

Abstract
Background
Cholesterol deficiency (CD), a newly identified autosomal recessive genetic defect in Holstein cattle, is associated with clinical signs of diarrhea, failure to thrive, and hypocholesterolemia.
Hypothesis/Objectives
The objective is to describe the clinicopathological phenotype of affected Holstein cattle homozygous for the causative apolipoprotein B gene (APOB) mutation.
Animals
Six Holstein cattle, 5 calves with a clinical history of chronic diarrhea, and 1 heifer with erosions in the buccal cavity and neurologic symptoms were admitted to the Clinic for Ruminants.
Methods
This case review included a full clinical examination, a complete blood count, blood chemistry, and measurements of cholesterol and triglycerides. The animals were euthanized and necropsied. A PCR‐based direct gene test was applied to determine the APOB genotype.
Results
All 6 animals were inbred, could be traced back to the sire Maughlin Storm, and were confirmed homozygous for the APOB mutation. The clinical phenotype included poor development, underweight, and intermittent diarrhea in the calves, and neurologic signs in the heifer included hypermetria and pacing. Hypocholesterolemia and low triglycerides concentrations were present in all animals. The pathological phenotype of all animals was steatorrhea with enterocytes of the small intestine containing intracytoplasmic lipid vacuoles. The peripheral nervous system of the heifer displayed degenerative changes.
Conclusions and clinical importance
Suspicion of CD in Holstein cattle is based on the presence of chronic diarrhea with no evidence of primary infections. Confirmation of the associated APOB gene mutation is needed. Additionally, the heifer demonstrated primarily signs of neurologic disease providing an unexpected phenotype of CD.
Keywords: Apolipoprotein B, Diarrhea, Lipid metabolism, Neuropathy
Abbreviations
- ABL
abetalipoproteinemia
- APOB
apolipoprotein B gene
- APOB
apolipoprotein B protein
- CD
cholesterol deficiency
- CNS
central nervous system
- FHBL
familial hypobetalipoproteinemia
- LDL
low‐density lipoprotein
- PNS
peripheral nervous system
- TEM
transmission electron microscopy
- VLDL
very low‐density lipoprotein
An inherited autosomal recessive genetic defect in Holstein calves, named cholesterol deficiency (CD), was reported for the first time in the summer of 2015 in Germany.1, 2 Homozygous calves demonstrate clinical signs of diarrhea unresponsive to treatment and failure to thrive.1 They suffer marked hypocholesterolemia and hypolipidemia, indicating an inherited fat metabolism disorder. These animals usually die within the first 6 months of life, and it has been assumed that about 80% of homozygous affected calves do not survive more than 1 year.1 Heterozygous carrier animals do not show any clinical signs but have reduced levels of blood cholesterol and triglycerides.1 Breeding organizations in Switzerland and other countries have reported an increasing occurrence of cases in Holstein cattle. The causal mutation has recently been identified in the apolipoprotein B gene (APOB).3 A 1,299‐bp insertion of a transposable element located in exon 5 of bovine APOB was shown to be perfectly associated with the disease and leads to truncated transcripts and aberrant splicing.3 The Canadian artificial insemination Holstein sire Maughlin Storm, born in 1991, has been identified genetically to be the first carrier bull.2 A significant recent advancement has been the development of a PCR‐based direct gene test, allowing the detection of animals with CD without pedigree information.3
In human patients, truncating mutations in APOB give rise to familial hypobetalipoproteinemia (FHBL). The APOB encodes 2 proteins via a mRNA‐editing process: the APOB‐48 protein is required for chylomicron production in the small intestine, and the APOB‐100 protein is expressed in the liver. The APOB‐100 protein is a structural component of very low‐density lipoprotein (VLDL) and its metabolic products and serves as the ligand for low‐density lipoprotein (LDL) receptor–mediated endocytosis of lipid particles.4 The clinical consequences of the mutation are well described in human medicine, but have not been described to date in cattle. The aim of this report is to describe the clinical and pathological phenotype associated with the genetic defect of APOB causing this new disease entity in Holstein cattle.
Materials and Methods
Five Holstein calves (3 females, 2 males) and a 2.5‐year‐old heifer were referred to the Clinic for Ruminants of the Vetsuisse Faculty, University of Bern, Switzerland. The calves had a history of failure to thrive and intermittent diarrhea unresponsive to treatment since birth. The mean age of the calves was 86 days (min‐max: 18–224 days), and the mean weight was 69 kg (min‐max: 41–153 kg), as shown in Table 1. The heifer (472 kg) had shown poor development and intermittent diarrhea in the first months of life, but had no longer shown diarrhea later on; it was presented to the Clinic with a primary complaint of lesions in the buccal cavity. A complete clinical examination of all animals was performed, and a CBC and blood chemistry profile was performed for calves numbered (no.) 1, 2, and 3. In addition, cholesterol and triglycerides were measured in all animals and were compared to a group of 5 healthy Holstein calves and a group of 5 inappetent Holstein calves with other diseases than diarrhea (Table 1). Fecal samples were sent for routine viral, bacterial, and parasitological analyses to exclude the most common pathogens causing calf diarrhea (rotavirus, coronavirus, cryptosporidia, coccidia, E. coli, and Salmonella spp.). Dietetic causes for diarrhea were excluded by clinical history. The animals were tested for bovine viral diarrhea virus (BVDV) using an antigen‐ELISA1 on a skin biopsy (≤180 days in life) or an EDTA blood sample (>180 days in life). All animals were euthanized (Phenobarbitalum natricum, 100 mg/kg i.v.)2 1–14 days after admission to the Clinic, based on the poor prognosis and suspicion of CD. A complete necropsy was performed on all animals. Fresh tissue samples were immediately fixed in 4% neutral buffered formalin, embedded in paraffin, cut at 4 μm, and stained with hematoxylin and eosin (HE). As lipids are removed from tissues during routine histological processing, for the visualization of lipids, optimal cutting temperature compound (O.C.T.) (Tissue‐Tek)3 –embedded frozen sections of small and large intestine and liver were stained with Sudan stain. Selected brain, spinal cord, and nerve sections of the heifer were stained with Luxol fast blue and Bielschowsky, respectively. Electron microscopical analysis was performed on small intestinal villi and liver of animals no. 5 and 6. Formalin‐fixed tissue samples were postfixed with 1% OsO4 in 0.1 M cacodylate buffer for 1 hour at 4°C and embedded in Epon.4 Semi‐thin sections were stained with toluidine blue and used to narrow down further regions of interest. Sections were collected on collodion‐coated 200 mesh copper grids.5 Sections were double stained with 0.5% uranyl acetate6 for 30 minutes at 40°C and 3% lead citrate7 for 10 minutes at 20°C in an Ultrastain8 and examined in a Philips CM12 transmission electron microscope9 at an accelerating voltage of 80 kV. Micrographs were captured with a Mega View III camera using the iTEM software.10 The presence of the APOB mutation was demonstrated by a PCR‐based direct gene test described elsewhere.3
Table 1.
Comparison of cholesterol and triglyceride values between affected and control groups
| Animal | Age (days) | Weight (kg) | Cholesterol (mmol/L) | Triglycerides (mmol/L) | ||||
|---|---|---|---|---|---|---|---|---|
| Min‐max | Min‐max | Median | IQR | Min‐max | Median | IQR | Min‐max | |
| Affected calves (n = 5) | 18–224 | 41–153 | 0.18 | 0.12–0.24 | 0.09–0.24 | 0.06 | 0.04–0.06 | 0.04–0.16 |
| Healthy controls (n = 5) | 156–264 | 168–285 | 2.29 | 1.85–2.59 | 1.79–3.20 | 0.31 | 0.27–0.40 | 0.27–0.43 |
| Sick controlsa (n = 5) | 23–115 | 60–123 | 2.55 | 2.25–2.64 | 1.63–3.72 | 0.25 | 0.21–0.35 | 0.21–0.35 |
Inappetent Holstein calves affected with other diseases than CD.
IQR, interquartile range; min, minimum; max, maximum.
Results
Clinical Phenotype
The physical examination of the 5 calves revealed a slightly reduced general condition. The calves were severely emaciated with normal appetite (Fig 1). The weight of the affected animals and of healthy Holstein cattle at the same age is shown in detail in Table 2. Examination of the digestive system revealed normal activity of the gastrointestinal tract, and percussion and succession auscultation were negative in both flanks. The feces had a yellow to olive‐green color, normal smell, and, at variable intervals over an observation period of up to 14 days, a fecal consistency which changed between soft (normal) and liquid (intermittent diarrhea). In addition, the feces of calf no. 1 contained undigested fibers (>2 cm in length). Examination of the cardiovascular, respiratory, urinary, musculoskeletal, and neurologic systems showed no abnormalities. The calves' gait was normal. The blood concentrations of cholesterol and triglycerides were distinctly below the values obtained from the 5 healthy control calves and from 5 calves affected with diseases other than diarrhea (Table 1). No abnormalities were detected in the CBC and chemistry profiles performed for 3 calves. A moderate amount of acanthocytes was observed in the blood smear of calf no. 2. Calf no. 3 showed a slight anemia with a hematocrit of 17% (norm: 19–34%) and a low selenium concentration (47 μg/L, norm: >50 μg/L). Coprological examinations revealed the presence of few Eimeria bovis oocysts in calf no. 1 and Cryptosporidium parvum in calf no. 2. All calves tested negative for BVDV, rotavirus, and coronavirus, and bacterial cultures for E. coli and Salmonella spp. were also negative.
Figure 1.

Holstein cattle at 7.5 months of age with cholesterol deficiency. The affected Holstein cattle (no. 1, right) was poorly developed and underweight in comparison to a healthy animal of the same age (left) at the farm.
Table 2.
Weight of the CD‐affected animals and of healthy Holstein cattle at the same age
| Animal | Age (days) | Weight (kg) | Reference values (mean ± SD) for the corresponding age17 (kg) |
|---|---|---|---|
| Case 1 | 224 | 153 | 214.9 ± 39.4 |
| Case 2 | 36 | 55 | 77.2 ± 18.7 |
| Case 3 | 18 | 40 | 53.1 ± 8.7 |
| Case 4 | 34 | 41 | 77.2 ± 18.7 |
| Case 5 | 97 | 57 | 118.5 ± 20.8 |
| Case 6 | 921 | 472 | 528.9 ± 99.4 |
The 2.5–year‐old and 4‐month pregnant heifer (no. 6) presented with a complaint of anorexia and buccal lesions. Infectious causes were excluded, and the stomatitis was treated symptomatically. In addition, the animal was in a moderately reduced general condition and slightly emaciated (Table 2). Despite resolution of the digestive problem, restored appetite and activity of the gastrointestinal tract, hypermetria, and pacing, suggestive of a diffuse cortical lesion, remained apparent upon neurologic examination. The cerebrospinal fluid showed a slight pleocytosis (9.7 cells/μL, norm: 3 cells/μL) with 39% monocytes and 61% lymphocytes. The pandy test for proteins was slightly positive. The blood concentrations of cholesterol (0.11 mmol/L, norm: 1.20–3.84 mmol/L) and triglycerides (0.03 mmol/L, norm: 0.19–0.51 mmol/L) were similar to those of the affected calves (Table 1). The heifer tested negative for BVDV. Coprological examination revealed the presence of a few Trichuris spp. eggs. Diarrhea was not observed in this animal.
Pathological Phenotype
Macroscopical examination showed that all animals were emaciated. The anus and perianal region of calves no. 1–5 were soiled with feces, which was interpreted as a sign of diarrhea. Calf no. 1 and the heifer (no. 6) had a moderate amount of fat reserves, whereas calves no. 2–5 were cachectic.
The small intestine of all animals was diffusely filled with a moderate amount of bright yellow, beige to lime‐green, liquid, and partially foamy to fatty content (Fig 2A). The mucosa of the distal two thirds of the small intestine was severely edematous and appeared whitish (Fig 2A). The large intestine was filled with a yellow to green liquid with a partially foamy to fatty content, consistent with steatorrhea. The rectal contents of calf no. 1 and the heifer (no. 6) appeared less liquid than those from calves no. 2–5.
Figure 2.
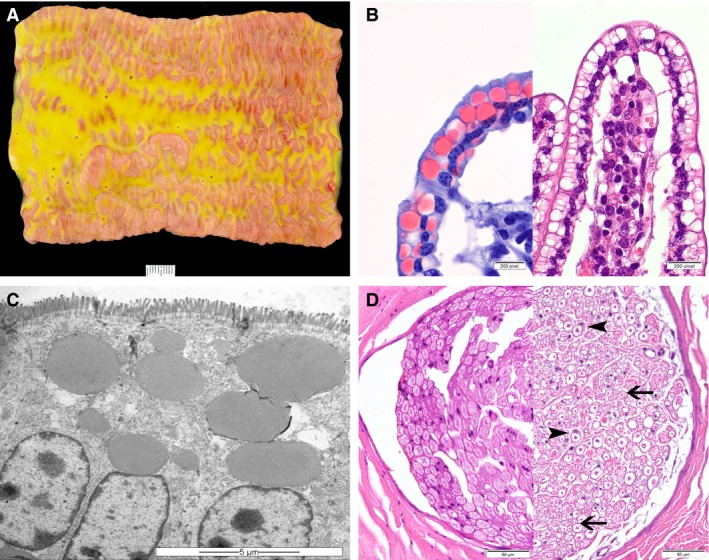
Lesions of congenital cholesterol deficiency in Holstein cattle. (A) Macroscopic appearance of the small intestine and its content of the heifer no. 6. The intestinal content is foamy and greasy, varying from light‐yellow to green in color, and the mucosa is edematous (scale bar = 1 cm). (B) In routinely processed histological sections of jejunum, many enterocytes contain optically empty cytoplasmic vacuoles, which are shown to represent lipid inclusions in Sudan‐stained frozen sections (left) (100×). (C) Electron micrograph of enterocytes containing fat vacuoles within the apical cytoplasm (scale bar = 5 μm). (D) Sciatic nerve of the heifer (no. 6) (right), showing thin and irregular myelin sheaths (arrow) with Schwann cell activation (arrowheads) in comparison to a control animal of the same age (left) (HE, 40×).
Histological changes were most prominent in the small intestine and accentuated in the jejunum in all animals. The enterocytes covering the tips of the villi contained large amounts of optically empty, round to oval vacuoles, ranging from 2 to 20 μm in diameter (Fig 2B). Multifocally, the nuclei of the affected enterocytes were displaced to the basal cell borders. In frozen sections, these intracytoplasmic vacuoles and large parts of the intestinal content were positively stained by Sudan stain indicating lipid origin (Fig 2B). Furthermore, multiple lacteals were moderately to severely dilated, correlating with the macroscopically visible edema. No pathomorphological or pathohistological lesions were observed in the liver. Transmission electron microscopy (TEM) corroborated the presence of large intracytoplasmic lipid droplets in the small intestinal enterocytes. The droplets filled most of the supranuclear cytoplasm but also occurred in an infranuclear position (Fig 2C). In other respects, organelles and intercellular spaces were typical. Furthermore, isolated lipid droplets were seen in hepatocytes but to a minor extent as compared with the enterocytes, excluding a hepatic steatosis on TEM level. Histologically, single dilated myelin sheaths containing fragmented axons and macrophages without any particular distribution pattern were observed in the central nervous system (CNS) of the heifer. The variation of nerve fiber size and the proportion of small nerve fibers appeared to be increased in the sciatic nerve of the heifer, compared to a control animal of the same age (Fig 2D). Myelin sheaths of numerous axons were decreased in thickness and, multifocally, hypertrophied Schwann cells enclosed the myelin sheaths. In the eyes, no retinal degeneration was present.
Genetic Analysis
Pedigree analysis indicated familial relationships among affected animals, and the distribution of affected animals in both genders was suggestive of an autosomal recessive genetic defect (Fig 3). Analysis of the genealogy of all 6 affected animals revealed a relation to the sire Maughlin Storm on the maternal and on the paternal side (Fig 3). This Holstein sire has been reported as the possible founder of the inherited CD disorder in previous investigations.1, 3 According to the Swiss cattle breeding organizations, approximately 25 cases in calves with diarrhea and inbreeding to this sire have been recorded in Switzerland in the last few months. Most of these calves died within a couple of days or weeks. An APOB mutation causing CD was recently identified partially based on the data provided in this investigation.3 The homozygous presence of the APOB insertion was subsequently confirmed in all 6 affected animals. The 4‐month‐old fetus of the heifer was tested heterozygous for the APOB insertion.
Figure 3.
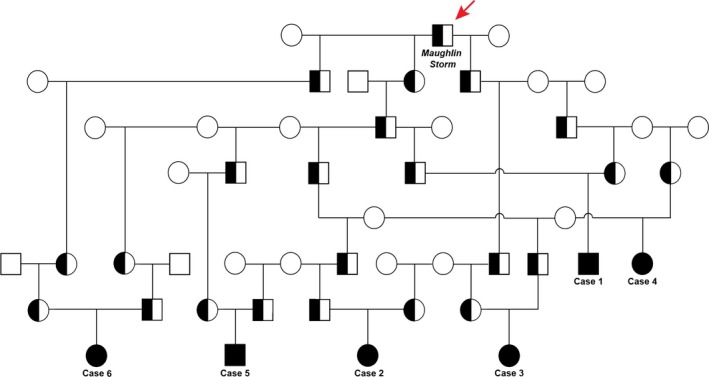
Pedigree of Holstein cattle with cholesterol deficiency. Males are represented by squares and females by circles. Affected animals are shown with full black symbols, while heterozygous carriers are indicated with a half‐filled symbol. Note that all 6 cases described in detail in this study can be traced back to a common ancestor confirmed as the carrier of the APOB mutation causing CD (red arrow).
Discussion
The most consistent clinical features in the CD calves were intermittent diarrhea and failure to thrive associated with hypocholesterolemia and low triglyceride concentrations. While the clinical signs were unspecific and of moderate intensity, the CD‐affected animals had distinctly lower blood cholesterol and triglyceride values than the control calves. The 2.5‐year‐old heifer showed similarly low values in cholesterol and triglycerides, but the most prominent clinical features (after resolution of the stomatitis) were the neurologic symptoms. However, this animal had also shown intermittent diarrhea as a calf, based on which the pedigree of the heifer was reviewed, and it was subjected to a genetic test. Indeed, the pathological phenotype of the homozygous APOB mutant heifer was comparable to the phenotype of the 5 affected calves. Taking the pathological findings of steatorrhea, the histological and TEM phenotype of lipid vacuole accumulation within the enterocytes of the tips of the villi in the small intestine and the clinical symptoms of hypocholesterolemia, low triglyceride concentration, and the neurologic symptoms into account, bovine CD is highly similar to human FHBL.4, 5 Acanthocytosis, described as one of the earliest laboratory features of FHBL in humans, was present in one calf.5, 6 Over 60 truncating APOB mutations have been identified as causes for FHBL.5 We previously demonstrated that the mutant proteins in the liver do not fulfill any physiological function in cattle homozygous for the bovine APOB mutation.3 Therefore, it is highly probable that both physiological APOB protein isoforms are missing. Currently, it is not known if mutant APOB proteins are expressed in CD‐affected cattle or not, and this should be subject to further investigation.3 Impaired function of human APOB‐100 protein results in decreased triglyceride export (by LDL receptor–mediated endocytosis) from the liver, which in turn leads to the development of fatty liver.7 None of the investigated cattle had signs of fatty liver (hepatic steatosis), leading to the assumption that hepatic steatosis due to APOB‐100 protein dysfunction is not a feature of CD in cattle. Alternatively, the absence of hepatic steatosis in our CD calves and heifer might be explained by the differences in fat metabolism between ruminants and humans. In humans, most of the de novo fatty acid synthesis takes place in the liver with glucose as a basic substrate.8, 9 In ruminating animals, acetate absorbed from the rumen is the substrate for direct de novo synthesis of fatty acids.8 Acetate is produced by microbial fermentation of cellulose in the rumen and stored in adipose tissue as well as used for milk fat synthesis in the mammary gland of lactating animals.8, 10 Thus, the main site of fatty acid synthesis in ruminants is the adipose tissue and the mammary gland, not the liver.8, 11 However, this applies to ruminating animals, ie, in our case animals no. 1 and 6, as calves no. 2–5 were still mostly fed with milk. Therefore, intestinal absorption may still have played a more important role in lipid intake and digestive processes in these calves may have been more comparable to the human fat metabolism. This explanation is supported by the fact that the calves on a diet milk based were cachectic, whereas the 2 ruminating animals had still some abdominal fat reserves. This might suggest that the specific metabolism of ruminants allows for the accumulation of some fat reserves in CD animals as soon as they start ruminating. In humans, truncation mutations in APOB affecting the APOB‐48 protein were shown to result in defective chylomicron formation in the small intestine.4, 5 This protein is exclusively synthesized in enterocytes of the small intestine.12 The histologically visible accumulation of lipid vacuoles within the enterocytes in the CD‐affected cattle indicates that the enterocytes of these animals are capable of resorbing fat from the ingesta. The hypocholesterolemia and low triglycerides concentrations, however, demonstrate that the transfer of these lipids from the enterocytes into the blood is impaired or even absent. Thus, these histological findings further support a chylomicron formation defect due to the lack of the APOB‐48 protein, as it has been described in FHBL.4, 12 In human patients, FHBL is additionally characterized by malabsorption of lipid‐soluble vitamins (A, D, E, K), leading to retinal degeneration, neuropathy, and coagulopathy.6 These neurologic disorders are associated with cerebellar dysfunction and compromising of the posterior column function with demyelination in the CNS and peripheral nervous system (PNS).5, 6 We assume that the examined calves were too young to present clinical and pathological signs of chronic cholesterol and lipid deficiency and/or chronic lipid‐soluble vitamin deficiencies. In contrast, the hypermetria observed in the heifer, which was first interpreted as a sign of cortical dysfunction in combination with abnormal gait (pacing), might also have been due to cerebellar dysfunction.13 In the heifer, the histological lesions were mild in general, but were most prominent in the PNS and only minimal in the CNS. Peripheral neuropathy has been described in humans in association with abetalipoproteinemia (ABL), a similar genetic fat metabolism disorder.14 However, this needs further study of additional older affected animals, including examination of semithin sections and ultrastructural studies. We assume that the mild changes in the sciatic nerve observed in the heifer might be a consequence of APOB mutation. Therefore, it represents a possible further histological feature of APOB‐associated CD in cattle. Because no clinical PNS symptoms were present, the question whether the histological lesions are of clinical relevance remains open. Severe demyelination within the CNS as described in FHBL in humans5, 6 was not visible by light microscopy in the heifer. Therefore, we cannot exclude a functional cause of the clinical CNS symptoms. Calves with CD are described to usually die within the first 6 months of life.1 Thus, the case of the 2.5‐year‐old heifer is a rare example of an adult animal surviving the initial phase of CD. The heifer may have established chronic cholesterol and lipid deficiency and/or chronic lipid‐soluble vitamin deficiencies as described in FHBL. Upon treatment, young human patients with FHBL can expect to return to a normal growth rate, although they may not reach their full growth potential.4 In fact, it is proposed that FHBL might be a longevity syndrome, as a result of low LDL cholesterol in the bloodstream,15 leading to absence of atherosclerosis.16 The treatment described for humans, ie, total fat restriction and lipid‐soluble vitamin supplementation,5, 6 is not applicable in cattle. Exceptionally older CD cattle, like the heifer in our study, are particularly interesting animals for further investigation concerning the pathogenesis of the disease. The possibility of a natural large animal model for human FHBL could be explored with further investigations.
The main limitations for the diagnosis of CD in cattle are the unspecific clinical symptoms of diarrhea and failure to thrive in young calves. It is important, as demonstrated by the affected animals described here which belong to an age group most susceptible for various diarrhea‐causing pathogens, to first exclude common agents causing diarrhea. However, as shown by the findings of 2 calves excreting oocysts of Eimeria bovis and Cryptosporidium parvum and, in the case of the heifer, Trichuris spp., CD can be present concomitantly with pathogens causing diarrhea. In case of intermittent diarrhea resistant to treatment in young Holstein cattle, analysis of total cholesterol and triglycerides can lead to a suspicion of CD. Pathological investigations are only diagnostic, if samples of the small intestine are immediately fixed in formalin (within minutes after euthanasia). Otherwise, the lipid vacuoles in the enterocytes of the villi tips are lost due to immediate autolytic changes in the small intestine. A clinical or pathological suspicion of CD should be confirmed using the gene test detecting the APOB mutation after consideration of pedigree information indicating inbreeding linked to the founder sire Maughlin Storm.3 This newly developed diagnostic tool will allow a reduction in unnecessary treatment costs, use of antibiotics, and time‐consuming care of affected animals. The early diagnosis of CD will enable targeted eradication of the APOB mutation from the Holstein population and thus prevent economical losses in the future.
Acknowledgments
The authors are grateful to all farmers involved in the project and to the referring veterinarians for notification of the affected animals, as well as the Agroscope in Posieux, Switzerland, for providing blood samples from healthy controls. The authors thank Swissgenetics and the Swiss cattle breeding associations (www.swissherdbook.ch and www.holstein.ch) for supporting the sampling.
Funding: No external funding was used for the project.
Conflict of Interest Declaration: Authors declare no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
This work was performed at the Clinic for Ruminants, the Institute of Animal Pathology, the Institute of Genetics, the Division of Veterinary Anatomy, and the Division of Neurological Sciences of the Vetsuisse Faculty, University of Bern, Switzerland.
Footnotes
IDEXX BVDV Ag/Serum Plus; IDEXX Diavet AG, Baech, Switzerland
Esconarkon ad us. vet., Streuli Pharma AG, Uznach, Switzerland
Tissue‐Tek O.C.T. Compound, Biosystems Switzerland AG, Muttenz, Switzerland
Epon, FLUKA, Buchs, Switzerland
Copper grids, Electron Microscopy Sciences, Hatfield, PA
Uranyl acetate, Sigma Aldrich, Steinheim, Germany
Lead citrate, Laurylab, Saint Fons, France
Ultrastain, Leica, Vienna, Austria
Philips CM12 transmission electron microscope, FEI, Eindhoven, Holland
iTEM software, version 5.2; Olympus Soft Imaging Solutions GmbH, Münster, Germany
References
- 1. Kipp S, Segelke D, Schierenbeck S, et al. A new Holstein Haploytpe affecting calf survival. Interbull Bull 2015;49:49–53. [Google Scholar]
- 2. Vanraden P, Null D. Holstein Haplotype for Cholesterol Deficiency (HCD) [Internet]. Council on Dairy Cattle Breeding; 2015. [cited 2015 Dec 20]. Available at: https://www.cdcb.us//reference/changes/HCD_inheritance.pdf.
- 3. Menzi F, Besuchet‐Schmutz N, Fragnièr M, et al. A transposable element insertion in APOB causes cholesterol deficiency in Holstein cattle. Anim Genet 2016;47:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hooper AJ, van Bockxmeer FM, Burnett JR. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit Rev Clin Lab Sci 2005;42:515–545. [DOI] [PubMed] [Google Scholar]
- 5. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014;25:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis 2014;37:333–339. [DOI] [PubMed] [Google Scholar]
- 7. Tanoli T, Yue P, Yablonskiy D, Schonfeld G. Fatty liver in familial hypobetalipoproteinemia: roles of the APOB defects, intra‐abdominal adipose tissue, and insulin sensitivity. J Lipid Res 2004;45:941–947. [DOI] [PubMed] [Google Scholar]
- 8. Vernon RG. Lipid metabolism in the adipose tissue of ruminant animals. Prog Lipid Res 1980;19:23–106. [DOI] [PubMed] [Google Scholar]
- 9. Loeffler G. Integration und hormonelle Regulation des Energiestoffwechsels In: Heinrich PC, Müller M, Graeve L, eds. Löffler/Petrides Biochemie und Pathobiochemie, 9th ed Berlin: Springer; 2014:474. [Google Scholar]
- 10. Bell AW. Lipid metabolism in liver and selected tissues and in the whole body of ruminant animals. Prog Lipid Res 1980;18:117–164. [DOI] [PubMed] [Google Scholar]
- 11. Moore J, Christie W. Lipid metabolism in the mammary gland of ruminant animals. Prog Lipid Res 1979;17:347–395. [DOI] [PubMed] [Google Scholar]
- 12. Kane JP, Hardman DA, Paulus HE. Heterogeneity of apolipoprotein B: isolation of a new species from human chylomicrons. Proc Natl Acad Sci U S A 1980;77:2465–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Metre DC, Mackay RJ. Localization and differentiation of neurologic diseases In: Large Animal Internal Medicine, 5th ed St‐Louis, Missouri: Mosby Elsevier; 2015: 107–130. [Google Scholar]
- 14. Wichman A, Buchthal F, Pezeshkpour GH, Gregg RE. Peripheral neuropathy in abetalipoproteinemia. Neurology 1985;35:1279–1289. [DOI] [PubMed] [Google Scholar]
- 15. Glueck CJ, Gartside P, Fallat RW, et al. Longevity syndromes: familial hypobeta and familial hyperalpha lipoproteinemia. J Lab Clin Med 1976;88:941–957. [PubMed] [Google Scholar]
- 16. Kahn JA, Glueck CJ. Familial hypobetalipoproteinemia. Absence of atherosclerosis in a postmortem study. J Am Med Assoc 1978;240:47–48. [DOI] [PubMed] [Google Scholar]
- 17. Heinrichs AJ, Losinger WC. Growth of Holstein dairy heifers in the United States. J Anim Sci 1998;76:1254–1260. [DOI] [PubMed] [Google Scholar]