

Abstract
This article describes a rapid UPLC‐MS/MS method to quantitate novel bile acids in biological fluids and the evaluation of their diagnostic potential in Niemann‐Pick C (NPC). Two new compounds, NPCBA1 (3β‐hydroxy,7β‐N‐acetylglucosaminyl‐5‐cholenoic acid) and NPCBA2 (probably 3β,5α,6β‐trihydroxycholanoyl‐glycine), were observed to accumulate preferentially in NPC patients: median plasma concentrations of NPCBA1 and NPCBA2 were 40‐ and 10‐fold higher in patients than in controls. However, NPCBA1 concentrations were normal in some patients because they carried a common mutation inactivating the GlcNAc transferase required for the synthesis of this bile acid. NPCBA2, not containing a GlcNAc moiety, is thus a better NPC biomarker.
Keywords: bile acids, Biomarkers, GlcNAc transferase, Niemann‐Pick C, screening, UPLC‐MS/MS
Abbreviations
GC‐MS, gas chromatography‐mass spectrometry
LC‐MS, liquid chromatography‐mass spectrometry
NPC, Niemann‐Pick C
SRM, single reaction monitoring
UPLC‐MS/MS, ultra performance liquid chromatography linked to a tandem mass spectrometer
Niemann‐Pick C (NPC) is a severe autosomal recessive lipid storage disorder that can give rise to liver disease during infancy 1, 2 and produce neurological dysfunction starting in childhood/early adult life. To date, mutations in two genes have been identified: NPC1 (OMIM 607623), in ~ 95% of NPC patients, and NPC2 (OMIM 601015) in 5% 3, 4. Although the functions of NPC1 and NPC2 proteins have not been completely elucidated, their critical involvement in cholesterol export from the lysosome/late endosomal cellular compartment has been recognized 5, 6, 7, 8. Indeed, the abnormal trafficking and metabolism of cholesterol evident in NPC has proved to be useful in the diagnosis of the disorder. Lysosomal accumulation of unesterified cholesterol can be detected by filipin staining of cultured fibroblasts 9 and cholesterol is converted into auto‐oxidation products (oxysterols; 7‐oxocholesterol and cholestane‐3β,5α,6β‐triol) whose levels are elevated in plasma 10, 11, 12, 13, 14, 15. Filipin staining requires culturing of fibroblasts and so is not a viable rapid screening test. For measurement of oxysterols, blood needs to be taken into EDTA tubes and the plasma needs to be rapidly separated. Delays result in the ex vivo conversion of cholesterol to oxysterols with the potential for false positive results. Most methods for oxysterol quantitation require derivatization for analysis by liquid chromatography‐ (LC‐MS) or gas chromatography‐mass spectrometry (GC‐MS). There is a need for a simple test using samples that are easily sent to the laboratory such as dried blood spots or plasma spotted onto paper – samples that can be sent by post even from another country. This could be used at the earliest stages of the disease process, for example, infantile cholestasis or childhood splenomegaly, which often precede the characteristic neurological symptoms, including ataxia, vertical supranuclear gaze palsy, dystonia and dementia 16, 17, 18. In addition, a rapid and simple method could be used to screen patients with the first hint of neurological dysfunction or psychiatric disease, a nonspecific presentation of NPC in adolescents and adults that often remains undiagnosed and, consequently, untreated for months or years 19, 20.
A prompt diagnosis of NPC is important because, in Europe, a licensed treatment (Miglustat) is available for this disorder 21, 22, 23 and studies on animal models indicate that early treatment offers the best chance of limiting neurological damage 24. Other treatments for NPC are currently undergoing clinical trials (2‐hydroxypropyl‐β‐cyclodextrin 25, histone deacetylase inhibitors 26 and arimoclomol).
We have investigated bile acids as potential biomarkers for NPC. Alvelius and coworkers, in 2001, demonstrated the presence of a unique set of unusual bile acids in the urine of a cholestatic infant with NPC 27. These were unsaturated C24 bile acids (5‐cholenoic acids) with a sulfated 3β‐hydroxyl group, a 7β‐hydroxyl group, either free or conjugated with N‐acetylglucosamine (GlcNAc) and the C24 carboxyl group either free or conjugated with glycine or taurine. The discovery in 2010, of increased production of 7‐oxocholesterol in NPC, probably as a result of free radical attack on accumulating cholesterol 10, provided a possible explanation for the origin of these ‘NPC bile acids’; they could be produced by hepatic metabolism of 7‐oxocholesterol, probably via 3β,7β‐dihydroxy‐5‐cholenoic acid and its 7β‐N‐acetylglucosamine conjugate. Subsequently, Maekawa et al. developed methods for quantitation of three NPC bile acids in urine, having synthesized the reference compounds, 3β‐sulfoxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid and its glycine and taurine conjugates 28, 29. Initial results suggested that the urinary excretion of these pooled metabolites was 400‐fold greater than controls. Unfortunately, further analyses have shown overlap between NPC and controls, limiting the use of the test in the diagnostic setting (M. Maekawa, unpublished data). We considered a possible cause of failure to detect the bile acids conjugated with N‐acetyl‐glucosamine in NPC patients. A common homozygous polymorphism/mutation in the UGT3A1 gene, c.T361G, leads to an amino acid substitution (p.C121G) and absence of activity of the encoded 7β‐hydroxy bile acid UDP N‐acetylglucosaminyl transferase 30, 31: homozygosity for c.T361G is present in about 20% of Asian and Caucasian populations.
7‐Oxocholesterol is not the only oxysterol produced by nonenzymatic auto‐oxidation of accumulating cholesterol in NPC. Another oxysterol whose production is greatly increased is cholestane‐3β,5α,6β‐triol 10. We reasoned that this oxysterol would probably also be metabolized to a bile acid in the liver, producing an isomer of glycocholic and/or taurocholic acid. Thus, we investigated the use of ultra performance liquid chromatography linked to a tandem mass spectrometer (UPLC‐MS/MS) to quantitate, in body fluids, the following potential biomarkers for NPC: (a) bile acids derived from 7‐oxocholesterol (looking at nonsulfated and sulfated bile acids for the first time) and (b) an unusual isomer of glycocholic acid (probably derived from cholestane‐3β,5α,6β‐triol).
Materials and methods
Chemicals and reagents
Cholic acid (CA), chenodeoxycholic acid (CDCA), ursodeoxycholic acid (UDCA), hyocholic acid (HCA), hyodeoxycholic acid (HDCA), deoxycholic acid (DCA), lithocholic acid (LCA) and the glycine and taurine conjugates of these bile acids (GCA, TCA, GCDCA, TCDCA, GUDCA, TUDCA, GDCA, TDCA, GLCA, TLCA) were purchased from Steraloids (Newport, RI, USA)), and all the deuterium labelled internal standards used (D4‐CA, D4‐CDCA, D4‐GCA, D5‐TCA, D4‐GCDCA and D4‐TCDCA) were obtained from CDN Isotopes (Pointe‐Claire, QC, Canada).
HPLC‐grade methanol was from Merck, water was purified using a Milli‐Q system (Millipore UK Ltd, Watford, ND, USA) and formic acid was from Fluka. 3β‐Sulfoxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid and its glycine and taurine conjugates were synthesized 28.
Instrumentation and experimental conditions
The UPLC‐MS/MS instrument consisted of a Waters ACQUITY UPLC coupled to a Xevo TQ‐S triple quadrupole mass spectrometer with an electrospray ionization source. The mass spectrometer was operated in negative ion mode and data were acquired using masslynx V4.1 software. Chromatographic separations were achieved using a Waters ACQUITY UPLC™ BEH (Waters UK, Herts, UK) C18 column (1.7 μm, 2.1 × 50 mm) maintained at 40 °C. Binary gradient profiles were developed using water with 0.01% formic acid (A) and methanol (B) at a flow rate of 500 μL·min−1. Separations were conducted under the following chromatographic conditions: 60% solvent A for 0.5 min, decreased to 1% over 1 min, maintained for 1 min at 1% before being increased to 60% over 0.1 min. Column equilibration time was 0.9 min, with a total run time of 3.5 min. The injection volume was 20 μL. Mass spectrometric conditions were as follows: capillary voltage 2.7 kV, desolvation temperature 600 °C, cone gas flow 150 L·h−1, desolvation gas flow 1200 L·h−1, collision gas flow 0.15 L·h−1 and nebulizer gas flow 7 bar. Dwell time was set as 3 ms for each analyte. The quantitation of the different analytes was then performed using the single reaction monitoring (SRM) parameters described in Table 1.
Table 1.
Retention time, cone voltage, collision energy, precursor and product ion used in identification of normal and abnormal bile acids detected in healthy participants and in patients with inborn errors of metabolism
| Bile acida | Conjugationb [Abbreviation] | Retention time (min) | Cone voltage (V) | Collision energy (V) | Precursor ion (m/z) | Product ion (m/z) |
|---|---|---|---|---|---|---|
| Chenodeoxycholic acid (3α,7α‐diOH‐5β) | Gly [GCDCA] | 1.76 | 88 | 31 | 448.4 | 74.0 |
| D4‐chenodeoxycholic acid | Gly [D4‐GCDCA] | 1.76 | 88 | 31 | 452.4 | 74.0 |
| Deoxycholic acid (3α,12α‐diOH‐5β) | Gly [GDCA] | 1.80 | 88 | 33 | 448.4 | 74.0 |
| Ursodeoxycholic acid (3α,7β‐diOH‐5β) | Gly [GUDCA] | 1.59 | 88 | 31 | 448.4 | 74.0 |
| Cholic acid (3α,7α,12α‐triOH‐5β) | Gly [GCA] | 1.67 | 86 | 33 | 464.4 | 74.0 |
| D4‐cholic acid | Gly [D4‐GCA] | 1.67 | 86 | 33 | 468.4 | 74.0 |
| Lithocholic acid (3α‐OH‐5β) | Gly [GLCA] | 1.86 | 88 | 31 | 432.3 | 74.0 |
| Chenodeoxycholic acid (3α,7α‐diOH‐5β) | Tau [TCDCA] | 1.64 | 110 | 59 | 498.4 | 80.0 |
| D4‐chenodeoxycholic acid | Tau [D4‐TCDCA] | 1.64 | 110 | 59 | 502.4 | 80.0 |
| Deoxycholic acid (3α,12α‐diOH‐5β) | Tau [TDCA] | 1.65 | 116 | 59 | 498.4 | 80.0 |
| Ursodeoxycholic acid (3α,7β‐diOH‐5β) | Tau [TUDCA] | 1.49 | 110 | 59 | 498.4 | 80.0 |
| Cholic acid (3α,7α,12α‐triOH‐5β) | Tau [TCA] | 1.56 | 116 | 59 | 514.4 | 80.0 |
| D5‐cholic acid | Tau [D5‐TCA] | 1.56 | 116 | 59 | 519.4 | 80.0 |
| Lithocholic acid (3α‐OH‐5β) | Tau [TLCA] | 1.72 | 110 | 59 | 482.3 | 80.0 |
| Chenodeoxycholic acid (3α,7α‐diOH‐5β) | None [CDCA] | 1.87 | 86 | 5 | 391.4 | 391.4 |
| D4‐chenodeoxycholic acid | None [D4‐CDCA] | 1.87 | 86 | 5 | 395.4 | 395.4 |
| Deoxycholic acid (3α,12α‐diOH‐5β) | None [DCA] | 1.88 | 86 | 5 | 391.4 | 391.4 |
| Ursodeoxycholic acid (3α,7β‐diOH‐5β) | None [UDCA] | 1.72 | 86 | 5 | 391.4 | 391.4 |
| Cholic acid (3α,7α,12α‐triOH‐5β) | None [CA] | 1.77 | 86 | 5 | 407.4 | 407.4 |
| D4‐cholic acid | None [D4‐CA] | 1.77 | 86 | 5 | 411.4 | 411.4 |
| Lithocholic acid (3α‐OH‐5β) | None [LCA] | 1.97 | 86 | 5 | 375.3 | 375.3 |
| Hyocholic acid (3α,6α,7α‐triOH‐5β) | None [HCA] | 1.72 | 86 | 5 | 407.4 | 407.4 |
| Hyodeoxycholic acid (3α,6α‐diOH‐5β) | None [HDCA] | 1.75 | 86 | 5 | 391.4 | 391.4 |
| Characteristic of 3β‐hydroxy‐Δ5‐C27‐steroid dehydrogenase deficiency | ||||||
| 3β,7α,12α‐triOH‐Δ5 | None | 1.61 | 86 | 5 | 405.3 | 405.3 |
| 3β,7α‐diOH‐Δ5 | Gly | 1.58 | 88 | 31 | 446.0 | 74.0 |
| 3β,7α,12α‐triOH‐Δ5 | Gly | 1.47 | 86 | 33 | 462.0 | 74.0 |
| 3β,7α‐diOH‐Δ5 | Sulf | 1.58 | 87 | 32 | 469.0 | 97.0 |
| 3β,7α,12α‐triOH‐Δ5 | Sulf | 1.47 | 110 | 59 | 485.0 | 97.0 |
| 3β,7α‐diOH‐Δ5 | Sulf, Gly | 1.47 | 110 | 59 | 526.0 | 97.0 |
| 3β,7α,12α‐triOH‐Δ5 | Sulf, Gly | 1.37 | 86 | 33 | 542.0 | 97.0 |
| 3β,7α‐diOH‐Δ5 | GlcNAc | 1.62 | 80 | 33 | 591.9 | 389.0 |
| Characteristic of Δ4‐3‐oxosteroid 5β‐reductase deficiency | ||||||
| 7α‐OH‐3‐oxo‐Δ4 | Gly | 1.21 | 86 | 33 | 444.0 | 74.0 |
| 7α,12α‐diOH‐3‐oxo‐Δ4 | Gly | 1.46 | 86 | 33 | 460.0 | 74.0 |
| 7α‐OH‐3‐oxo‐Δ4 | Tau | 1.55 | 110 | 59 | 494.0 | 80.0 |
| 7α,12α‐diOH‐3‐oxo‐Δ4 | Tau | 1.41 | 110 | 59 | 510.0 | 80.0 |
| Characteristic of oxysterol 7α‐hydroxylase deficiency | ||||||
| 3β‐OH‐α5 | Sulf, Gly | 1.70 | 86 | 33 | 510.0 | 74.0 |
| Characteristic of Niemann‐Pick C | ||||||
| 3β,7β‐diOH‐Δ5 | Gly | 1.65 | 86 | 33 | 446.0 | 74.0 |
| 3β,7β‐diOH‐Δ5 | GlcNAc | 1.59 | 80 | 33 | 591.9 | 389.0 |
| 3β,7β‐diOH‐Δ5 | GlcNAc, Gly | 1.44 | 88 | 31 | 649.4 | 74.0 |
| 3β,7β‐diOH‐Δ5 | GlcNAc, Tau | 1.33 | 116 | 59 | 699.4 | 80.0 |
| 3β,7β‐diOH‐Δ5 | Sulf, GlcNAc | 1.48 | 80 | 34 | 672.4 | 97.0 |
| 3β,5α,6β‐triOH | Gly | 1.42 | 86 | 33 | 464.4 | 74.0 |
| 3β,7β,ζ‐triOH‐Δ5 | Gly | 1.49 | 86 | 33 | 462.4 | 74.0 |
Positions of hydroxyl groups and double bonds in precursor bile acid in brackets. 5β is 5β‐cholanoic acid, Δ5 is 5‐cholenoic acid, Δ4 is 4‐cholenoic acid.
Gly, glycine; Taur, taurine, Sulf, sulfate; GlucA, glucuronic acid; GlcNAc, N‐acetylglucosamine. Abbreviation in square brackets.
Preparation of stock solutions
Stock solutions of standard bile acids (50 μm) were prepared in methanol before being combined and further diluted into a series of working solutions, used for the calibration standards (prior to the addition of the deuterated internal standards, final concentration: 20 nm). Calibration curves were plotted on the day of analysis across the concentration range of 0.1–250 nm for dried plasma and blood spots, and 0.25–250 μm for urine.
Clinical samples
Plasma samples, stored at − 80 °C, were available from previous NPC biomarker studies 32. The NPC1 genotypes of the patients included in the study are shown in Table 1 of the supporting information. For seven of the NPC1 patients, exome sequencing data were available, allowing determination of their status with regard to the c.T361G (pC121G) mutation/polymorphism in UGT3A1 (two homozygous for the mutation, two heterozygotes and three wild‐type). The samples included untreated NPC1 patients (mutations listed in Table S1), NPC1 heterozygotes and age‐matched controls. They were aged between 6 months and 54 years, with total clinical score 20 between 0 and 35 and annual severity increment score (ASIS) 33 between 0 and 6.21. New plasma, blood spot and random urine samples were obtained from patients attending clinics at Great Ormond Street Hospital and at National Hospital for Neurology and Neurosurgery (London). Ethical consent for biomarker discovery in NPC was obtained from NRES Committee South Central – Southampton A (Ref13/SC/0109) and informed consent was obtained from all the participants. Blood spots, plasma and random urine samples from patients with suspected or confirmed disorders of bile acid synthesis were analysed to evaluate the specificity and selectivity of the proposed test (Table 1).
Sample preparation
Ten microlitres of plasma was spotted onto neonatal screening cards (Whatman 903™; GE Healthcare Life Science, Buckinghamshire, UK). The whole sample was punched out and eluted by sonicating for 15 min with 300 μL of methanol containing the deuterated bile acid internal standards (concentration: 20 nm).
Blood was collected onto screening cards (Whatman 903™; GE Healthcare Life Science) and allowed to dry overnight prior to storage in a sealed bag with dessicant at − 80 °C. A 6 mm disc was punched out and sonicated as described for plasma spots above.
Fifty microlitres of urine was diluted with an equal volume of methanol containing 20 nm internal standards and the results were normalized to creatinine concentration 34.
Correlation between NPC bile acids and oxysterols in plasma
The correlation between the levels of NPCBA1, NPCBA2 and their oxysterol precursors (7‐oxocholesterol and cholestane‐3β,5α,6β‐triol) was investigated in eight samples from untreated NPC1 patients who had had oxysterols analysed on the same sample 10, 33.
Method validation
The method was fully validated, in accordance with the ICH Q2(R1) guidelines 35, by evaluating accuracy, precision, limit of detection (LOD), limit of quantitation (LOQ), linearity and matrix effect. Accuracy and precision were assessed using nine determinations, covering the selected concentration range (three concentrations/three replicates each), whereas LOD and LOQ were established by means of a method based on the calibration curve, where the standard deviation of y‐intercept was used as standard deviation. Matrix effect(s) were determined by comparing the linearity of working solutions prepared from extracted human blood/plasma spots, urine samples to linearity in methanol.
Statistical analysis
Statistical evaluation was performed on the data sets by Kruskal–Wallis test followed by Dunns, significance level = 0.05 (*P < 0.05; **P < 0.01; ***P < 0.001).
Results
Synthesis of 3β‐hydroxy‐7β‐N‐acetylglucosaminyl‐5‐cholenoic acid (NPCBA1) reference standard and hydrophilic NPCBA1 metabolites
We looked for evidence, in NPC samples, of 3β‐hydroxy‐7β‐N‐acetylglucosaminyl‐5‐cholenoic acid (which we called Niemann‐Pick C bile acid 1, NPCBA1). This compound was prepared by solvolysis 31 of the 3‐sulfate 28 and used as a reference standard for the UPLC‐MS/MS analyses. Solvolysis of 3β‐sulfoxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid 28 produced a bile acid with [M−H]− of m/z 592, consistent with the expected 3β‐hydroxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid and a main product ion of m/z 389, after the neutral loss of 203 mass units attributable to the cleavage of the O‐GlcNAc bond.
The glycine and taurine conjugates of 3β‐hydroxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid were also prepared by solvolysis of the respective sulfates 28. Their mass spectra were characterized by precursor ions of 649 and 699 m/z and main fragments of 74 and 80 m/z, due to the loss of glycine and taurine respectively.
Investigation of NPCBA1 accumulation in NPC1
NPCBA1 was one of the major abnormal bile acids detected in plasma from patients with NPC1. The difference between the mean plasma concentration in NPC1 patients and controls was highly significant (P < 0.001). Nevertheless, a considerable overlap was observed between some NPC1 patients, NPC1 heterozygotes and controls (Fig. 1A). For two NPC1 patients, in whom the GlcNAc‐conjugated bile acid was undetectable, whole exome sequencing (WES) data were available. These individuals were found to be homozygous for the inactivating missense variant p.C121G (c.T361G) in the UGT3A1 gene, which encodes the 7β‐hydroxy bile acid UDP N‐acetylglucosaminyl transferase 30. Conversely, analysis of WES data from five NPC1 patients whose NPCBA1 levels were found to be well above the control range, revealed that two of these individuals were heterozygotes for c.T361G while the others did not carry the c.T361G UGT3A1 variant.
Figure 1.
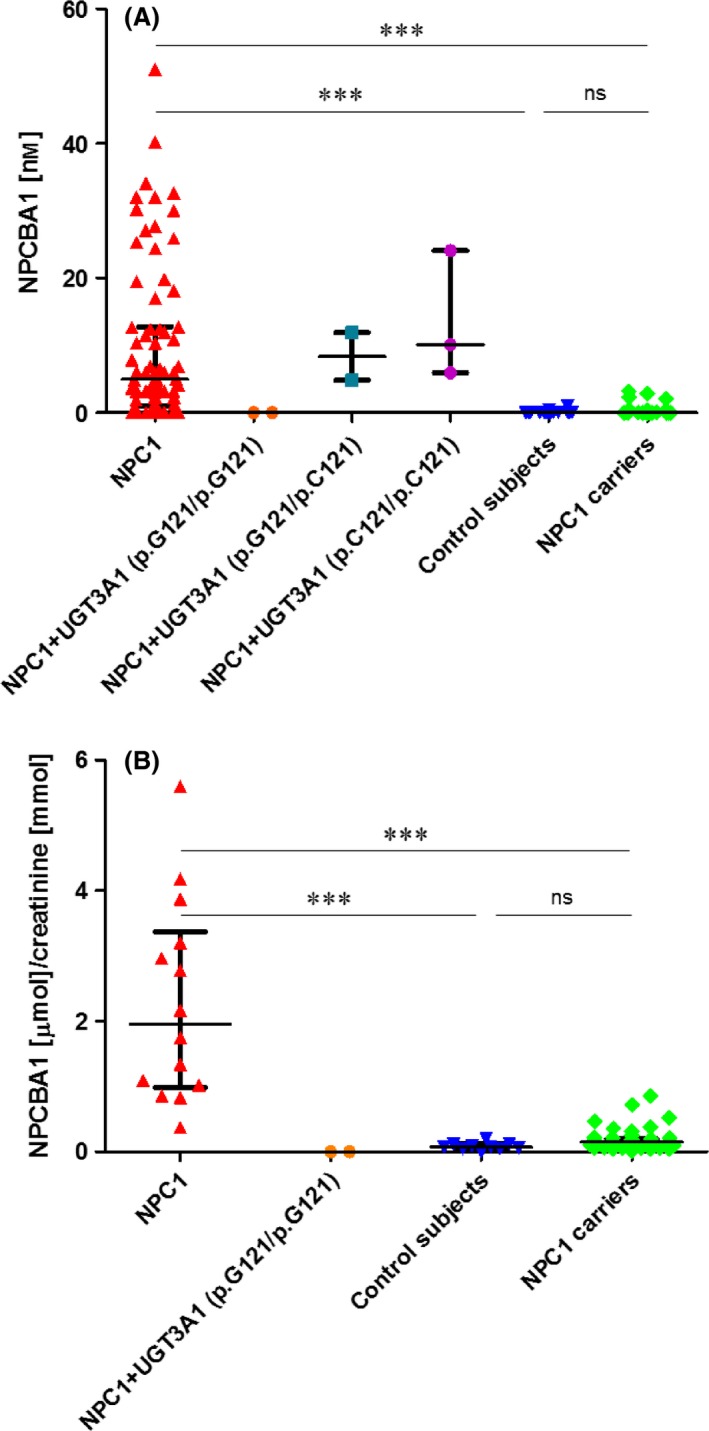
NPCBA1 in dried plasma spots (A) and random urine samples (B). (A) From left to right: plasma samples from individuals with NPC1 disease (confirmed by genotyping) but with unknown UGT3A1 status (n = 73); patients with NPC1 but also homozygous (n = 2), heterozygous (n = 2) and wild‐type (n = 3) for the T361G (p.C121 > G) mutation/polymorphism in UGT3A1 that inactivates the GlcNAc transferase, controls (n = 84) and NPC1 heterozygotes (n = 70). (B) From left to right: urine samples from individuals with NPC1 disease (confirmed by genotyping) but with unknown UGT3A1 status (n = 14), patients with NPC1 but also homozygous for the T361G (p.C121 > G) mutation/polymorphism in UGT3A1 that inactivates the GlcNAc transferase (n = 2), controls (n = 10) and NPC1 heterozygotes (n = 47). Data are expressed as median with interquartile range. (***P < 0.001).
The urinary excretion of NPCBA1, normalized to creatinine, was assessed in random urine specimens. NPCBA1 was markedly increased in most NPC1 patients, showing results above the range for healthy controls (P < 0.001) (Fig. 1B); however, NPCBA1 was not increased in the two patients shown by WES to have the defect in UGT3A1.
Results on dried blood spots confirmed the data obtained from plasma and urine although levels of NPCBA1 were below or very close to the LOQ, making a precise quantitation impossible.
Urinary excretion of hydrophilic metabolites of 3β,7β‐dihydroxy‐cholenoic acid and its GlcNAc conjugate (NPCBA1)
3β,7β‐dihydroxy‐5‐cholenoic acid (derived from7‐oxocholesterol) can potentially be modified in a number of ways leading to increased urinary excretion 28. These include conjugations with sulfate in position 3, GlcNAc in 7, taurine or glycine on the carboxylic group in 24, and the introduction of additional hydroxyl groups on the sterane ring (Fig. 2).
Figure 2.
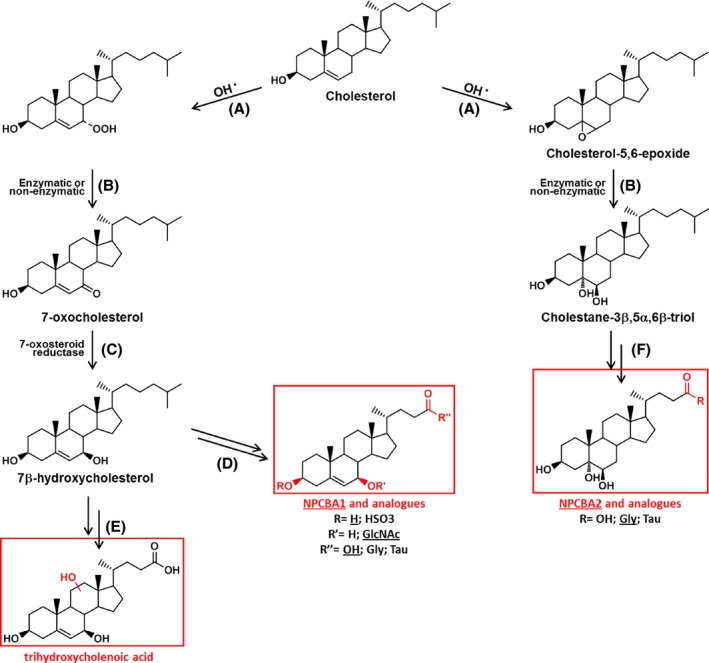
Proposed pathway for the hepatic synthesis of ‘NPC bile acids’. Hepatic formation of NPCBA1, NPCBA1 analogues (left pathway) and NPCBA2 (right pathway). Endolysosomal accumulation of unesterified cholesterol, associated with cellular oxidative stress (A,B), leads to the production of oxysterol molecules (7‐oxocholesterol and cholestane‐3β,5α,6β‐triol). The keto group of the 7‐oxocholesterol can be reduced by 7‐oxosteroid reductase (C) forming 7β‐hydroxycholesterol which, upon further enzymatic modifications, can lead to the series of NPCBA1 (D) or, alternatively to the formation of a trihydroxycholenoic acid (E). In a similar way, NPCBA2 series can be produced, via enzymatic reactions (F), from cholestane‐3β,5α,6β‐triol.
These bile acids, being highly conjugated and thus extremely hydrophilic, were easily quantified by UPLC‐MS/MS in urine of NPC1 patients but were not detectable or only present in traces in plasma and blood samples. Specifically, we observed urinary excretion of the following bile acids conjugated to N‐acetyl glucosamine (SRM transitions in square brackets): 3β‐hydroxy‐7β‐glucosaminyl‐5‐cholenoic acid (NPCBA1) [591.9 > 389.0], glycine conjugate of 3β‐hydroxy‐7β‐glucosaminyl‐5‐cholenoic acid (NPCBA1‐Gly) [649.4 > 74.0], taurine conjugate of 3β‐hydroxy‐7β‐glucosaminyl‐5‐cholenoic acid (NPCBA1‐Taur) [699.4 > 80.0], 3β‐sulfoxy‐7β‐glucosaminyl‐5‐cholenoic acid (NPCBA1‐Sulf) [672.4 > 97.0] and glycine conjugate of 3β‐sulfoxy‐7β‐glucosaminyl‐5‐cholenoic acid (NPCBA1‐Sulf‐Gly) [729.4 > 74.0]. Furthermore, an unsaturated compound (rt 1.49 min) characterized by the transition [462.4 > 74.0] was found to be elevated in NPC urine compared with controls or NPC carriers. Although the identity of the compound has not yet been fully established, it could possibly be the glycine conjugate of a trihydroxy‐5‐cholenoic acid and be produced by metabolism of 7‐oxocholesterol. When compared with the urinary measurement of NPCBA1, the pooled measurement of the 6 bile acids derived from 7‐oxocholesterol demonstrated an improvement in the separation between NPC1 patients and carriers (P < 0.001) and between NPC1 patients and controls (P < 0.001) (Fig. 3).
Figure 3.
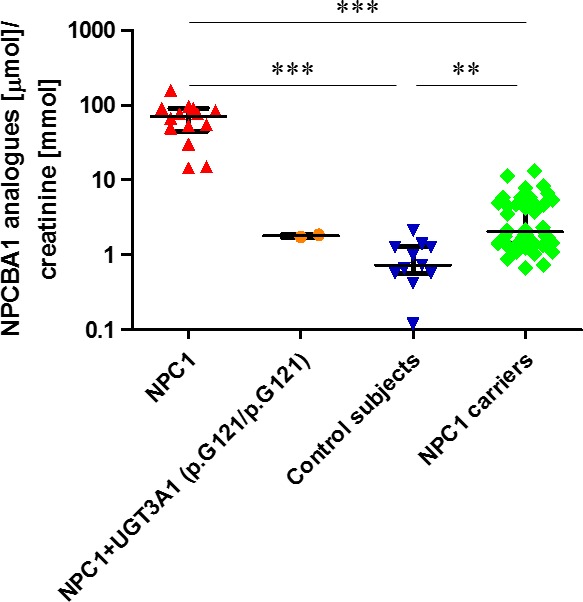
Urinary excretion of NPCBA1 analogues. Urinary excretion of NPCBA1 analogues in (from left to right): individuals with NPC1 disease (confirmed by genotyping) but with unknown UGT3A1 status (n = 14), patients with NPC1 but also homozygous for the T361G (p.C121 > G) mutation/polymorphism in UGT3A1 that inactivates the GlcNAc transferase (n = 2), controls (n = 10), and NPC1 heterozygotes (n = 47). Data are expressed as median with interquartile range. (***P < 0.001). Note: Log scale.
Investigation of NPCBA2 (probably 3β,5α,6β‐trihydroxycholanoyl‐glycine) in biological fluids from NPC1 patients
In the light of the data obtained for NPCBA1, we assumed that cholestane‐3β,5α,6β‐triol would be converted into a bile acid in a similar way to 7‐oxocholesterol (Fig. 2). The expected cholestane‐3β,5α,6β‐triol‐derived bile acid is an isomer of cholic acid which could be further conjugated with glycine or taurine to produce isomers of glyco‐ and tauro‐cholic acid. Therefore, we investigated the accumulation of these hypothesized molecules in biological fluids, via UPLC‐MS/MS. Interesting results were obtained by the examination of the SRM chromatograms (transitions in square brackets) of glycocholic acid and isobaric species [464.4 > 74.0]. As expected, an isobaric species of glycocholic acid, NPCBA2 (rt 1.44 min compared with glycocholate 1.67 min) was seen in samples from NPC patients in greater amounts than in samples from heterozygotes and controls (Fig. S1A).
Measurement of the plasma level of NPCBA2 (Fig. 4A) demonstrated a significant accumulation in NPC1 patients compared with NPC1 carriers and controls. In fact, the plasma concentration of the compound in affected participants (median = 251 nm; mean = 321 nm) was more than 10× higher than in healthy controls (median = 20 nm; mean = 21 nm) and no overlap was observed between the two groups (P < 0.001). Comparison of the NPC1 patients with the carriers indicated a small overlap but still a highly significant difference between the two groups (P < 0.001). Similar results have been obtained for dried blood spot analysis (Fig. 4B), although no overlap between NPC patients and carriers was observed here (albeit with smaller numbers). Moreover, a good correlation (R 2 = 0.96) was demonstrated (Fig. S2) between the amount of NPCBA2 extracted from a 10 μL plasma spot and a 6 mm blood spot, estimated to contain 12.4 μL of blood 33. Finally, the analysis of random urine samples showed a higher urinary excretion of NPCBA2 in NPC1 patients than in carriers (P < 0.001) and controls (P < 0.001) but with a small overlap between the groups, as seen with the plasma results (panel C, Fig. 4). The plasma concentration of NPCBA2 (which is produced without GlcNAc conjugation) was found to be above the control ranges in all NPC1 patients, including the two with the homozygosity for p.C121G (Fig. 4A), consistent with the hypothesis that the failure to detect an increase in NPCBA1 and analogues in NPC1 patients can be due to the presence of a common mutation in the UGT3A1 gene.
Figure 4.
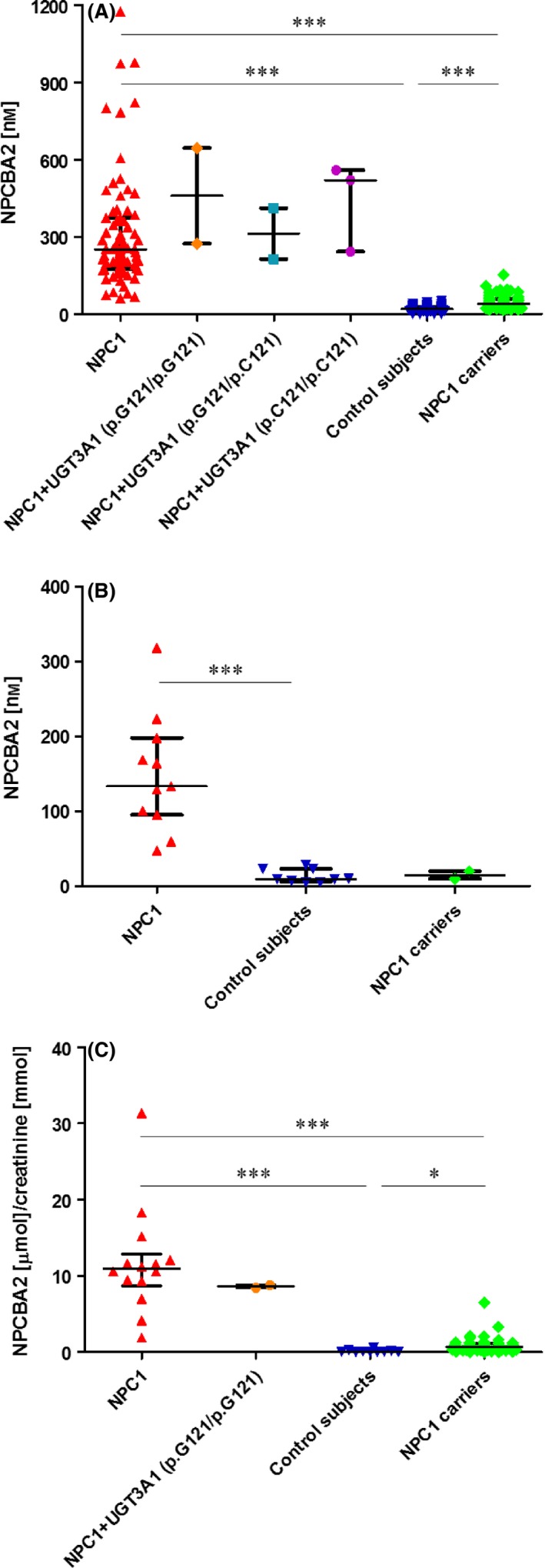
NPCBA2 in dried plasma spots (A), dried blood spots (B) and random urine samples (C). (A) Concentration (nmol·L−1) of NPCBA2 in plasma samples from individuals with NPC1 disease (confirmed by genotyping) with unknown UGT3A1 status (n = 73), patients with NPC1 and known UGT3A1 status: homozygous (n = 2), heterozygous (n = 2) and wild‐type (n = 3) for the T361G (p.C121 > G) mutation/polymorphism in UGT3A1 that inactivates the GlcNAc transferase, controls (n = 84) and NPC1 heterozygotes (n = 70). (B) Concentration (nmol·L−1) of NPCBA2 in dried blood spots from individuals with NPC1 disease (confirmed by genotyping) and unknown UGT3A1 status (n = 11), controls (n = 9) and NPC1 heterozygotes (n = 2). (C) Excretion of NPCBA2 (μmol·mmol−1 creatinine) in random urine samples from individuals with NPC1 disease (confirmed by genotyping) with unknown UGT3A1 status (n = 14), patients with NPC1 but also homozygous for the T361G (p.C121 > G) mutation/polymorphism in UGT3A1 that inactivates the GlcNAc transferase (n = 2), controls (n = 10) and NPC1 heterozygotes (n = 47). Data are expressed as median with interquartile range. (*P < 0.05; ***P < 0.001).
Correlation between NPC bile acids and oxysterols in plasma
The correlations between the levels of NPCBA1 and NPCBA2 and their putative respective precursors, 7‐oxocholesterol and cholestane‐3β,5α,6β‐triol, were assessed on eight plasma samples from untreated patients (Fig. 5). The oxysterol concentrations in these samples had been measured by HPLC‐MS/MS 10 in a previous study 32. The magnitude of the correlation coefficients were 0.608 and 0.585, respectively, indicating that the bile acids and oxysterols are moderately correlated. This result is in keeping with the hypothesis that NPCBA1 and NPCBA2 are generated by metabolism of oxysterols (Fig. 2). However, Fig. 5A shows that one patient had elevated 7‐oxocholesterol but normal NPCBA1; the likely explanation is that this patient had the GlcNAc transferase defect; we did not have WES data to confirm or refute this. Other factors that could contribute to the relatively loose correlation between NPCBA1 and 2 and their oxysterol precursors include genetic and environmental factors affecting oxysterol and bile acid metabolism. Cytochrome P450 hydroxylases and sulfatases show genetic variation and are inducible by drugs and xenobiotics. Conjugation of bile acids with taurine as opposed to glycine is affected by age and diet. Nonetheless, the results strongly suggest that NPCBA1 and NPCBA2 are the main plasma metabolites of 7‐oxocholesterol and cholestane‐3β,5α,6β‐triol respectively.
Figure 5.
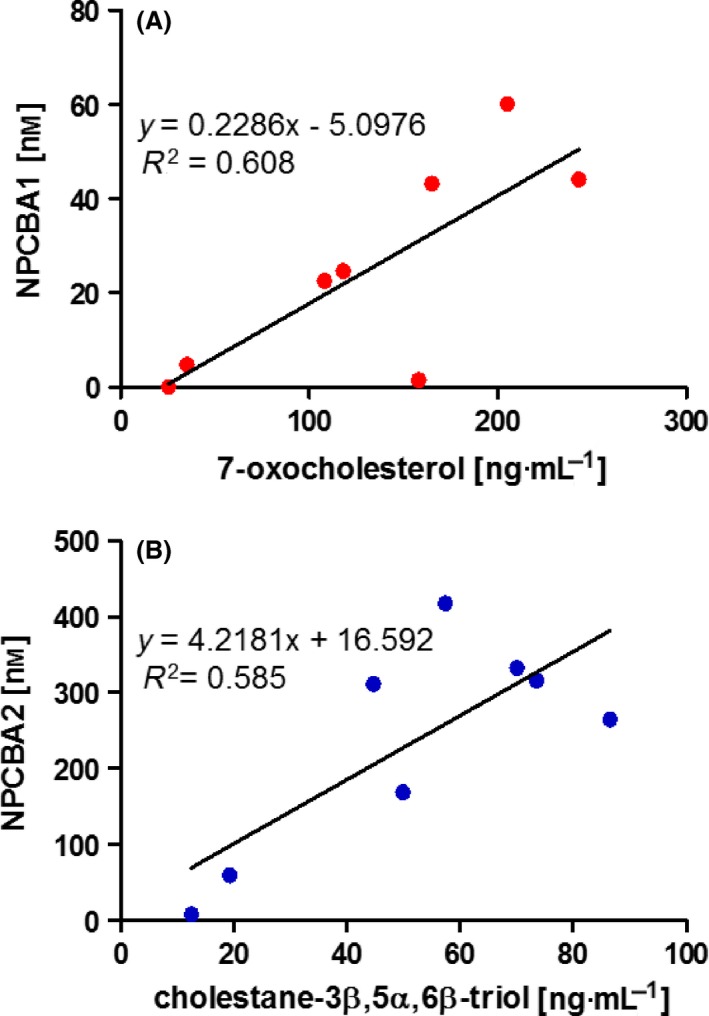
Correlation between NPC bile acids and oxysterols. (A) Correlation between plasma concentration of NPCBA1 (3β‐hydroxy,7β‐N‐acetylglucosaminyl‐5‐cholenoic acid) and its precursor 7‐oxocholesterol and (B) between NPCBA2 (3β,5α,6β‐trihydroxycholanoyl‐glycine) and cholestane‐3β,5α,6β‐triol, assessed on eight samples from untreated NPC patients.
Method validation
The diagnostic method described in this article has been validated according to the International Conference of Harmonisation (ICH) guidelines, section Q2(R1) ‘Validation of analytical procedures: text and methodology’ 35. The UPLC‐MS/MS identifiers (retention times, SRM transitions and acquisition parameters) for the internal standards and compounds that could be unequivocally identified in patient samples are listed in Table 1 and summaries of the validation parameters for quantitation are reported in Table 2. The method showed a good linearity for all the analytes over the range 0.1–250 nm, as demonstrated by the R 2 values. Limit of detection (LOD) and limit of quantitation (LOQ) values were found to be satisfactory and appropriate for the analysis of the samples with values lower than 10 nm for urine (Table 2), equivalent to 300 nm for plasma because of the 30‐fold dilution. As reported in Table 2, the analytical method is highly accurate, precise and reproducible, with an intra‐ and interday coefficient of variation lower than 20% and, in some cases, lower than 10%. In addition, matrix effects, which can be observed as ion enhancement or ion suppression, have been excluded by comparing the linearity of the response in methanol with that obtained in plasma, blood spots or urine samples (Fig. S3).
Table 2.
Validation results of the UPLC‐MS/MS method. Lower limit of detection (LOD), lower limit of quantitation (LOQ) and linearity assessed by inter‐ and intraday R 2 values. Accuracy, precision and intermediate precision assessed at three levels of concentration (15, 50 and 150 nm)
| GCDCA | TCDCA | GCA | TCA | |
|---|---|---|---|---|
| LOD (nm) | 2 | 2 | 2 | 2 |
| LOQ (nm) | 7 | 6 | 8 | 6 |
| R 2 intraday | 0.997 | 0.998 | 0.997 | 0.998 |
| R 2 interday | 0.988 | 0.995 | 0.988 | 0.995 |
| Accuracy % low level (15 nm) | 99 | 95 | 98 | 96 |
| Accuracy % medium level (50 nm) | 93 | 87 | 92 | 91 |
| Accuracy % high level (150 nm) | 98 | 94 | 95 | 90 |
| RDS% precision low level (15 nm) | 1.3 | 0.5 | 1.5 | 1.6 |
| RDS% precision medium level (50 nm) | 0.8 | 1.4 | 0.8 | 2.4 |
| RDS% precision high level (150 nm) | 3.2 | 2.7 | 5.1 | 5.3 |
| RDS% intermediate precision low level (15 nm) | 14.7 | 6.1 | 6.9 | 3.5 |
| RDS% intermediate medium level (50 nm) | 16.5 | 4.7 | 3.3 | 2.7 |
| RDS% intermediate high level (150 nm) | 9.1 | 3.6 | 8.0 | 6.0 |
Finally, as bile acids are a class of structurally related molecules sharing the presence of the sterane nucleus and often differing only in the position or orientation of substituents, the specificity and selectivity of the diagnostic test were investigated. An efficient and high‐resolution chromatographic separation was crucial in order to accurately and unequivocally quantify the novel set of bile acids and discriminate between them and their regio‐ or stereoisomers, which are often characteristic of other bile acid‐related disorders that can also present initially with cholestatic liver disease. Samples from patients affected by inborn errors of bile acid synthesis were used as controls to exclude a possible misdiagnosis (Table 1). In order to definitively diagnose NPC, it was essential to distinguish between NPCBA2 and its regioisomer glycocholic acid (Fig. S1A), as well as between NPCBA1 and its stereoisomer, 3β‐hydroxy‐7α‐glucosaminyl‐5‐cholenoic acid (3β,7α‐diOH‐Δ5,GlcNAc conjugate, Table 1), observed as a minor component of the bile acid profile in patients with 3β‐HSDH deficiency (Fig. S1B). Indeed, although urinary levels of NPCBA1 in patients with 3β‐HSDH deficiency fell within the NPC range, the presence of 3β‐hydroxy‐7α‐glucosaminyl‐5‐cholenoic acid (rt 1.64 min) was found to be specifically diagnostic for β‐HSDH deficiency. Retention times of additional isobaric species observed in different pathological conditions are reported in Table 1. Furthermore, a full bile acid profile was performed for these samples, in order to evaluate the possibility of using the proposed UPLC‐MS/MS method, not only for the diagnosis of NPC, but also for the detection of inborn errors of bile acid synthesis (Table 1). Some abnormal bile acids needed to be identified by using patient samples as the source of the reference compound. As an example: a urine spectrum from a patient with mutations in AKR1D1 suggested that a major component was a taurine‐conjugated oxo‐dihydroxy‐cholenoic acid (precursor ion m/z 510 with major product ion m/z 80). GC‐MS analysis, following deconjugation, showed the major bile acid was 7α,12α‐dihydroxy‐3‐oxo‐4‐cholenoic acid (by comparing with the synthesised compound). The retention time and optimized MS/MS parameters of the compound in this urine sample were used to identify taurine‐conjugated 7α,12α‐dihydroxy‐3‐oxo‐4‐cholenoic acid in subsequent UPLC‐MS/MS analyses.
Discussion
Mutations in the NPC1 gene usually cause a severe neurodegenerative disorder associated with cholestatic liver disease during infancy and persistent splenomegaly. Although diagnostic tests are currently available, sample collection, handling and analysis are not quick and simple; this combined with a variety of clinical presentations contributes to the diagnostic delay. To increase the chances that treatment will prevent the devastating neurological manifestations, it is vital to develop sensitive diagnostic methods that can be applied to samples that are easily obtained and easily sent to the reference laboratory. This article demonstrates the potential diagnostic value of novel bile acids derived from hepatic metabolism of 7‐oxocholesterol and cholestane‐3β,5α,6β‐triol, as depicted in Fig. 2. The 7‐oxocholesterol family comprises NPCBA1 (3β‐hydroxy‐7β‐N‐acetyl‐glucosaminyl‐5‐cholenoic acid), conjugated forms of this bile acid (3β‐hydroxyl group conjugated to sulfate and/or carboxyl group amidated with glycine or taurine) and a trihydroxycholenoic acid (precise structure not yet determined) present as the glycine conjugate. Measurement of NPCBA1 in a dried plasma/blood spot or a random urine sample is a useful rapid screening test for NPC1 but a ‘false‐negative’ result can be obtained for NPC1 patients who are homozygous for the missense polymorphism/mutation p.C121G (c.T361G) in UGT3A1 because these patients cannot make GlcNAc‐conjugated bile acids. Although measurement in urine of all the 7‐oxocholesterol derived bile acids produces good separation of the majority of NPC1 patients from controls and NPC1 heterozygotes, as do analyses of NPCBA1 in plasma and dried blood spots, the UGT3A1 variant homozygotes are not differentiated from controls. However, quantitation of NPCBA2 (the bile acid derived from cholestane‐3β,5α,6β‐triol, 3β,5α,6β‐trihydroxy‐cholanoyl glycine) in a dried plasma/blood spot is capable of complete separation between NPC1 patients and controls, although there is some overlap with NPC1 heterozygotes. The results obtained for patients whose whole exome sequencing data were available indicated that there are no false‐negative results for the UGT3A1 variant homozygotes when NPCBA2 is assayed. If a patient has a high suspicion index for NPC but normal level of NPCBA1, genetic testing could be carried out to look for the UGT3A1 mutation.
One of the main advantages of the proposed method, when compared with existing tests, is the stability of the analytes in blood spots, plasma spots (for stored samples) and urine samples. Thus, they can be obtained easily and transported easily to the analytical laboratory. A serious potential problem for oxysterol analysis is that blood and plasma samples contain large amounts of cholesterol which can be converted to 7‐oxocholesterol and cholestane‐3β,5α,6β‐triol by ex vivo auto‐oxidation; there is no potential precursor of the abnormal bile acids that could give rise to them outside the body. The proposed test, fully validated in accordance to the ICH Q2(R1) guidelines, is suitable not only for NPC, but for the simultaneous and selective determination of a variety of structurally related bile acids and thus for the diagnosis and treatment monitoring of several bile acid disorders.
This simple single step assay involving extraction of bile acids from dried blood or plasma spots by means of a 15 min sonication or simple dilution of urine samples is suitable for translation into routine clinical diagnostic laboratories being both robust and versatile, (capable of accurately and precisely measuring analytes over the concentration range of three orders of magnitude) as well as being rapid (UPLC run: 3.5 min) and cost‐effective. The simplified protocols make it possible not only to reduce turnaround times and increase the sample throughput, but also minimize experimental bias and human error.
Ideally, before being used clinically, a diagnostic test should be subjected to external validation; however, in the case of a rare disease such as NPC (incidence 1 in 100 000), it will take a considerable amount of time to collect samples for a validation set; the derivation data included 73 samples from untreated NPC patients collected from both Europe and USA over a period of 2 years.
Conflict of interest
This was an investigator‐led project but the funding (including the salary of F. M.) was provided by Actelion Pharmaceuticals UK Ltd, who market miglustat which is licensed in Europe for the treatment of Niemann‐Pick C disease.
Author contributions
The project was conceived by PTC. The early steps in setting up the method were undertaken by SC supervised by KM, PM and PC. The major body of work on method development, analysis and interpretation of the data and the writing of the article was undertaken by FM supervised by PTC. Samples from the previously studied cohort together with genotypes and severity scores were supplied by FP and FDP. The exome data (on the UGT3A1 variant) for these patients were provided by ERN, DV and CW. New samples from families attending Great Ormond Street were supplied by PG who was also instrumental in obtaining funding for the project. MM supplied sulfated bile acid standards. PM undertook a detailed review of the manuscript suggesting additional experiments.
Funding
Actelion Pharmaceuticals UK Ltd.
Supporting information
Fig. S1. Chromatographic separation between NPCBA2 and GCA (A) and between NPCBA1 and 3β‐hydroxy,7α‐N‐acetylglucosaminyl‐5‐cholenoic acid (B).
Fig. S2. Correlation between the concentrations of NPCBA2 in plasma and DBS.
Fig. S3. Matrix effect evaluated in plasma and urine.
Table S1. NPC1 genotypes of patients included in the study.
Acknowledgements
This project depended on a unique collection of plasma and urine samples from well‐characterized patients with NPC1, samples collected by an international team of clinicians listed in reference 33, to whom and to whose patients we are greatly indebted.
Edited by Ned Mantei
References
- 1. Kelly DA, Portmann B, Mowat AP, Sherlock S and Lake BD (1993) Niemann‐Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J Pediatr 123, 242–247. [DOI] [PubMed] [Google Scholar]
- 2. Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Ashmead JW and Wenger DA (2002) Niemann‐pick disease type C in neonatal cholestasis at a North American Center. J Pediatr Gastroenterol Nutr 35, 44–50. [DOI] [PubMed] [Google Scholar]
- 3. Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB et al (1997) Niemann‐Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277, 228–231. [DOI] [PubMed] [Google Scholar]
- 4. Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M and Lobel P (2000) Identification of HE1 as the second gene of Niemann‐Pick C disease. Science 290, 2298–2301. [DOI] [PubMed] [Google Scholar]
- 5. Peake KB and Vance JE (2010) Defective cholesterol trafficking in Niemann‐Pick C‐deficient cells. FEBS Lett 584, 2731–2739. [DOI] [PubMed] [Google Scholar]
- 6. Ory DS (2000) Niemann‐Pick type C: a disorder of cellular cholesterol trafficking. Biochim Biophys Acta 1529, 331–339. [DOI] [PubMed] [Google Scholar]
- 7. Walkley SU and Suzuki K (2004) Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta 1685, 48–62. [DOI] [PubMed] [Google Scholar]
- 8. Sturley SL, Patterson MC and Pentchev P (2009) Unraveling the sterol‐trafficking defect in Niemann‐Pick C disease. Proc Natl Acad Sci USA 106, 2093–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S and Brady RO (1985) A defect in cholesterol esterification in Niemann‐Pick disease (type C) patients. Proc Natl Acad Sci USA 82, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, Platt FM, Fujiwara H, Scherrer DE, Zhang J et al (2011) A sensitive and specific LC‐MS/MS method for rapid diagnosis of Niemann‐Pick C1 disease from human plasma. J Lipid Res 52, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette‐Mackie JE and Pentchev PG (2001) Niemann‐Pick disease type C: a lipid trafficking disorder In The Metabolic and Molecular Bases of Inherited Disease (Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW. and Vogelstein B, eds), pp. 3611–3634. McGraw Hill, New York, NY. [Google Scholar]
- 12. Ioannou YA (2000) The structure and function of the Niemann‐Pick C1 protein. Mol Genet Metab 71, 175–181. [DOI] [PubMed] [Google Scholar]
- 13. Ioannou YA (2001) Multidrug permeases and subcellular cholesterol transport. Nat Rev Mol Cell Biol 29, 657–668. [DOI] [PubMed] [Google Scholar]
- 14. Zhang JR, Coleman T, Langmade SJ, Scherrer DE, Lane L, Lanier MH, Feng C, Sands MS, Schaffer JE, Semenkovich CF et al (2008) Niemann‐Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J Clin Invest 118, 2281–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tint GS, Pentchev P, Xu G, Batta AK, Shefer S, Salen G and Honda A (1998) Cholesterol and oxygenated cholesterol concentrations are markedly elevated in peripheral tissue but not in brain from mice with the Niemann‐Pick type C phenotype. J Inherit Metab Dis 21, 853–863. [DOI] [PubMed] [Google Scholar]
- 16. Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO and Pentchev PG (1988) Niemann‐Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet 33, 331–348. [DOI] [PubMed] [Google Scholar]
- 17. Hulette M, Earl NL, Anthony DC and Crain BJ (1992) Adult onset Niemann‐Pick disease type C presenting with dementia and absent organomegaly. Clin Neuropathol 11, 293–297. [PubMed] [Google Scholar]
- 18. Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, Patterson MC, Wraith JE and Kolb SA (2012) Development of a suspicion index to aid diagnosis of Niemann‐Pick disease type C. Neurology 78, 1560–1567. [DOI] [PubMed] [Google Scholar]
- 19. Dvorakova L, Sikora J, Hrebicek M, Hulkova H, Bouckova M, Stolnaja L and Elleder M (2006) Subclinical course of adult visceral Niemann‐Pick type C1 disease. A rare or underdiagnosed disorder? J Inherit Metab Dis 29, 591. [DOI] [PubMed] [Google Scholar]
- 20. Yanjanin NM, Vélez JI, Gropman A, King K, Bianconi SE, Conley SK, Brewer CC, Solomon B, Pavan WJ, Arcos‐Burgos M et al (2010) Linear clinical progression, independent of age of onset in Niemann‐Pick disease type C. Am J Med Genet B Neuropsychiatr Genet 153B, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walterfang M, Yu‐Chien C, Imrie J, Rushton D, Schubiger D and Patterson MC (2012) Dysphagia as a risk factor for mortality in Niemann‐Pick disease type C: systematic literature review and evidence from studies with miglustat. Orphanet J Rare Dis 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patterson MC, Vecchio D, Prady H, Abel L and Wraith JE (2007) Miglustat for treatment of Niemann‐Pick C disease: a randomised controlled study. Lancet Neurol 6, 765–772. [DOI] [PubMed] [Google Scholar]
- 23. Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT and Wijburg F (2012) Recommendations for the diagnosis and management of Niemann‐Pick disease type C: an update. Mol Genet Metab 106, 330–344. [DOI] [PubMed] [Google Scholar]
- 24. Zervas M, Somers KL, Thrall MA and Walkley SU (2001) Critical role for glycosphingolipids in Niemann‐Pick disease type C. Curr Biol 11, 1283–1287. [DOI] [PubMed] [Google Scholar]
- 25. Ottinger EA, Kao ML, Carrillo‐Carrasco N, Yanjanin N, Shankar RK, Janssen M, Brewster M, Scott I, Xu X, Cradock J et al (2014) Collaborative development of 2‐hydroxypropyl‐β‐cyclodextrin for the treatment of Niemann‐Pick type C1 disease. Curr Top Med Chem 14, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helquist P, Maxfield FR, Wiech NL and Wiest O (2013) Treatment of Niemann‐pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alvelius G, Hjalmarson O, Griffiths WJ, Björkhem I and Sjövall J (2001) Identification of unusual 7‐oxygenated bile acid sulfates in a patient with Niemann‐Pick disease, type C. J Lipid Res 42, 1571–1577. [PubMed] [Google Scholar]
- 28. Iida T, Kakiyama G, Hibiya Y, Miyata S, Inoue T, Ohno K, Goto T, Mano N, Goto J, Nambara T et al (2006) Chemical synthesis of the 3‐sulfooxy‐7‐N‐acetylglucosaminyl‐24‐amidated conjugates of 3β,7β‐dihydroxy‐5‐cholen‐24‐oic acid, and related compounds: unusual, major metabolites of bile acid in a patient with Niemann‐Pick disease type C1. Steroids 71, 18–29. [DOI] [PubMed] [Google Scholar]
- 29. Maekawa M, Misawa Y, Sotoura A, Yamaguchi H, Togawa M, Ohno K, Nittono H, Kakiyama G, Iida T, Hofmann AF et al (2013) LC/ESI‐MS/MS analysis of urinary 3β‐sulfooxy‐7β‐N‐acetylglucosaminyl‐5‐cholen‐24‐oic acid and its amides: new biomarkers for the detection of Niemann‐Pick type C disease. Steroids 78, 967–972. [DOI] [PubMed] [Google Scholar]
- 30. Mackenzie PI, Rogers A, Treloar J, Jorgensen BR, Miners JO and Meech R (2008) Identification of UDP glycosyltransferase 3A1 as a UDP N‐acetylglucosaminyl‐transferase. J Biol Chem 283, 36205–36210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marschall HU, Griffiths WJ, Götze U, Zhang J, Wietholtz H, Busch N, Sjövall J and Matern S (1994) The major metabolites of ursodeoxycholic acid in human urine are conjugated with N‐acetylglucosamine. Hepatology 20, 845–853. [DOI] [PubMed] [Google Scholar]
- 32. te Vruchte D, Speak AO, Wallom KL, Al Eisa N, Smith DA, Hendriksz CJ, Simmons L, Lachmann RH, Cousins A, Hartung R et al (2014) Relative acidic compartment volume as a lysosomal storage disorder‐associated biomarker. J Clin Invest 124, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prinsen HC, Holwerda‐Loof NE, de Sain‐van der Velden MG, Visser G and Verhoefen‐Duif NM (2013) Reliable analysis of phenylalanine and tyrosine in a minimal volume of blood. Clin Biochem 46, 1272–1275. [DOI] [PubMed] [Google Scholar]
- 34. Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, Hemingway C, Marlow N, Rennie J, Baxter P et al (2010) Genotypic and phenotypic spectrum of pyridoxine‐dependent epilepsy (ALDH7A1 deficiency). Brain 133, 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. ICH (2005) Validation of analytical procedures: text and methodology Q2(R1). http://www.ich.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Chromatographic separation between NPCBA2 and GCA (A) and between NPCBA1 and 3β‐hydroxy,7α‐N‐acetylglucosaminyl‐5‐cholenoic acid (B).
Fig. S2. Correlation between the concentrations of NPCBA2 in plasma and DBS.
Fig. S3. Matrix effect evaluated in plasma and urine.
Table S1. NPC1 genotypes of patients included in the study.