sugar heads of GM1;16 thus the strong interaction between Aβ and ganglioside can lead to high surface concentrations of peptide and accelerate aggregation simply due to general surface effects.18
In summary, we demonstrate that Sph is a specific trigger of oligomerization of Aβ40 and that the effect is counteracted by physiological concentrations of GM1. The peptide oligomerizes in DOPC/Chol/Sph and DOPC/Sph bilayers, but not in DOPC, DOPC/Chol, or DOPC/DSPC bilayers (Figure 3 a). The presence of Sph creates significant changes in the bilayers’ properties, as shown by our experiments and simulations (Figures S16–S19). Interestingly, DOPC/Chol/Sph ternary mixtures exhibit transient nanoheterogeneities despite being in the Ld phase. Simulations show that Aβ seemingly adopts a conformation with a higher amount of β‐sheet structure in the DOPC/Sph bilayer compared to pure DOPC membrane. Knowing β‐sheet structures are important in supramolecular assembly, a conformational change of Aβ caused by Sph can explain the dramatic differences in oligomerization: virtually zero in DOPC, DOPC/Chol, or DOPC/DSPC bilayers, and occurring within few hours in membranes containing Sph (Figure 3 a,b).
Figure 3.
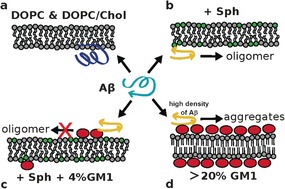
Proposed model. a) Aβ does not oligomerize when bound to DOPC and DOPC/Chol bilayers. The C‐terminus of Aβ has no secondary structure (dark blue representation). b) Oligomerization of Aβ occurs in the presence of Sph, for both DOPC/Sph and DOPC/Chol/Sph membranes. The C‐terminus of monomeric Aβ acquires β‐sheet structure (yellow representation) in bilayers containing Sph. c) Binding of Aβ to the headgroup of GM1 (red ellipses) sequesters the peptide and prevents it from forming oligomers. d) High density of both GM1 and Aβ facilitates aggregation of the peptide and fibril formation (from literature),5 which can be explained by generic surface effects.18
The binding of Aβ to GM1 can accelerate or decelerate oligomerization depending on ganglioside concentration.18a Full coverage of the membrane by negatively charged GM1 sugar heads16 and reduction of dimensionality, from 3D to 2D, can increase the rate of aggregation due to general surface effects18b,18c (Figure 3 d). However, at low GM1 levels—like the physiological amounts used (2–4 %)—the binding of Aβ to GM1 does not cause aggregation of the peptide. In fact, the presence of GM1 inhibits Aβ’s oligomerization (Figure 3 c). The sequestering of Aβ by the ganglioside seems to be the cause for the arrest of spontaneous oligomerization, which is otherwise observed in Sph‐containing membranes.
Our observations are the first molecular evidence for GM1 as an inhibitor of the oligomerization of Aβ40 and thus bring forward brand‐new insight into molecular mechanism(s) possibly involved in AD. Our findings suggest that decreasing GM1 levels (reported to occur with age)7, 8a, 19 could lead to reduced protection from the oligomerization‐triggering effect of Sph and thus contribute to spark AD's onset. The provided molecular insights support reports on the neuroprotective effects of GM1 in cell cultures and rat models of AD.9, 20 Our results help to rationalize data found in vivo and can help build the basis for a better understanding of amyloid diseases.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support: GACR (P208/12/G016; 14‐12598S), MEYS, European Regional Development Fund (CZ.1.05/1.1.00/02.0068 CEITEC), CAS for Praemium Academie award. Computational resources provided by MetaCentrum (LM2010005) and CERIT‐SC (CZ.1.05/3.2.00/08.0144). We thank M. Šulc for analytical ultracentrifugation results, J. Humpolíčková for contributing to FRET experiments, M.Manna for initial parametrization of Aβ and GM1, and A. Hermetter and M. Cebecauer for helpful discussions.
M. Amaro, R. Šachl, G. Aydogan, I. I. Mikhalyov, R. Vácha, M. Hof, Angew. Chem. Int. Ed. 2016, 55, 9411.
Contributor Information
Dr. Mariana Amaro, Email: amaro@jh-inst.cas.cz.
Prof. Martin Hof, Email: hof@jh-inst.cas.cz.
References
- 1.
- 1a. Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M., Nature 2002, 416, 507–511; [DOI] [PubMed] [Google Scholar]
- 1b. Shankar G. M., Li S., Mehta T. H., Garcia-munoz A., Nina E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A. et al., Nat. Med. 2008, 1 4, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Amaro M., Birch D. J. S., Rolinski O. J., Phys. Chem. Chem. Phys. 2011, 13, 6434–6441; [DOI] [PubMed] [Google Scholar]
- 2b. Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., Klenerman D., Nat. Struct. Mol. Biol. 2011, 19, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Almeida R. F. M., Fedorov A., Prieto M., Biophys. J. 2003, 85, 2406–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Devanathan S., Salamon Z., Lindblom G., Gröbner G., Tollin G., FEBS J. 2006, 273, 1389–1402; [DOI] [PubMed] [Google Scholar]
- 4b. Chi E. Y., Ege C., Winans A., Majewski J., Wu G., Kjaer K., Lee K. Y. C., Proteins Struct. Funct. Bioinf. 2008, 72, 1–24; [DOI] [PubMed] [Google Scholar]
- 4c. Williams T. L., Johnson B. R. G., Urbanc B., Jenkins A. T. A., Connell S. D. A., Serpell L. C., Biochem. J. 2011, 439, 67–77. [DOI] [PubMed] [Google Scholar]
- 5. Yanagisawa K., J. Neurochem. 2011, 116, 806–812. [DOI] [PubMed] [Google Scholar]
- 6. Ledeen R. W., J. Supramol. Struct. 1978, 8, 1–17. [DOI] [PubMed] [Google Scholar]
- 7. Tettamanti G., Anastasia L. in Handb. Neurochem. Mol. Neurobiol. (Eds.: A. Lajtha, G. Tettamanti, G. Goracci), Springer, Boston, 2010, pp. 99–171. [Google Scholar]
- 8.
- 8a. Ariga T., McDonald M. P., Yu R. K., J. Lipid Res. 2008, 49, 1157–1175; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Mocchetti I., Cell. Mol. Life Sci. 2005, 62, 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Kreutz F., Frozza R. L., Breier A. C., Oliveira V. A., Horn A. P., Pettenuzzo L. F., Netto C. A., Salbego C. G., Trindade V. M. T., Neurochem. Int. 2011, 59, 648–655; [DOI] [PubMed] [Google Scholar]
- 9b. Yang R., Wang Q., Min L., Sui R., Li J., Liu X., Neurol. Sci. 2013, 34, 1447–1451; [DOI] [PubMed] [Google Scholar]
- 9c. Kreutz F., Scherer E. B., Ferreira A. G. K., Petry F. D. S., Pereira C. L., Santana F., de Souza Wyse A. T., Salbego C. G., Trindade V. M. T., Neurochem. Res. 2013, 38, 2342–2350. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Göttfert F., Wurm C. A., Mueller V., Berning S., Cordes V. C., Honigmann A., Hell S. W., Biophys. J. 2013, 105, L01–L03; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Sevcsik E., Schütz G. J., BioEssays 2016, 38, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Macháň R., Hof M., Biochim. Biophys. Acta Biomembr. 2010, 1798, 1377–1391. [DOI] [PubMed] [Google Scholar]
- 12. Sachl R., Amaro M., Aydogan G., Koukalová A., Mikhalyov I. I., Boldyrev I. A., Humpolíčková J., Hof M., Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 850–857. [DOI] [PubMed] [Google Scholar]
- 13. Amaro M., Sachl R., Jurkiewicz P., Coutinho A., Prieto M., Hof M., Biophys. J. 2014, 107, 2751–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cohen S. I. A., Vendruscolo M., Welland M. E., Dobson C. M., Terentjev E. M., Knowles T. P. J., J. Chem. Phys. 2011, 135, 065105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Kakio A., Nishimoto S. I., Yanagisawa K., Kozutsumi Y., Matsuzaki K., J. Biol. Chem. 2001, 276, 24985–24990; [DOI] [PubMed] [Google Scholar]
- 15b. Kakio A., Nishimoto S., Yanagisawa K., Kozutsumi Y., Matsuzaki K., Biochemistry 2002, 41, 7385–7390. [DOI] [PubMed] [Google Scholar]
- 16. Sagle L. B., Ruvuna L. K., Bingham J. M., Liu C., Cremer P. S., Van Duyne R. P., J. Am. Chem. Soc. 2012, 134, 15832–15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Devarajan S., Sharmila J. S., J. Mol. Liq. 2014, 195, 59–64; [Google Scholar]
- 17b. Manna M., Mukhopadhyay C., PLoS One 2013, 8, e71308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Vácha R., Linse S., Lund M., J. Am. Chem. Soc. 2014, 136, 11776–11782; [DOI] [PubMed] [Google Scholar]
- 18b. Minton A. P., Biophys. Chem. 2000, 86, 239–247; [DOI] [PubMed] [Google Scholar]
- 18c. Minton A. P., Biophys. J. 2001, 80, 1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kracun I., Rosner H., Drnovsek V., Vukelic Z., Cosovic C., Trbojevic-Cepe M., Kubat M., Neurochem. Int. 1992, 20, 421–431. [DOI] [PubMed] [Google Scholar]
- 20. Sokolova T. V., Zakharova I. O., Furaev V. V., Rychkova M. P., Avrova N. F., Neurochem. Res. 2007, 32, 1302–1313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary